Aqueous-based route toward noble metal nanocrystals: morphology-controlled synthesis and their applications
Qiang
Yuan
ab and
Xun
Wang
*a
aDepartment of Chemistry, Tsinghua University, Beijing, 100084, P. R. China. E-mail: wangxun@mail.tsinghua.edu.cn
bDepartment of Chemistry, Guizhou University, Guiyang, Guizhou province 550025, P. R. China
First published on 27th August 2010
Abstract
Noble metal nanocrystals with controlled morphologies play important role in many fields, such as catalysis and SERS, etc. To date, solution-based methods developed to synthesize nanocrystals mainly exploit organic reagents as solvents including polyol, oleic acid/oleylamine, toluene, diphenyl ether and so on. In organic solvent systems, expensive organometallic precursors and toxic organic solvents are often used, bringing about substantial environmental issues. In this article, based on our recent endeavors, we will summarize facile, general aqueous methods to synthesize monodisperse, uniform, single and binary noble metal nanostructures and their applications in liquid fuel cells. We believe this review article will be useful to those devoted to the catalysis and nanocrystal fields.
 Qiang Yuan | Dr Qiang Yuan is currently a postdoctoral research fellow in Professor Xun Wang's group. He received his BS degree from department of chemical engineering, Fuzhou University in 1997, and MS and PhD degree from department of chemistry, Xiamen University in 2001 and 2006, respectively. He joined the faculty of chemistry department and was appointed as associate professor of Guizhou University in 2006. His current research interests include controllable synthesis, understanding formation mechanism and application in catalysis of nanocrystals. |
 Xun Wang | Professor Xun Wang received his PhD degree from the Department of Chemistry, Tsinghua University in 2004. He joined the faculty of the Department of Chemistry, Tsinghua University in 2004, and was promoted to associate professor and full professor in 2005 and 2007, respectively. His main awards include National Science Fund for Distinguished Young Scholars (2007) and IUPAC Prize for Young Chemists (2005). His current research interests include synthetic methodology, self-assembly and properties of inorganic nanocrystals. |
Introduction
Noble metal nanocrystals have been paid great attention because of their many important properties in fields such as catalysis,1–7 optics,8–13 sensors,14–16 and medical therapy.17–20 Furthermore, previous work has shown that the special properties of noble metal nanocrystals were often size- and shape-dependent.21–25 For example, in the case of a catalytic study for ethylene and pyrrole hydrogenation, the Pt nanocrystals exhibit obvious size- and shape-dependent catalytic properties.26 Ethylene hydrogenation rates over the Pt nanocrystals were independent of both size and shape and comparable to Pt single crystals. For pyrrole hydrogenation, the nanocubes enhanced the ring-opening ability and thus showed a higher selectivity to n-butylamine as compared to nanopolyhedra. Au and Ag nanocrystals often exhibit unique optical properties in the visible region. Commonly, symmetric spherical crystals have a single scattering peak. However, the rods,27 triangular prisms28 and cubes,29 which expose more corners and edges compared with spherical crystals, displayed multiple scattering peaks. In recent years, binary noble metal nanostructures have been attracting more extensive interest because of their superior or tunable properties relative to their mono-component counterparts.30–36 In binary metal system, the physical and chemical properties can be tailored by tuning the composition of crystals. For instance, the optical extinction mode can be tuned by bimetallic Au and Ag nanostructures37 and Pt–Cu alloys displayed composition-dependent electrocatalytic activity for formic acid oxidation.38 These reported cases have shown that the physical and chemical properties of nanocrystals can be tuned by elaborate control of the size, shape and composition of the crystal. Therefore, in summary, how to control precisely the size, shape and composition of noble metal nanocrystals to maximise the utilization and exposure of atoms is thought-provoking in chemistry and materials science due to their scarcity.Up to now, many synthesis methods have been developed to synthesize nanomaterials. Whereas the synthesis strategies can be mainly summarized as two approaches: physical (top-down) approach (e.g. evaporation and laser ablation) and chemical (bottom-up) approach. Among them, the bottom-up, solution-based approach is widely used to produce monodisperse, uniform and sub-10 nm nanostructures.39,40 The solvent plays a key role in the solution-based approaches. To date, solution-based methods developed to synthesize nanocrystals that mainly exploit organic reagents as solvents including polyol, oleic acid/oleylamine, toluene, diphenyl ether and so on. In organic solvent systems, expensive organometallic precursors and toxic organic solvents are often concerned, bringing about substantial environmental issues.
Water, as an environmentally friendly solvent with the most abundant resources, can avoid these concerns as most metal chlorides and nitrates can be dissolved in it. However, compared with organic solvent systems, limited successful examples have been achieved to produce uniform noble metal nanocrystals in an aqueous solution.28,30,36,41–43 In this article, based on our recent endeavors, we summarize facile, general aqueous methods to synthesize monodisperse, uniform, single and binary noble metal nanostructures and their applications in liquid fuel cells.
1. Single noble metal nanocrystals
The final shape and size of nanocrystals is determined by an integrative process, which concerns the combined effect of reagents (precursor, ligand, reducing reagent, additive and solvent) and reaction temperature and time. Furthermore, it is well-known that the size and shape of crystals are mainly controlled by thermodynamic and kinetic factors. Spherical particles are often formed by thermodynamic control processes in colloidal chemistry. To obtain a variety of nonspherical crystal morphologies, inorganic/organic additives were often introduced into the synthesis process to tune the crystal growth through kinetic control. Studies have shown that additives such as Fe3+,44 Ag+,45 Cu2+,46 Cl−,47 Br−,41 I−,48 CTAB49 and ligands50–52 played a very important role in controlling the particle morphology in different synthesis systems. Our recent work41 showed high-quality Pd nanocubes with a mean diameter of 7.6 ± 0.4 nm and a selectivity beyond 95% was achieved in an aqueous solution containing PdCl2, PVP, sodium lauryl sulfate (SLS) and NaBr (Fig. 1a and b). These results indicated that SLS mainly acts as both shape and size controllers. Meanwhile, Br− also plays the role of shape controller and PVP acts as both reducing agent and stabilizing agent. These suggestions were supported by control experiments. In the absence of SLS, the product consists of mixed morphologies including rods, tetrahedra, triangles, plates and other polyhedra (Fig. 1c) and the size of mixed nanocrystals cannot be controlled. Nevertheless, without NaBr, the product consists of networked nanowires, short nanorods and nanoparticles with a relative uniform diameter below 10 nm (Fig. 1d). The Rh nanocubes with size of 3–4 nm and selectivity beyond 95% also can be acquired in the present system (Fig. 1e and f). | ||
| Fig. 1 (a) TEM and (b) HRTEM images of Pd nanocubes. The inset in (b) is the FT pattern of the selected area; TEM images of Pd nanocrystals synthesizes without SLS (c) and without NaBr (d); TEM (e) and HRTEM (f) images of Rh nanocubes. Reproduced with permission from ref. 41. Copyright 2009 the Royal Society of Chemistry. | ||
Interestingly, high-selectivity Rh nanocrystals with a variety of morphologies can be tuned by halogen anions (F−, Cl−, Br−, I−). Rh nanodendrites (Fig. 2a and b) with nearly 100% selectivity and narrow size distribution from 18 to 25 nm have been achieved in an aqueous solution containing RhCl3, PVP, SLS and NaF. The HAADF-STEM image (Fig. 2d) reveals the nanodendrites consist of many branches, each branch is along different direction, and the maximum number of branches can be beyond twenty. The HRTEM image (Fig. 2c) shows each branch of a single rhodium nanodendrite is shaped like a rice grain with sharp tip and an edge-to-edge width W of about 4.8 nm, and an apex-to-apex length L around 7.3 nm. The interval between two lattice fringes was examined to be 0.220 nm, closed to the (111) lattice spacing of the fcc rhodium. Moreover, the lattice fringes of each branch are along the same direction, which indicates the branch is single crystalline.
 | ||
| Fig. 2 The TEM and HRTEM images of rhodium nanostructures synthesized in the presence of NaF, NaCl, NaBr and NaI, respectively: (a)–(d) dendrites, NaF; (e) horned particles, NaCl; (f) cubes, NaBr; (g) and (h) network-shaped wires, NaI. Reproduced with permission from ref. 43. Copyright 2010 American Chemical Society. | ||
The dendritic structure is one of the most complex structures synthesized by colloidal chemistry processes thus far and was produced mainly via kinetic control,30,53 because their anisotropic shapes give them larger surface areas, which is metastable and energetically unfavorable for thermodynamic control. The formation mechanism of rhodium dendrites was deduced as following. In the first stages, the Rh3+ ions were reduced by PVP at elevated temperature and nucleated to form seeds. The seeds grew and sprouted in random directions to form rice-shape or horned particles with size of ∼5 nm or so by Ostwald ripening. Then the rice-shaped and horned particles were attached or aggregated to branched structures (dendrites). It was supported by the intermediates of rhodium dendrites (Fig. 3).
 | ||
| Fig. 3 The TEM images of intermediates of Rh nanodendrites at primary growth stage. Reproduced with permission from ref. 43. Copyright 2010 American Chemical Society. | ||
The other high-selectivity morphologies of Rh nanostructures such as horned particles, cubes and network-shaped wires (Fig. 2e–g) can be produced by replacement F− with equal mole quantity of other halogen anion (Cl−, Br− and I−). Among these nanostructures, the rhodium network-shaped wires were first reported by us. The formation mechanism of network-shaped wires is suggested by interface–defect and tip-connection attachments (Fig. 2h)
In this system, it is proposed that the electronegativity and ionic radii of different halogen anions (F−, Cl−, Br− and I−) should play the most important role in these processes, which would affect the coordination strength/number of rhodium and halogen. Maybe these factors integrate to control the reduction, nuclei and growth rate of Rh crystals. Meanwhile, the effect of SLS is also important to obtain rhodium crystals with highly selective morphologies.
2. Binary noble metal nanocrystals
The synthesis methods that have been developed to synthesize bimetallic noble metal nanostructures mainly include co-deposition of bimetallic precursors on support,54 co-decomposition/co-reduction in organic solvent or oil systems55,56,57 and seed-mediated epitaxial growth methods,58,59etc. However, the synthesis of bimetallic alloys with controlled size and shape has had limited success in aqueous solution systems. Pd–Pt random alloys (two metals atoms are mixed statistically according to the overall concentration) with a tunable composition have been reported in our previous work.36 Monodisperse Pd–Pt alloys with an average diameter of 7.1 ± 0.2 nm and a selectivity of more than 90% have been achieved by co-reduction of PdCl2 and K2PtCl6 in an aqueous solution in the presence of SLS, PVP and Br− (Fig. 4). The result of EDS line scanning profiles of a single nanocube showed the cube is made of Pd and Pt (Fig. 5). XPS results indicated that the internal composition of a Pd–Pt alloy can be harnessed in a wide range, which was supported by the regular variation of the relative intensity of Pd and Pt. (Fig. 6). The results of ICP-OES and XPS showed the at% of Pt can be tuned from zero to 34.0%.36 | ||
| Fig. 4 TEM images of representative Pd–Pt nanocube alloys. (a) Pd90.6Pt9.4 (b) Pd80.8Pt19.2, (c) Pd74.4Pt25.6, (d) Pd66.0Pt34.0. (The inset is HRTEM and FT patterns of single nanocube.) Reproduced with permission from ref. 36. | ||
 | ||
| Fig. 5 STEM image and EDS line scanning profiles of a single nanocube alloy. Reproduced with permission from ref. 36. | ||
 | ||
| Fig. 6 The Pd and Pt XPS spectra of Pd–Pt nanocubes alloys. Reproduced with permission from ref. 36. | ||
Furthermore, one-dimensional binary noble metal nanostructures, Pd/Pt and Rh/Pt ultrathin wires have been achieved by a seed-mediated method in aqueous solution containing only Pd or Pt seeds, PVP and Pt precursor. Seed-mediated epitaxial growth method is often exploited to synthesize binary metallic heterostructures. The morphologies of final products are mainly determined by both the shapes of the seeds and the lattice mismatch of binary metals. Accordingly, the morphologies of the products are commonly similar to those of seeds. In our synthetic strategy, we made use of Pt epitaxial growth on Pd or Rh (the Pd and Pt have a very small lattice mismatch of only 0.77%, the lattice mismatch of Rh/Pt is about 3.2%) nanocube seeds to produce heterostructured ultrathin nanowires.41,42
Fig. 7a and b show the selectivity of Pd/Pt nanowires is nearly 100%. The length of the wires can exceed 300 nm. The wires are made of nodes and stems, the diameter of Pt stems is about 3.1 ± 0.1 nm and that of nodes is below 5.0 nm (Fig. 7c). The STEM image and selected-area element analysis maps of Pt and Pd are shown in Fig. 8a, the results indicated that Pt atoms distribute in the whole area (green), whereas, the Pd atoms nearly distribute in the portion of nodes (orange). The results of the EDS line profiles taken from the single node and the single stem (Fig. 8c and d) indicate the node is made of Pt and Pd atoms, nevertheless, the stem is almost made of Pt atoms. Meanwhile, the EDS line profile (Fig. 8c) of single node indicates the node has the core–shell structure. The distance of surface lattice fringes of a node is 0.227, which corresponds to (111) of fcc platinum (Fig. 7d). It showed the node has Pd@Pt structure. The above-mentioned evidence indicated that the heterostructured nanowires are made of core–shell Pd@Pt structured nodes (the heavy dark dots) and Pt stems.
 | ||
| Fig. 7 TEM (a, b) and HRTEM (c) and (d) images of Pd/Pt heterostructured nanowires. Reproduced with permission from ref. 41. | ||
 | ||
| Fig. 8 STEM images (a) and (b) of Pd/Pt heterostructured nanowires and selected-area (the red rectangle) element analysis maps of Pt (green) and Pd (orange) and (c, d) cross-sectional compositional EDS line profiles. Reproduced with permission from ref. 41. | ||
With same synthesis strategy, high-yield Rh/Pt bimetallic ultrathin nanowires have been synthesized42 (Fig. 9). However, the results of EDS line scanning and elemental analysis maps have shown that the Rh atoms distribute discretely in the wires.42 At the same time, the nodes of Rh/Pt wires are not core–shell structures. The reason suggested is the difference of size of seeds. For Pd/Pt wires and Rh/Pt wires, the size of Pd seeds and Rh seeds is about 8.0 nm and 3.3 ± 0.2 nm, respectively. It is hard for the Pt atoms to fully displace the inner Pd atoms of Pd seeds via diffusion, which makes the core–shell Pd@Pt nodes. According to the presence of intermediates such as elongated particles and short rods, the formation mechanism of bimetallic Pd/Pt and Rh/Pt ultrathin wires was described as following (Scheme 1). The first step was that the Pt4+ ions diffused the to surface of Pd and Rh seeds were and reduced by surface atoms of the seeds via galvanic replacement, at the same time, the Pt nucleated and grew along the lowest energy facet orientation (111) to form Pt particles. Then, the rest of Pt ions and re-oxidized Pd or Rh ions were reduced by an autocatalytic process and grew anisotropically along the (111) facets of the Pt shell to form the short rods. Finally, the rods grew into wires by oriented attachment at the ends of each short rod. The growth mode led to the formation of wire structures with heavy nodes and stems.
 | ||
| Fig. 9 TEM and HRTEM images of Rh/Pt ultrathin nawires. Reproduced with permission from ref. 42. Copyright 2010 American Chemical Society. | ||
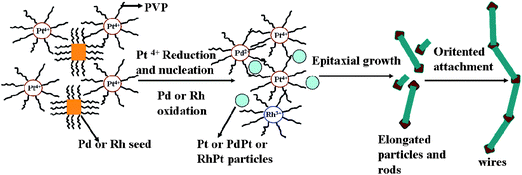 | ||
| Scheme 1 Proposed illustration of formation modes of Pd/Pt and Rh/Pt bimetallic nanowires. | ||
3. Applications in fuel cells
Pd, Pt and Rh nanoparticles are often used as catalysts to catalyze organic small molecules such as formic acid, methanol and ethanol in liquid fuel cells. Their electrocatalytic properties toward ethanol oxidation of Rh particles such as horn-, cube- and dendrite-shape particles are shown in Fig. 10. It is clear that the Rh particles display a shape-dependent catalytic performance. The activity increased, in order, with cubes, horned particles and dendrites. The Rh dentrites have maximum catalytic activity, which is about twice that of cubes. This may be ascribed to their greater number of corners/edges. Pd–Pt random alloys demonstrated composition-dependent properties for formic acid oxidation (Fig. 11a and b). The peak potential decreases with increasing Pt content (Fig. 11b), but the peak current of the Pd–Pt nanocubes alloy increased at a Pt content below 25.6 at%, then decreased (Fig. 11a). The peak current of all Pd–Pt nanocube alloys is higher than that of Pd nanocubes and commercial Pd black. Furthermore, the maximum peak potential decrease and peak current increase is 0.11 V and 3.1 mA cm−2 compared with the commercial Pd black. The catalytic activity indicated that the Pt favored the enhancement of the capability of Pd for formic acid oxidation owing the synergetic effect of Pd atoms and Pt atoms. | ||
| Fig. 10 The cyclic voltammetric curves (CVs) of Rh cubes (black), horned particles (red) and nanodendrites (blue) in a 0.1 M ethanol +0.1 M HClO4 solution. Reproduced with permission from ref. 43. Copyright 2010 American Chemical Society. | ||
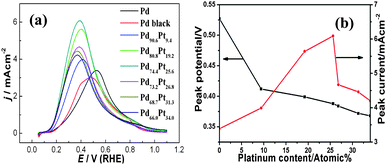 | ||
| Fig. 11 (a) The comparison of electrocatalytic activity of PdPt nanocubes alloys, Pd nanocubes and commercial Pd black. (b) The dependence of the peak potential and current on Pt content. Reproduced with permission from ref. 36. | ||
The electrocatalytic properties of the Rh/Pt bimetal alloys, Rh nanocubes and commercial Pt black were shown in Fig. 12. This showed that Rh favors a decreased peak potential compared with Pt black. However, the peak current decreased with increasing Rh content in the order Rh/Pt 3, Rh/Pt 2, Rh/Pt 1 and Rh cubes. Meanwhile, the ultrathin Rh/Pt 3nanowires exhibited maximum catalytic activity. There are two peaks in Fig. 12. Commonly, Peak A is ascribed to the oxidation of ethanol to acetaldehyde, acetic acid and CO2, and Peak B is almost completely ascribed to acetic acid.60,61 Furthermore, the peak intensity of Peak B is obviously higher than that of Peak A on commercial Pt catalysts. This means that the acetic acid is the main “end-product” for pure Pt catalysts. The partial oxidation of ethanol tremendously lowered the efficiency of energy utilization and impeded the use of ethanol in fuel cells. However, peak B is much lower than peak A on Rh/Pt bimetallic catalysts. This means that the Rh can enhance the selectivity of ethanol oxidation to CO2.
 | ||
| Fig. 12 The cyclic voltammetric curves (CVs) of Rh cubes, commercial Pt black and Rh/Pt bimetal alloys in a 0.1 M ethanol + 0.1 M HClO4 solution. Reproduced with permission from ref. 42. Copyright 2010 American Chemical Society. | ||
In situ FTIR spectroscopic studies are commonly used to identify the intermediates and products of ethanol oxidation. The in situ FTIR spectra of commercial Pt and Rh/Pt bimetal catalysts are shown in Fig. 13a–d. The band at 2345 cm−1 is the signature peak for the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO asymmetric stretch vibration of CO2, which reflects the cleavage of the C–C bond in ethanol oxidation. The band at ∼1715 cm−1 is the stretching vibration of the CO bond in acetic acid and/or acetaldehyde because of possible overlap at this location. A well-defined band at 1285 cm−1 is the characteristic absorption of C–O stretching in acetic acid, which is usually used for quantitative analysis of acetic acid. The band intensities of acetic acid and/or acetaldehyde are obviously stronger than those of CO2 on commercial Pt black at the potential beyond 0.4 V (Fig. 13a). Comparison of the ratio of band intensity of CO2 to acetic acid (located at 1285 and 1715 cm−1) for in situ FTIR spectra of ethanol oxidation on the commercial Pt black and Rh/Pt heterostructured catalysts at 0.80 V is illustrated in Fig. 13e. Obviously, much more CO2 and much less acetic acid was formed on Rh/Pt alloys catalysts as compared with commercial Pt black. For example, the band intensities of CO2 at 2345 cm−1 and acetic acid at 1285 cm−1 are 0.0107 and 0.0023 respectively on the Rh/Pt 3 catalysts (nanowires) at 0.80 V (Fig. 13f), and corresponding values are 0.0090 and 0.0052 on the commercial Pt black.42 The ratio value of band intensities of CO2 to acetic acid on the Rh/Pt 3 (nanowires) is 2.69 times that on the commercial Pt black (i.e., 4.65 vs. 1.73). Besides, the ratio value of band intensities of CO2 to that located at 1715 cm−1 on the Rh/Pt 3 (nanowires) is 2.27 times to that on the commercial Pt black (i.e., 1.43 vs. 0.63). It implies that the introduction of Rh favors the splitting of the C–C bond and enhances the CO2 selectivity. Among these samples, Rh/Pt 3 ultrathin nanowires showed the best activity and selectivity toward oxidizing ethanol to CO2 (Fig. 12 and 13e).
CO asymmetric stretch vibration of CO2, which reflects the cleavage of the C–C bond in ethanol oxidation. The band at ∼1715 cm−1 is the stretching vibration of the CO bond in acetic acid and/or acetaldehyde because of possible overlap at this location. A well-defined band at 1285 cm−1 is the characteristic absorption of C–O stretching in acetic acid, which is usually used for quantitative analysis of acetic acid. The band intensities of acetic acid and/or acetaldehyde are obviously stronger than those of CO2 on commercial Pt black at the potential beyond 0.4 V (Fig. 13a). Comparison of the ratio of band intensity of CO2 to acetic acid (located at 1285 and 1715 cm−1) for in situ FTIR spectra of ethanol oxidation on the commercial Pt black and Rh/Pt heterostructured catalysts at 0.80 V is illustrated in Fig. 13e. Obviously, much more CO2 and much less acetic acid was formed on Rh/Pt alloys catalysts as compared with commercial Pt black. For example, the band intensities of CO2 at 2345 cm−1 and acetic acid at 1285 cm−1 are 0.0107 and 0.0023 respectively on the Rh/Pt 3 catalysts (nanowires) at 0.80 V (Fig. 13f), and corresponding values are 0.0090 and 0.0052 on the commercial Pt black.42 The ratio value of band intensities of CO2 to acetic acid on the Rh/Pt 3 (nanowires) is 2.69 times that on the commercial Pt black (i.e., 4.65 vs. 1.73). Besides, the ratio value of band intensities of CO2 to that located at 1715 cm−1 on the Rh/Pt 3 (nanowires) is 2.27 times to that on the commercial Pt black (i.e., 1.43 vs. 0.63). It implies that the introduction of Rh favors the splitting of the C–C bond and enhances the CO2 selectivity. Among these samples, Rh/Pt 3 ultrathin nanowires showed the best activity and selectivity toward oxidizing ethanol to CO2 (Fig. 12 and 13e).
 | ||
| Fig. 13
In situ FTIR spectra of samples for ethanol oxidation in a 0.1 M ethanol + 0.1 M HClO4 solution. (a) Commercial Pt black; (b) Rh/Pt 1; (c) Rh/Pt 2; (d) Rh/Pt 3 (nanowires). (e) Band intensity comparisons of CO2 with 1280 and 1715 cm−1 of commercial Pt black and Rh/Pt heterostructures at 0.8 V. (f) Band intensities as a function of potential associated with CO2, 1715 and 1280 cm−1 of ethanol oxidation on ultrathin wires surfaces. Note: Black squares: 2345 cm−1 band of CO2; blue triangles: 1715 cm−1 CO stretch vibration in acetaldehyde and/or acetic acid; red cycles: 1280 cm−1 band of acetic acid. The Rh content decreased in the order Rh/Pt 1, Rh/Pt 2 and Rh/Pt 3. Reproduced with permission from ref. 42. Copyright 2010 American Chemical Society. | ||
5. Summary and outlook
The article summarizes our recent progress on the synthesis of single and binary noble metal nanocrystals by a one-pot, aqueous solution method and their applications in liquid fuel cells. The synthesis strategy demonstrated a facile, general route to produce nonspherical, single and binary nanocrystals. In particular, the concept of seed displacement, the epitaxial synthesis of bimetallic one-dimensional ultrathin nanowires, favors the development of bimetallic one-dimensional structures. The quest for new synthesis strategies and insight into the true nature of crystal nucleation and growth are always fascinating for chemists and material scientists. The exploration of novel structures and properties and understanding of the correlation of structure and property for nanocrystals are important and promising.Acknowledgements
This work was supported by NSFC (20725102 and 20921001), the Fok Ying Tung Education Foundation (111012), Natural science foundation of Guizhou Province (20072013) and the State Key Project of Fundamental Research for Nanoscience and Nanotechnology (2006CB932301).References
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60 CrossRef CAS.
- H. Lee, S. E. Habas, S. Kweskin, D. Butcher, G. A. Somorjai and P. D. Yang, Angew. Chem., Int. Ed., 2006, 45, 7824 CrossRef CAS.
- J. Y. Park, Y. W. Zhang, M. Grass, T. F. Zhang and G. A. Somorjai, Nano Lett., 2008, 8, 673 CrossRef CAS.
- C. Wang, H. Daimon, T. Onodera, T. Koda and S. Sun, Angew. Chem., Int. Ed., 2008, 47, 3588 CrossRef CAS.
- N. Tian, Z. Y. Zhou, S. G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732 CrossRef CAS.
- T. Ming, W. Feng, Q. Tang, F. G. Wang, L. Sun, J. Wang and C. Yan, J. Am. Chem. Soc., 2009, 131, 16350 CrossRef CAS.
- N. Li, W. Wang, D. Tian and H. Cui, Chem. Commun., 2010, 46, 1520 RSC.
- Q. Zhang, J. Ge, T. Pham, J. Goebl, Y. Hu, Z. Lu and Y. D. Yin, Angew. Chem., Int. Ed., 2009, 48, 3516 CrossRef CAS.
- A. Tao, P. Sinsermsuksakul and P. Yang, Nat. Nanotechnol., 2007, 2, 435 CrossRef CAS.
- C. J. Orendorff, A. Gole, T. K. Sau and C. J. Murphy, Anal. Chem., 2005, 77, 3261 CrossRef CAS.
- R. Alvarez-Puebla, R. Contreras-Caceres, I. Pastoriza-Santos, J. Perez-Juste and L. M. Liz-Marzan, Angew. Chem., Int. Ed., 2009, 48, 138 CrossRef CAS.
- W. Li, P. H. C. Camargo, X. Lu and Y. Xia, Nano Lett., 2009, 9, 485 CrossRef CAS.
- P. H. C. Camargo, M. Rycenga, L. Au and Y. Xia, Angew. Chem., Int. Ed., 2009, 48, 2180 CrossRef CAS.
- H. Bai, M. Han, Y. Du, J. Bao and Z. Dai, Chem. Commun., 2010, 46, 1739 RSC.
- B. K. Jena and C. R. Raj, Chem.–Eur. J., 2006, 12, 2702 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS.
- C. J. Murphy, A. M. Gole, J. W. Stone, P. N. Sisco, A. M. Alkilany, E. C. Goldsmith and S. C. Baxter, Acc. Chem. Res., 2008, 41, 1721 CrossRef CAS.
- X. Yang, S. E. Skrabalak, Z.-Y. Li, Y. Xia and L. V. Wang, Nano Lett., 2007, 7, 3798 CrossRef CAS.
- K. H. Song, C. Kim, C. M. Cobley, Y. Xia and L. V. Wang, Nano Lett., 2009, 9, 183 CrossRef CAS.
- K. H. Park, K. Jang, H. J. Kim and S. U. Son, Angew. Chem., Int. Ed., 2007, 46, 1152 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Am. Chem. Soc., 2004, 126, 7194 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097 CrossRef CAS.
- P. N. Njoki, I. S. Lim, D. Mott, H.-Y. Park, B. Khan, S. Mishra, R. Sujakumar, J. Luo and C.-J. Zhong, J. Phys. Chem. C, 2007, 111, 14664 CrossRef CAS.
- C.-K. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816 CrossRef CAS.
- Y. Y. Yu, S. S. Chang, C. L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 6661 CrossRef CAS.
- R. Jin, Y. Cao, C. A. Mirkin, K. L. Kelly, G. C. Schatz and J. G. Zheng, Science, 2001, 294, 1901 CrossRef CAS.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176 CrossRef CAS.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302 CrossRef CAS.
- H. Lee, S. E. Habas, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2008, 130, 5406 CrossRef CAS.
- Z. Peng and H. Yang, J. Am. Chem. Soc., 2009, 131, 7542 CrossRef CAS.
- S. Alayoglu, A. U. Nilekar, M. Mavrikakis and B. Eichhorn, Nat. Mater., 2008, 7, 333 CrossRef CAS.
- V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross, N. Markovic, J. Rossmeisl, J. Greeley and J. K. Noskov, Angew. Chem., Int. Ed., 2006, 45, 2897 CrossRef CAS.
- M. Saruyama, M. Kanehara and T. Teranishi, Chem. Commun., 2009, 2724 RSC.
- Q. Yuan, Z. Zhou, J. Zhuang and X. Wang, Chem. Commun., 2010, 46, 1491 RSC.
- E. C. Cho, P. H. C. Camargo and Y. N. Xia, Adv. Mater., 2010, 22, 744 CrossRef CAS.
- D. Xu, S. Bliznakov, Z. Liu, J. Fang and N. D. imitrov, Angew. Chem., Int. Ed., 2010, 49, 1282 CrossRef CAS.
- X. Huang, H. Zhang, C. Guo, Z. Zhou and N. Zheng, Angew. Chem., Int. Ed., 2009, 48, 4808 CrossRef CAS.
- X. Wang, J. Zhuang, Q. Peng and Y. D. Li, Nature, 2005, 437, 121 CrossRef CAS.
- Q. Yuan, J. Zhuang and X. Wang, Chem. Commun., 2009, 6613 RSC.
- Q. Yuan, Z. Zhou, J. Zhuang and X. Wang, Chem. Mater., 2010, 22, 2395 CrossRef CAS.
- Q. Yuan, Z. Zhou, J. Zhuang and X. Wang, Inorg. Chem., 2010, 49, 5515, DOI:10.1021/ic100249t.
- B. Lim, X. Lu, M. Jiang, P. H. C. Camargo, E. C. Cho, E. P. Lee and Y. N. Xia, Nano Lett., 2008, 8, 4043 CrossRef CAS.
- S. E. Skrabalak, L. Au, X. Li and Y. Xia, Nat. Protoc., 2007, 2, 2182 Search PubMed.
- J. Sun, M. Guan, T. Shang, C. Gao, Z. Xu and J. Zhu, Cryst. Growth Des., 2008, 8, 906 CrossRef CAS.
- S. G. A. Filankembo, I. Lisiecki and M. P. Pileni, J. Phys. Chem. B, 2003, 107, 7492 CrossRef.
- J. E. Millstone, W. Wei, M. R. Jones, H. Yoo and C. A. Mirkin, Nano Lett., 2008, 8, 2526 CrossRef CAS.
- J. H. Song, F. Kim, D. Kim and P. Yang, Chem.–Eur. J., 2005, 11, 910 CrossRef CAS.
- K.-S. Choi, Dalton Trans., 2008, 5432 RSC.
- X. Kou, S. Zhang, Z. Yang, C.-K. Tsung, G. D. Stucky, L. Sun, J. Wang and C. Yan, J. Am. Chem. Soc., 2007, 129, 6402 CrossRef CAS.
- Y. Xiong, J. M. McLellan, Y. Yin and Y. Xia, Angew. Chem., Int. Ed., 2007, 46, 790 CrossRef CAS.
- L. Wang and Y. Yamauchi, J. Am. Chem. Soc., 2009, 131, 9152 CrossRef CAS.
- X. Li and I. M. Hsing, Electrochim. Acta, 2006, 51, 3477 CrossRef CAS.
- D. Xu, Z. Liu, H. Yang, Q. Liu, J. Zhang, J. Fang, S. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217 CrossRef CAS.
- Z. Liu, J. E. Hu, Q. Wang, K. Gaskell, A. I. Frenkel, G. S. Jackson and B. Eichhorn, J. Am. Chem. Soc., 2009, 131, 6924 CrossRef CAS.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989 CrossRef CAS.
- F. R. Fan, D. Y. Liu, Y. Wu, S. Duan, Z. X. Xie, Z. Y. Jiang and Z. Q. Tian, J. Am. Chem. Soc., 2008, 130, 6949 CrossRef CAS.
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. D. Yang, Nat. Mater., 2007, 6, 692 CrossRef CAS.
- H. Hitmi, E. M. Belgsir, J.-M. Leger, C. Lamy and R. O. Lezna, Electrochim. Acta, 1994, 39, 407 CrossRef CAS.
- S.-C. Chang, L.-W. H. Leung and M. J. Weaver, J. Phys. Chem., 1990, 94, 6013 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |