One-pot synthesis of triangular gold nanoplates allowing broad and fine tuning of edge length†
Adelaide
Miranda
a,
Eliana
Malheiro
a,
Elżbieta
Skiba‡
a,
Pedro
Quaresma
a,
Patrícia A.
Carvalho
b,
Peter
Eaton
a,
Baltazar
de Castro
a,
John A.
Shelnutt
cd and
Eulália
Pereira
*a
aREQUIMTE/Departamento de Química e Bioquímica, Faculdade de Ciências, Universidade do Porto, Rua do Campo Alegre, 687, 4169-007, Porto, Portugal. E-mail: eulalia.pereira@fc.up.pt; Fax: +351 220 402 959; Tel: +351 220 402 588
bDepartamento de Engenharia dos Materiais do Instituto Superior Técnico, Av. Rovisco Pais, 1049-001, Lisboa, Portugal
cAdvanced Materials Laboratory, Sandia National Laboratories, 1001 University Blvd SE, Albuquerque, NM 87185-1349, USA
dDepartment of Chemistry, University of Georgia, Athens, GA 30602, USA
First published on 16th August 2010
Abstract
A photocatalytic approach was used to synthesize triangular nanoplates in aqueous solution. The synthesis is based on the reduction of a gold salt using a tin(IV) porphyrin as photocatalyst, cetyltrimethylammonium bromide (CTAB) as a stabilizing agent, and triethanolamine (TEA) as the final electron donor. The average edge length of the triangular nanoplates can be easily changed in the range 45–250 nm by varying the concentration of photocatalyst, and fine-tuning of the average edge length is achieved by varying the concentration of CTAB. Study of the mechanism of formation of the nanoplates by UV-vis and by transmission electron microscopy (TEM) shows that there is a first stage where formation of 5 nm seeds takes place, further growth is probably by fusion and by direct reduction of gold onto the preformed nanoparticles. The nanoparticles formed during the photocatalytic reduction of the gold precursor show an irregular shape that evolves to regular triangular nanoplates after ripening in solution for 24 h.
Introduction
The search for chemical methods to prepare metal nanoparticles with controlled shape and size is one of the major current challenges in nanosciences and nanotechnology. In particular, the synthesis of nanotriangles or nanoplates has been the subject of great interest, mainly because these 2-D nanostructures show a strong plasmon band in the NIR region that is highly sensitive to small morphological changes and to changes in its close environment.1–5 Nanoparticles with bands in the NIR region of the spectrum have potential use in cancer hyperthermia, biological assays, cell imaging, and optical coating for solar energy converters.6–8 Additionally, nanotriangles show anisotropic electrical conductivity,9 and strong enhancement of electric field at the vertices,10 which is expected to make them useful in photonics and optoelectronics, information storage, optical sensing, imaging, metal enhanced fluorescence,11 and surface enhanced Raman scattering.12–15 It has also been recently reported that nanotriangles can be more efficient as catalysts in the oxidation of small organic substrates than spherical nanoparticles16 or nanowires.17Synthesis of Au nanotriangles has been reported using biological reducing agents,6,10,18 seeding19,20 and thermal methods,21–23 photoreduction,24 and the polyol method,25 but most of these methods yield mixtures of different shapes, and high dispersion of size. It has been proposed that the capping agent, most commonly CTAB or PVP, has a key role in the asymmetric growth of nanoparticles leading to nanotriangles. Nevertheless, the nature of this directing effect is still not clear. Some authors claim that it is due to preferred absorption of the capping agent to the [111] surface of fcc Au,26,27 while others propose that the formation of stacking faults during the first nucleation events is the main factor for asymmetric growth of nanoparticles.28,29 In addition, it has been reported that factors that influence the rate of nucleation and growth of nanoparticles may also play a key role in the formation of the nanotriangles.30 In addition, a recent review on shape selection in wet chemical methods proposes that the thermodynamic driving force of the reaction controls the formation of a particular shape. In the case of nanotriangles, for which it is necessary a stabilization of [111] facets and a break in the symmetry of the nanocrystal, a low driving force of the chemical reduction reaction was proposed as the main controlling factor.31,32
There are still many open questions regarding the formation mechanism, and thus it is difficult to rationally design synthetic strategies to optimize the synthesis of nanotriangles. For instance, a few methods to prepare nanotriangles with tuned size have been reported,6,18,19,21,22,33,34 but several different factors appear to be dominant in each case. Size variation was achieved either by changing the capping agent concentration,6,18,21,22,34 pH,33 reaction time,21,22 or by sequential growing steps,19 but often poor control of size and tuning range was achieved.
Here we have used a photocatalytic reaction to reduce the Au(III) precursor in order to understand the influence of the reaction kinetics on the formation of the Au nanoplates. This photocatalytic reaction has been previously described for size controlled growth of spherical or dendritic noble metal nanoparticles35–44but to our knowledge it is the first time that it is used to synthesize anisometric nanoparticles. The reaction is based on a photocatalytic cycle that can be represented by the following simplified equations:
| SnP + hν → SnP* |
| SnP* + TEA → SnP−• + TEAox |
| SnP−• + Au(III) → SnP + Au(II) |
Experimental section
Synthesis of triangular nanoplates
Hydrogen tetrachloroaurate(III) (30 wt% solution in dilute HCl, 99.99%), triethanolamine (TEA; 98%), and hexadecyltrimethylammonium bromide (CTAB) were obtained from Aldrich and used as received. Sn(IV) meso-tetra(N-methyl-4-pyridyl)porphine tetratosylate chloride (SntMepyP) was obtained from Porphyrin Products. All solutions were prepared with Millipore water (18.2 MΩ cm−1) and filtered through 0.22 μm pore size syringe filters. All glassware was washed in HNO3 conc./HCl conc. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (V:V) and thoroughly rinsed with water before use.
:3 (V:V) and thoroughly rinsed with water before use.
For the preparation of Au nanoplates, 20 μL of hydrogen tetrachloroaurate(III) solution (20 mM) were added to 500 μL of TEA (0.15 M)/ammonia (0.1 M) (pH 7.0 adjusted with HCl 1 M). The solution turned colorless immediately. Then, 500 μL of CTAB (10 mM), 2100 μL of water, and 100 μL of SntMepyP (3.0 μM) was added and the solution was irradiated for at least 2 h using a Osram discharge lamp HQI-T 250W D−1 E 40 12X1.
Characterization of nanoparticles
UV/vis absorption spectra were obtained using a TECHCOMP UV-VIS 8500 spectrophotometer; TEM characterization was performed on a HITACHI H-8100 electron microscope operating at 200 kV, in carbon copper grids. AFM imaging was carried out in tapping mode and contact mode with a Molecular Imaging PicoLE atomic force microscope. Tapping mode images were obtained using rectangular silicon cantilevers of resonant frequency ca. 75 kHz. For contact mode measurements a triangular cantilever of nominal force constant 0.12 N m−1 and tip radius <10 nm was used. Calibration of the photodetector of the AFM was achieved by measuring force curves on a stiff surface (mica). Samples were extensively washed by at least 5 cycles of ultrafiltration (Centricon YM-10) and dilution in ultrapure water (water for molecular biology, Sigma). Samples were then prepared by, depositing a drop onto freshly cleaved mica, and air-dried. Photoelectron spectroscopy (XPS) experiments were performed in a VG Scientific ESCALAB 200A spectrometer using non-monochromatic Al Kα radiation (1486.6 eV, Centro de Materiais da Universidade do Porto, CEMUP). Corrections for sample charge were made by taking the C 1s band at 285.0 V as internal standard. The colloidal solutions for XPS measurements were previously centrifuged at 60000 rpm for 10 min at room temperature and the pellet was deposited on a graphite support.
Results and discussion
Synthesis and characterization of Au nanoplates
Gold nanoplates were obtained by the photocatalytic reduction of HAuCl4, using SntMepyP as photocatalyst, TEA as electron-donor and CTAB as capping agent, at pH 6.7–7.0, following a previously proposed approach for the synthesis of platinum nanodendrites,35 with some modifications. In a typical experience, the sample was irradiated for 120 min and was kept in solution for at least 24 h before preparing the samples for microscopy characterization. The initial solution is light pink due to the tin(IV) porphyrin, and slowly turns to violet due to the appearance of two plasmon bands, one at 540 nm typical of gold nanospheres, and another band at 820 nm, that is typical of the in-plane dipole plasmon resonance of Au nanotriangles.1,3,45Typical TEM and SEM images of the samples thus obtained are shown in Fig. 1. The sample contains mainly triangular nanoplates (>84%) with slightly truncated corners and an average length of 137 ± 43 nm, with few smaller particles (average diameter of ≈40 nm) with irregular quasi-spherical shapes. Although, most of the synthesis of triangular nanoplates reported do not indicate quantitatively the shape selectivity of the method, based on a visual comparison of TEM images, the present yield >84% seems to be remarkably good. Selected Area Electron Diffraction (SAED) patterns of single nanotriangles (inset in Fig. 1A) show the typical {220} reflection for the lattice planes of gold, and inner circle spots corresponding to 1/3 {422} reflections. These spots, that are forbidden in a single-crystal fcc metal, can be attributed to parallel twin plates, and are typically observed in Au and Ag nanoplates.28,29 Pilleni et al.46 carried out a detailed analysis of the growth of silver nanotriangles, and proposed that the fcc forbidden 1/3 {422} reflections are due to (111) stacking fault(s) lying parallel to the (111) surface and extending across the entire planar particle. In our samples, cross-sectional observations of nanotriangles showed the characteristic bright/dark contrast of adjacent twin domains (Fig. 1C), but these were not observed in all cases.
 | ||
| Fig. 1 Electron microscopy images of Au nanoparticles obtained with a SnP concentration of 9.3 nM. (A) TEM image; (B) Selected area electron diffraction (SAED) pattern of a single nanotriangle (inset: TEM image of the nanotriangle used) showing the typical {220} reflections (marked in the Figure), and inner circle spots corresponding to forbidden 1/3 {422} reflections (arrows); (C) representative SEM image of a sample obtained in similar synthetic conditions; (D) TEM image of a cross-section view of a single nanotriangle showing the characteristic bright/dark contrast of adjacent twin domains. | ||
The nanotriangles were further characterized by X-ray photoelectron spectroscopy (XPS, detailed results are in the ESI†). The XPS spectrum of the gold nanotriangles exhibit doublet peaks located at 84.0 and 87.7 eV that can be assigned to Au 4f7/2 and 4f5/2, respectively, typical of Au(0) oxidation state. No peaks corresponding to Au(I) or Au(III) compounds were detected. The full spectrum also shows peaks for Br, N, and C with binding energies similar to those assigned to CTAB in CTAB stabilized Au NPs.47 One additional N1s peak was detected with a binding energy of 400.2 eV, assigned to the amine group of TEA,48 showing that the amine is also partially adsorbed at the NP surface. Peaks typical of chloride (197.8 and 199.6 eV) were also detected. Quantitative analysis of XPS data after correction for signal sensitivity showed similar amounts of both chloride and bromide anions.
An example AFM image of an isolated Au nanoplate is shown in Fig. 2A, along with a height profile over the triangle (Fig. 2B). Height profiles confirmed that the nanoplates were broad and flat, with the height seemingly independent of the triangle length. The nanoplates heights measured varied between 15–19 nm, even though the length of the nanotriangles varied much more, from 133 nm to 427 nm. In addition, it was seen that small irregular bumps covered the surfaces. These were extremely small, typically measuring just 1–2 nm in height, in accordance with the expected size for a hemi-micelle of CTAB.49,50 Almost all triangles seen had these features.
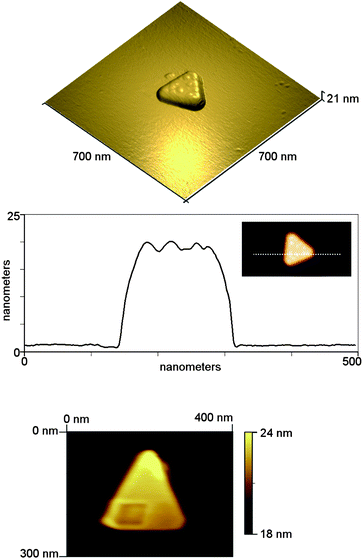 | ||
| Fig. 2 (Top) Tapping mode AFM image of isolated nanotriangle, and (middle) height profile over a triangle showing 1–2 nm bumps on the surface. (Bottom) Contact mode image of isolated nanotriangle after scratching experiment. The scratching was carried out in a square region in the lower half of the triangle. | ||
In order to confirm that the triangles had an organic coating, nano-scratching experiments were carried out by AFM to assess the ease of removal of the surface coating of the nanotriangles. A simple scratching measurement was performed by switching to contact mode and finding an isolated nanotriangle. This was initially scanned at a low force to determine the initial shape and then an image using a larger setpoint (i.e. a larger imaging force) was obtained in a small square area on top of the triangular crystal. The crystal was then imaged again at the lower force to see if any changes occurred. The applied forces were not calibrated; however based on the nominal spring constant of the cantilever, and using the calibrated sensitivity of the photodetector, the low (imaging) force was estimated as 2.2 nN, and the higher (scratching) force as 7.2 nN. It can clearly be seen in the bottom panel of Fig. 2 that in the square region where the scratching was carried out, some material was removed from the surface of the crystal.
Analysis of line profiles across this region show that approximately 0.5 nm of material was removed. However, given that the force applied by the tip to the sample was not calibrated fully, we cannot rule out that the AFM tip scratched the gold surface of the triangle without further control experiments. The scratching results suggest that there was a thin regular film of organic soft material (most probably CTAB, based on the XPS results) at the surface of the metal.
Mechanism of nanoparticle formation
To further understand the mechanism involved in the formation of the Au nanotriangles, we have followed the reaction by UV/vis spectroscopy (Fig. 3) and TEM (Fig. 4). Before irradiation the solution is colorless, and the UV/vis spectrum shows only a strong absorption in the UV (<250 nm) and a weak absorption due to the Soret band of the porphyrin. The disappearance of the typical color of [AuCl4]− is detected immediately after addition of TEA/NH3, and it is probably due to the formation of colorless hydroxo complexes. The speciation of Au(III) in the presence of chloride and bromide is quite complex, with possible formation of all mixed halide complexes, and mixed hydroxo–halide complexes, but taking into account the stability constants available, the main species are probably the colorless complexes [AuCl(HO)3]−, [Au(HO)4]−, and [AuBr2(HO)2]−.51,52 Another possible explanation for the color changes observed upon addition of TEA is the reduction of Au(III) to Au(I) or even Au (0). However, this does not seem to be the case, since lowering the pH restores the yellow color typical of Au(III)–halide complexes. Moreover, the addition of an oxidizing agent (10% H2O2) does not induce any color changes in the solution, as expected if part of the gold precursor would have been reduced, thus further confirming the +3 oxidation state for gold.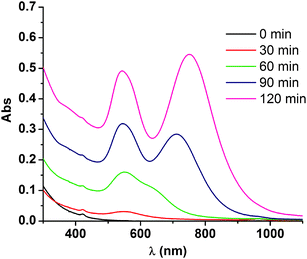 | ||
| Fig. 3 UV/vis spectra of the reaction solution obtained before irradiation (0 min), and after 30, 60, 90 and 120 min of irradiation. | ||
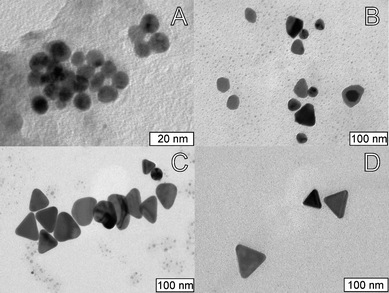 | ||
| Fig. 4 Representative TEM images of samples obtained after irradiation for (A) 30 min; (B) 60 min; and (C) 120 min. (D) Representative TEM image of the nanotriangles obtained after 120 min irradiation and ripening for 24 h in the dark. | ||
In order to verify if the reduction reaction proceeds only by the photocatalytic pathway, or by direct chemical reduction, we have studied the reaction in the absence of photocatalyst, either with irradiation or in the dark, and followed the reaction by UV/vis spectroscopy. In both cases, the typical plasmon bands for the nanotriangles were observed, but the reaction proceeds very slowly, and even after 24 h the absorbance of the band at higher wavelength is about half of that observed by the photocatalytic pathway after 120 min. In addition, the plasmon band shifts to higher wavelengths, typical of nanotriangles with a larger size. These results show that, when the photocatalytic pathway is not operative, the reduction of the Au(III) precursor still occurs by direct chemical reaction, but with an overall slower rate. In fact, we have found that Au(III) chemical reduction by TEA is very fast at pH lower than 6, probably because in this pH range the most abundant Au(III) species in solution are the bromide and chloride complexes. In this case, however, Au microparticles are obtained. On the other hand, reduction of the Au(III) precursor by the photocatalytic pathway using concentrations of SnP higher than 1 μM is much faster, but the NPs obtained have a spherical shape, and show only the typical plasmon band at 520 nm (data not shown).
In order to evaluate the influence of the capping agent, we have used cetylpyridinium bromide (CPyB) instead of CTAB in the experimental procedure, but for all the concentrations tested the final nanoparticles were spherical. Both surfactants are cationic, and have a similar alkyl chain, but show a different behavior in the adsorption to a metal surface due to the differences in headgroup properties.53,54 This different adsorption behavior is probably a major factor in the growth of triangular nanoplates.
We have also found that the formation of nanotriangles is only observed in the simultaneous presence of chloride and bromide. Replacement of CTAB by its chloride analog, CTAC, or replacement of the hydrochloric acid by hydrobromic acid also inhibits the formation of nanotriangles.
The formation of nanotriangles is sufficiently slow to enable its study by UV/vis spectroscopy (Fig. 3) and TEM (Fig.4). Samples irradiated for 30 min show a low intensity band around 540 nm, indicating that only a small amount of the Au(III) initially present was reduced. Accordingly, TEM images show few spherical Au nanoparticles with diameters 5–12 nm, some of them clearly undergoing fusion (Fig. 4A). After 60 min, most of the small nanoparticles have grown into irregular shaped nanoparticles with diameters in the range 30–80 nm. Some of these nanoparticles already show a form resembling a triangle, but with very irregular edges (Fig. 4B). UV/vis spectra show an increase in the intensity of both plasmon bands until ca. 120 min of irradiation. Samples taken after 120 min show that most of the nanoparticles have triangular shapes with irregular edges (Fig. 4C). After this irradiation time, there are no differences between samples kept in the dark and samples irradiated up to 24 h, and there are no significant changes in the intensity of the plasmon bands, indicating that no further reduction of Au(III) is taking place. To confirm that the entire gold precursor was depleted, we added a strong reducing agent (NaBH4) to the solution after 120 min of irradiation, and no significant changes in the intensity of any of the plasmon bands were observed, showing that the reduction of Au(III) was completed after 2 h. UV/vis spectra taken after 24 h in solution showed a slight red shift of the nanotriangle plasmon band, but after this, no more changes are observed even after 1–2 months in solution. Comparing the TEM images obtained at 120 min (Fig. 4C) and after 24 h in solution (Fig. 4D), it is clear that the nanoplates have evolved to a more regular triangular shape. In addition, most of the small spherical nanoparticles observed at 120 min have disappeared. This results suggest that the nanotriangles undergo Ostwald ripening, where the edge faults are slowly occupied at the expense of the smaller nanoparticles in solution.
Taken together, these results indicate that the chemical reduction of the Au(III) precursor yields very small nanoparticles (“seeds”) that then grow either by fusion or by reduction of Au(III) at their surface. This seeding/growing ends after ≈2 h of irradiation due to depletion of Au(III), yielding triangular NPs with irregular edges. The formation of regular triangular nanoplates proceeds then by a slow reorganization process, independent of light. The morphological changes observed throughout this study are completely inhibited by addition of a small amount of a strongly binding capping agent (11-mercaptoundecanoic acid), showing that the stronger interaction of the thiol group with the Au surface prevents the shape evolution mechanism observed, and indicating that CTAB is crucial for the formation of regular triangular nanoplates.
Control of the average length of nanotriangles
One of the advantages of using a photocatalyst in the present method is the possibility to control the rate of the reduction of Au(III) by changing the concentration of the photocatalyst used. Previous studies with platinum(IV) reduction have shown that for higher concentrations of the photocatalyst, smaller nanoparticles are obtained.35 This behavior was explained by a seeding/autocatalytic growth, where the role of the photocatalyst was especially relevant in the initial seed formation. Since the growth of the nanoparticles is fast compared to the formation of crystallization nuclei, it is expected that when the seeds reach a certain critical size, the reduction of the metal precursor at the seeds will be significantly faster that the formation of new seeds. The particles will then grow at the same rate, and will stop when the metal precursor is depleted from solution. Thus, an increase in the concentration of photocatalyst will increase the number of seeds competing for the reduction of the metal precursor, leading to a decrease in the average size of the nanoparticles.Fig. 5 shows TEM images of Au nanoparticles obtained with different concentrations of the photocatalyst. The expected behavior is observed for the concentration range 0–0.1 μM, allowing the synthesis of Au nanoplates with average edge lengths of 246 ± 170 nm, 136 ± 43 nm, 82 ± 22 nm, and 45 ± 10 nm (Fig. 5 A–D). Nanoplates with an average length of 246 nm were obtained in the absence of photocatalyst, but in this case the size homogeneity obtained is poor, with a standard mean deviation of 70% of the average size, and the reaction is very slow, taking more than 2 days. For the photocatalytic synthesis, the standard mean deviations are 20–30% of the average size, showing that the presence of the photocatalyst not only allows controlling the average size of the final nanoparticles, but also yields a better size dispersion, in accordance with the proposed in situ seeding mechanism.
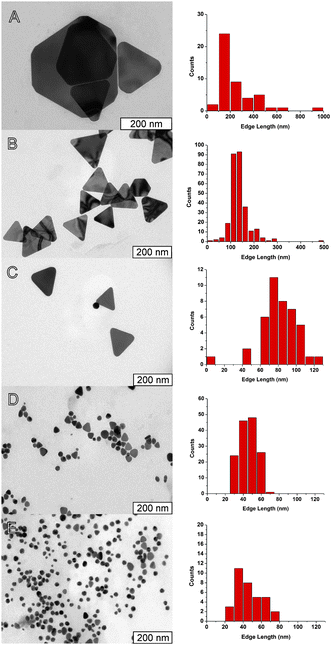 | ||
| Fig. 5 Representative TEM images (left) and corresponding histograms of distribution of side lengths (right) of samples obtained with the following photocatalyst concentrations: (A) without photocatalyst (average length 246 ± 170 nm); (B) 10 nM (average length 137 ± 43 nm); (C) 40 nM (average length 82 ± 22 nm); (D) 0.10 μM (average length 45 ± 10 nm); and (E) 1.0 μM (average length 46 ± 15 nm). | ||
In addition to the decrease in size with the increase in concentration of photocatalyst, the corners of the nanoplates are more truncated, and for concentrations of porphyrin of 1.0 μM (Fig. 5E) or higher most of the nanoparticles are spherical, indicating that the formation of nanotriangles is limited to a slow growth step.
Another common method to control the size of nanoparticles is to change the concentration of the capping agent. In this case, the usual behavior observed is a decrease of the average size as the concentration of capping agent increases, due to the hindering effect of the surface capping agent on the growth of nanoparticles. For the present synthesis, we have changed the concentration of CTAB in the range 0–50 mM. In the absence of CTAB, reduction of the gold precursor is very fast, even in the dark, and yields aggregates of irregular NPs with high dispersion of sizes (50–200 nm). Regular nanotriangles are obtained for a concentration of CTAB between 0.8 and 3.1 mM, and the expected decrease of the average length with increasing CTAB concentration is observed (Fig. 6). For higher concentrations of the capping agent, the amount of nanotriangles relative to other irregular shapes decreases. Further increase of the concentration of CTAB to 6.2 mM leads to the formation of branched NPs with lengths of 100–300 nm (see Fig. S2 in the ESI†). It is to be noted that the range of average sizes obtained by changing the concentration of CTAB is significantly smaller than that obtained by changing the porphyrin concentration. Thus a model emerges for the size control of the nanotriangles, in which the changes in the concentration of CTAB may be used to fine tune the size of the nanotriangles for a selected concentration of photocatalyst.
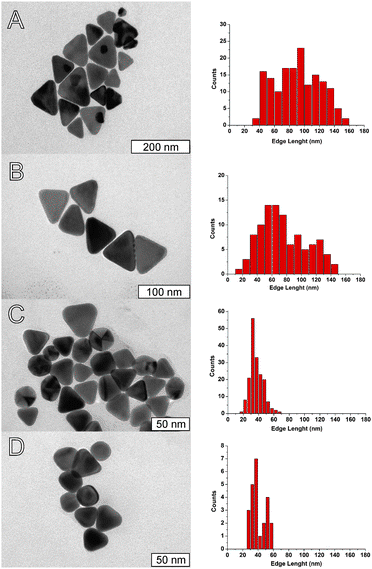 | ||
| Fig. 6 Representative TEM images (left) and corresponding histograms of distribution of side lengths (right) of nanoplates obtained with a fixed concentration of porphyrin (50 nM) and varying the concentration of CTAB: A) 0.80 mM (average length 90 ± 30 nm); B) 1.6 mM (average length 65 ± 36 nm); C) 2.3 mM (average length 36 ± 9 nm); and D) 3.1 mM (average length 42 ± 16 nm). | ||
Discussion on the formation of triangular nanoplates
CTAB has been widely used in the synthesis of non-spherical Au NPs, and it has been suggested that this surfactant plays a critical role in directing the asymmetric growth of NPs by preferential adsorption to a specific crystal face. Mirkin et al.5 in a recent review about gold and silver nanoprisms summarize the main crystallographic and chemical factors that are important for plate-like growth. It has been proposed that the most important crystallographic factor is the formation of twinning crystal defects during the initial nucleation events.28 Although the importance of the type of seeds seems undeniable, other chemical factors, like the concentration of capping agent, reductant, metal precursor, etc., are known to affect the shape of nanoparticles.5,55The importance of CTAB as a capping agent for the formation of nanotriangles is, in the present case, quite apparent, since the use of other related surfactants (CPyB or CTAC) does not result in the formation of triangular nanoparticles. The failure of CPyB to induce nanotriangle formation is particularly interesting in view of the similar structure and properties of CPyB and CTAB as both surfactants have similar values of cmc, and both form wormlike micelles in the same concentration range. This ability to form wormlike micelles has been suggested as a possible explanation for the shape-directing role of CTAB. Although some synthetic methods for Au nanotriangles use concentrations of CTAB much higher than cmc,21,22,56 where wormlike micelles are probably present, in our case and in some others57,58 nanotriangles are obtained at concentrations close to the cmc, where wormlike micelles are scarce.59 It thus seems more likely that the different results obtained with CTAB and CpyB are due to the different mode of adsorption of each of the cationic surfactants to the metal surface.53,54 Nevertheless, it is not clear at this time whether this influence of CTAB on the formation of nanotriangles is due to the formation of seeds with twinning faults or to the stabilization of a particular crystalline facet.
In addition to the role of the cationic part of the surfactant, our results show that the influence of CTAB is also to provide bromide in the concentration range 0.5–5 mM in order to obtain triangular nanoplates. The role of halide ions in the morphology of NPs is still poorly understood, and seems to depend not only on the type and concentration of halide anion, but also on the particular chemical reaction and experimental conditions used in the synthesis.60,61 The results obtained in this work also show that nanotriangles are formed only in a narrow range of bromide concentrations, with a large excess of chloride, but further studies are necessary in order to elucidate if this is due to stabilization of specific crystal facets, or to a kinetic effect.
In this study, the transformation of the seeds into nanotriangles proceeds through growth of the seeds into irregular nanoplates that then reorganize into more regular nanotriangles. The timescale for these two processes is different: while the growth step occurs during photocatalytic reduction (≈ 2 h, irradiation), the reorganization step is much slower (24 h) and is not affected by light. Two other mechanisms have been described, namely a self-repair mechanism of nanoporous plates,24,62 and aggregation and fusion of seeds first forming dendritic/radial nanoaggregates that then evolve into regular nanotriangles.21 Thus, the type of growth mechanism does not seem to be critical for the formation of regular nanotriangles. One common feature in all these mechanisms is that the formation of nanotriangles is very slow, a characteristic that may be indirectly related to a low driving force of the reaction used, in accordance with the mechanism proposed by Viswanath et al.31,32 Our results clearly show that a slow reaction is essential for the formation of nanoplates, since the ratio of spheres:triangles increases as the concentration of photocatalyst, and thus the reaction rate, increases. However, it is to be noted, that in the photocatalytic reaction used, part of the driving force for the reaction comes from the light absorbed, and thus the photocatalyst concentration in the excited state influences the rate of the reaction, but also the thermodynamic driving force of the reaction.
Conclusions
Triangular gold nanoplates with controllable size between 50–250 nm were synthesized by a one-pot chemical method, involving the reduction of a Au(III) precursor at pH 7, using a Sn(IV) porphyrin as a photocatalyst and TEA as the final electron donor, in the presence of CTAB as the capping agent. This preparation method allows highly selective production of nanoplates, with <20% contamination by nanospheres and other shapes. The method allows the control of the average size of the nanoplates by changing the concentration of the photocatalyst over a broad range of sizes (250–45 nm), and additionally it is possible to fine-tune the size of the nanotriangles for a specific concentration of photocatalyst by adjusting the concentration of CTAB. This one-pot synthetic method features a very simple implementation, excellent repeatability, and provides high yields of triangular nanoplates. In addition, the use of the photocatalyst allows independent control of the kinetics of reduction of gold and other experimental factors, such as the concentration of capping agent, pH, etc., providing a convenient way to further elucidate the mechanism of nanoplate growth. Studies on the influence of the concentration of chloride and bromide, as well as a detailed study of the kinetics of formation of nanoparticles are currently in progress. We believe that these studies will allow further understanding of the crystalline and chemical factors that influence the final shape of the nanoparticles, and will thus lead to the development of a rationale for the design of simple chemical methodologies for the synthesis of non-spherical nanoparticles.Acknowledgements
This work was funded in part by Fundação para a Ciência e Tecnologia through project PTDC/QUI/64484/2006 and by the Luso-American Foundation (FLAD). AM and PQ thank Fundação para a Ciência e Tecnologia for PhD grants BD/17566/2004 and BD/28209/2006, respectively. We thank Prof. António Fernando Silva and CIQUP, Laboratório de Química Analítica, Faculdade de Ciências, Universidade do Porto for the use of the AFM. We thank Yan Qiu and Yujiang Song for some early TEM images of the nanotriangles. Sandia is a multiprogram laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the United States Department of Energy's National Nuclear Security Administration under contract DE-AC04-94AL85000.Notes and references
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668–677 CrossRef CAS.
- L. M. Liz-Marzan, Langmuir, 2006, 22, 32–41 CrossRef CAS.
- J. E. Millstone, S. Park, K. L. Shuford, L. D. Qin, G. C. Schatz and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 5312–5313 CrossRef CAS.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- J. E. Millstone, S. J. Hurst, G. S. Metraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS.
- S. S. Shankar, A. Rai, A. Ahmad and M. Sastry, Chem. Mater., 2005, 17, 566–572 CrossRef CAS.
- X. D. Xu, M. Stevens and M. B. Cortie, Chem. Mater., 2004, 16, 2259–2266 CrossRef CAS.
- M. H. Rashid, R. R. Bhattacharjee and T. K. Mandal, J. Phys. Chem. C, 2007, 111, 9684–9693 CrossRef CAS.
- A. Singh, M. Chaudhari and M. Sastry, Nanotechnology, 2006, 17, 2399–2405 CrossRef CAS.
- S. S. Shankar, A. Rai, B. Ankamwar, A. Singh, A. Ahmad and M. Sastry, Nat. Mater., 2004, 3, 482–488 CrossRef CAS.
- K. Aslan, J. R. Lakowicz and C. D. Geddes, J. Phys. Chem. B, 2005, 109, 6247–6251 CrossRef CAS.
- J. P. Schmidt, S. E. Cross and S. K. Buratto, J. Chem. Phys., 2004, 121, 10657–10659 CrossRef CAS.
- J. T. Zhang, X. L. Li, X. M. Sun and Y. D. Li, J. Phys. Chem. B, 2005, 109, 12544–12548 CrossRef CAS.
- I. Pastoriza-Santos and L. M. Liz-Marzan, J. Mater. Chem., 2008, 18, 1724–1737 RSC.
- C. R. Yonzon, D. A. Stuart, X. Y. Zhang, A. D. McFarland, C. L. Haynes and R. P. Van Duyne, Talanta, 2005, 67, 438–448 CrossRef.
- R. N. Goyal, A. Aliumar and M. Oyama, J. Electroanal. Chem., 2009, 631, 58–61 CrossRef CAS.
- Y. Chen, W. Schuhmann and A. W. Hassel, Electrochem. Commun., 2009, 11, 2036–2039 CrossRef CAS.
- J. P. Xie, J. Y. Lee, D. I. C. Wang and Y. P. Ting, Small, 2007, 3, 672–682 CrossRef CAS.
- J. E. Millstone, G. S. Metraux and C. A. Mirkin, Adv. Funct. Mater., 2006, 16, 1209–1214 CrossRef CAS.
- Z. R. Guo, Y. Zhang, Y. Q. Mao, L. Huang and N. Gu, Mater. Lett., 2006, 60, 3522–3525 CrossRef CAS.
- W. L. Huang, C. H. Chen and M. H. Huang, J. Phys. Chem. C, 2007, 111, 2533–2538 CrossRef CAS.
- H. C. Chu, C. H. Kuo and M. H. Huang, Inorg. Chem., 2006, 45, 808–813 CrossRef CAS.
- Y. L. Luo, Mater. Lett., 2007, 61, 1346–1349 CrossRef CAS.
- S. C. Yang, T. W. Zhang, L. Zhang, Q. F. Wang, R. L. Zhang and B. J. Ding, Nanotechnology, 2006, 17, 5639–5643 CrossRef CAS.
- Y. J. Xiong, I. Washio, J. Y. Chen, H. G. Cai, Z. Y. Li and Y. N. Xia, Langmuir, 2006, 22, 8563–8570 CrossRef CAS.
- J. H. Lee, K. Kamada, N. Enomoto and J. Hojo, Cryst. Growth Des., 2008, 8, 2638–2645 CrossRef CAS.
- H. Kawasaki, K. Nishimura and R. Arakawa, J. Phys. Chem. C, 2007, 111, 2683–2690 CrossRef CAS.
- C. Lofton and W. Sigmund, Adv. Funct. Mater., 2005, 15, 1197–1208 CrossRef CAS.
- J. L. Elechiguerra, J. Reyes-Gasga and M. J. Yacaman, J. Mater. Chem., 2006, 16, 3906–3919 RSC.
- B. Lim, P. H. C. Camargo and Y. N. Xia, Langmuir, 2008, 24, 10437–10442 CrossRef CAS.
- B. Viswanath, P. Kundu, A. HaIder and N. Ravishankar, J. Phys. Chem. C, 2009, 113, 16866–16883 CrossRef CAS.
- B. Viswanath, P. Kundu and N. Ravishankar, J. Colloid Interface Sci., 2009, 330, 211–219 CrossRef CAS.
- R. Baigorri, J. M. Garcia-Mina, R. F. Aroca and R. A. Alvarez-Puebla, Chem. Mater., 2008, 20, 1516–1521 CrossRef CAS.
- C. S. Ah, Y. J. Yun, H. J. Park, W. J. Kim, D. H. Ha and W. S. Yun, Chem. Mater., 2005, 17, 5558–5561 CrossRef CAS.
- Y. J. Song, Y. Yang, C. J. Medforth, E. Pereira, A. K. Singh, H. F. Xu, Y. B. Jiang, C. J. Brinker, F. van Swol and J. A. Shelnutt, J. Am. Chem. Soc., 2004, 126, 635–645 CrossRef CAS.
- R. M. Garcia, Y. J. Song, R. M. Dorin, H. R. Wang, P. Li, Y. Qiu, F. van Swol and J. A. Shelnutt, Chem. Commun., 2008, 2535–2537 RSC.
- Y. Song, R. M. Garcia, R. M. Dorin, H. R. Wang, Y. Qiu, E. N. Coker, W. A. Steen, J. E. Miller and J. A. Shelnutt, Nano Lett., 2007, 7, 3650–3655 CrossRef CAS.
- Y. J. Song, S. R. Challa, C. J. Medforth, Y. Qiu, R. K. Watt, D. Pena, J. E. Miller, F. van Swol and J. A. Shelnutt, Chem. Commun., 2004, 1044–1045 RSC.
- Y. J. Song, R. M. Dorin, R. M. Garcia, Y. B. Jiang, H. Wang, P. Li, Y. Qiu, F. van Swol, J. E. Miller and J. A. Shelnutt, J. Am. Chem. Soc., 2008, 130, 12602 CrossRef CAS.
- Y. J. Song, R. M. Garcia, R. M. Dorin, H. R. Wang, Y. Qiu and J. A. Shelnutt, Angew. Chem., Int. Ed., 2006, 45, 8126–8130 CrossRef CAS.
- Y. J. Song, M. A. Hickner, S. R. Challa, R. M. Dorin, R. M. Garcia, H. R. Wang, Y. B. Jiang, P. Li, Y. Qiu, F. van Swol, C. J. Medforth, J. E. Miller, T. Nwoga, K. Kawahara, W. Li and J. A. Shelnutt, Nano Lett., 2009, 9, 1534–1539 CrossRef CAS.
- Y. J. Song, Y. B. Jiang, H. R. Wang, D. A. Pena, Y. Qiu, J. E. Miller and J. A. Shelnutt, Nanotechnology, 2006, 17, 1300–1308 CrossRef CAS.
- H. R. Wang, Y. J. Song, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2006, 128, 9284–9285 CrossRef CAS.
- P. Quaresma, L. Soares, L. Contar, A. Miranda, I. Osorio, P. A. Carvalho, R. Franco and E. Pereira, Green Chem., 2009, 11, 1889–1893 RSC.
- N. Felidj, J. Grand, G. Laurent, J. Aubard, G. Levi, A. Hohenau, N. Galler, F. R. Aussenegg and J. R. Krenn, J. Chem. Phys., 2008, 128, 094702 CrossRef CAS.
- V. Germain, J. Li, D. Ingert, Z. L. Wang and M. P. Pileni, J. Phys. Chem. B, 2003, 107, 8717–8720 CrossRef CAS.
- F. Hubert, F. Testard and O. Spalla, Langmuir, 2008, 24, 9219–9222 CrossRef CAS.
- A. Kumar, S. Mandal, P. R. Selvakannan, R. Pasricha, A. B. Mandale and M. Sastry, Langmuir, 2003, 19, 6277–6282 CrossRef CAS.
- S. Perkin, N. Kampf and J. Klein, J. Phys. Chem. B, 2005, 109, 3832–3837 CrossRef CAS.
- V. K. Aswal and P. S. Goyal, Chem. Phys. Lett., 2002, 364, 44–50 CrossRef CAS.
- W. Robb, Inorg. Chem., 1967, 6, 382 CrossRef CAS.
- I. V. Mironov, Russ. J. Inorg. Chem., 2008, 53, 655–659 CrossRef.
- S. M. Kreisig, A. Tarazona, E. Koglin and M. J. Schwuger, Langmuir, 1996, 12, 5279–5288 CrossRef CAS.
- E. Koglin, A. Tarazona, S. Kreisig and M. J. Schwuger, Colloids Surf., A, 1997, 123–124, 523–542 CrossRef CAS.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS.
- H. Min-Chen, R. S. Liu and D. P. Tsai, Cryst. Growth Des., 2009, 9, 2079–2087 CrossRef CAS.
- A. A. Umar, M. Oyama, M. M. Salleh and B. Y. Majlis, Cryst. Growth Des., 2009, 9, 2835–2840 CrossRef CAS.
- L. Zhang, C. Z. Huang, Y. F. Li and Q. Li, Cryst. Growth Des., 2009, 9, 3211–3217 CrossRef CAS.
- B. Jonsson, B. Lindman, K. Holmberg and B. Kronberg, Surfactants and Polymers in Aqueous Solution, John Wiley and Sons, Chichester, U.K., 1998 Search PubMed.
- T. H. Ha, H. J. Koo and B. H. Chung, J. Phys. Chem. C, 2007, 111, 1123–1130 CrossRef CAS.
- A. Rai, A. Singh, A. Ahmad and M. Sastry, Langmuir, 2006, 22, 736–741 CrossRef CAS.
- S. C. Yang, Y. P. Wang, Q. F. Wang, R. L. Zhang, Z. M. Yang, Y. Guo and B. J. Ding, Cryst. Growth Des., 2007, 7, 2258–2261 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional details concerning the synthesis of the nanotriangles in different experimental conditions and characterization by XPS and TEM. See DOI: 10.1039/c0nr00337a |
| ‡ Present Address: Institute of General and Ecological Chemistry, Technical University of Łódź, Żeromskiego 116, 90-924 Łódź, Poland |
| This journal is © The Royal Society of Chemistry 2010 |