Amplified energy transfer in conjugated polymer nanoparticle tags and sensors
Zhiyuan
Tian
,
Jiangbo
Yu
,
Changfeng
Wu†
,
Craig
Szymanski‡
and
Jason
McNeill
*
Department of Chemistry, Clemson University, Clemson, SC 29634, USA. E-mail: mcneill@clemson.edu
First published on 10th August 2010
Abstract
Nanoparticles primarily consisting of π-conjugated polymers have emerged as extraordinarily bright fluorescent tags with potential applications in biological imaging and sensing. As fluorescent tags, conjugated polymer nanoparticles possess a number of advantageous properties, such as small particle size, extraordinary fluorescence brightness, excellent photostability, and high emission rate. Exciton diffusion occurring in the nanoparticles results in amplified energy transfer, doubling the energy transfer efficiency in some cases. Amplified energy transfer has been exploited to obtain highly red-shifted emission, oxygen-sensing nanoparticles, and fluorescence photoswitching. Additional observed phenomena are attributable to amplified energy transfer in conjugated polymers, including superquenching by metal nanoparticles, and fluorescence modulation by hole polarons. This feature article presents an overview of recent investigations of optical properties and energy transfer phenomena of this relatively novel type of fluorescent nanoparticle with a viewpoint towards demanding fluorescence-based imaging and sensing applications.
 Zhiyuan Tian | Zhiyuan Tian graduated from Wuhan University, China with BSc in 1995. He joined Prof. Jiannian Yao's group at the Institute of Chemistry, Chinese Academy of Sciences (ICCAS), in 2000 and earned his PhD in physical chemistry in 2005. He is currently working as a postdoctoral researcher in Prof. Jason McNeill's group in the Department of Chemistry at Clemson University. His research interests include nanoscaled fluorescent tags and sensors based on π-conjugated polymers and their optical/photonic properties, photoswitchable fluorescent nanoprobes and their applications in super-resolution bioimaging and bioanalysis, nonlinear optical properties of organic nanomaterials. |
 Jiangbo Yu | Jiangbo Yu obtained his PhD degree in Chemistry from the Chinese Academy of Sciences (CAS) in 2005 and has since then worked as Assistant Professor in Changchun Institute of Applied Chemistry, CAS. In 2007, he went to University of Regensburg (Germany) and worked as a research assistant. Since 2008, he has worked as a postdoctoral researcher in Department of Chemistry, Clemson University. His current research interest focuses on studying the photophysical properties of fluorescent conjugated polymer nanoparticles based on the single molecule fluorescence microscopy. |
 Changfeng Wu | Changfeng Wu received his BS in material engineering from Shandong institute of light industry in 1998, his MS degree in material science from Jilin University in 2001, and his PhD in physics from the Chinese Academy of Sciences in 2004 with Prof. Weiping Qin. He obtained his PhD in chemistry from Clemson University in 2008 with Prof. Jason D. McNeill. He is currently conducting his postdoctoral research in the laboratory of Prof. Daniel T. Chiu at University of Washington. His research focuses on the development of semiconducting polymer nanoparticle probes for biological imaging and biosensors. |
 Jason McNeill | Jason McNeill earned his PhD in Physical Chemistry from the University of California, Berkeley in 1999. After postdoctoral research with Paul Barbara at the University of Texas, Austin, he joined the faculty of Department of Chemistry at Clemson University, where he currently holds the rank of Associate Professor. In 2006 he was the recipient of the CAREER award from the US National Science Foundation. His research interests include the development and characterization of fluorescent nanoparticles, single molecule spectroscopy, development of fluorescence-based imaging techniques, and applications in biology. |
1. Introduction
A wide variety of fluorescence-based spectroscopy and imaging techniques are used to explore biological and biochemical structure and dynamics, owing to the availability of a wide array of bright fluorophores and straightforward bioconjugation techniques, as well as the high sensitivity, low background, multiplexed detection, nanoscale structural information, and 3D imaging capabilities afforded by fluorescence-based methods. There has been a resurgence of interest in fluorescence-based methods due to the emergence of powerful tools such as multiphoton-excited fluorescence microscopy,1 single molecule spectroscopy,2–4 nanometre-resolution single particle tracking,5,6 and ultra-resolution fluorescence microscopy.7,8 The brightness and photostability of the fluorescent probes are often key limiting factors for these applications. The need for brighter and more photostable fluorescent tags has provoked intense interest in the development of fluorescent labels based on a wide variety of nanoparticles,9 including colloidal semiconductor quantum dots (QDs),10 and dye-doped silica or polymer nanoparticles.11–13Nanoparticles consisting primarily of conjugated polymers have emerged as a novel type of fluorescent nanoparticle with a variety of potentially useful optical properties. Fig. 1 shows the molecular structures of several conjugated polymers used to prepare nanoparticles, typical particle size and spectral data of the as-prepared polymer nanoparticles. Nanoparticles with controlled sizes ranging from a few nm (a single, collapsed conjugated polymer chain) to >50 nm can be formed by a nano-precipitation method.14,15 The resulting nanoparticles exhibit extraordinarily high fluorescence brightness as compared to other fluorescent nanoparticles of similar size.15,16Additionally, conjugated polymer nanoparticles (CPNs) exhibit extraordinarily high two-photon excitation cross-sections, which are promising for applications in two-photon-excited fluorescence imaging.17 CPNs display amplified energy transfer to fluorescent dye dopants, resulting in bright, highly red-shifted emission and other phenomena, such as sensing and photoswitching.18–21 Also, as nanoscale organic semiconductors, CPNs and single conjugated polymer chains exhibit a range of complex photophysical phenomena, such as exciton diffusion, exciton–polaron collisions, and (in some cases) single particle blinking.22–26 In this feature article, we will summarize a number of recent findings regarding this novel class of fluorescent nanoparticle. The key photophysical characteristics and parameters related to CPN fluorescence brightness will be summarized, establishing CPNs as arguably the brightest fluorescent nanoparticles to date in the 5–50 nm diameter size range. In addition, the role of exciton diffusion in complex photophysical phenomena such as amplified energy transfer, superquenching, single nanoparticle blinking, and possible applications of such phenomena are discussed. Demonstrations of oxygen sensing and photoswitching in dye-doped CPNs will be discussed. Finally, we discuss future prospects and current efforts to improve the photophysical properties or extend the range of applications of this promising class of fluorescent nanoparticles.
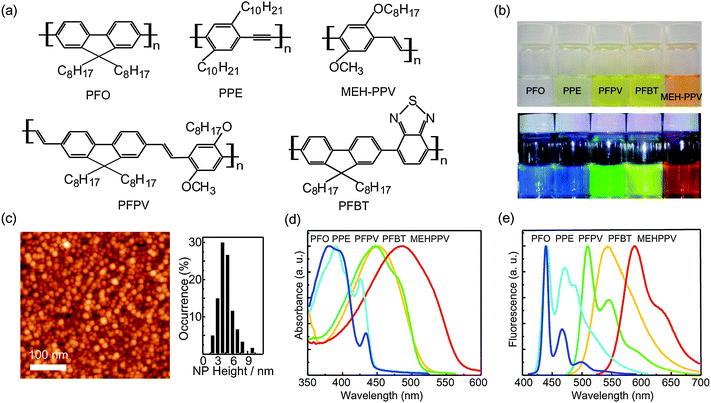 | ||
| Fig. 1 (a) Chemical structures of the conjugated polymer nanoparticles (CPNs). (b) Photographs of aqueous CPNs suspensions under room light (upper) and UV light (lower). (c) Typical AFM images of the densely packed PFO nanoparticles (left) and the corresponding histogram of the nanoparticle height (right). (d) Absorption spectra and (e) fluorescence spectra of various CPNs. Reproduced with permission from ref. 15 and 27. Copyright American Chemical Society, 2006 and 2008. | ||
2. Preparation, morphology, and structure of conjugated polymer nanoparticles
Conjugated polymer nanoparticles can be prepared using a variety of methods, including nano-precipitation,14–20,27–35 miniemulsion dispersion,36–39 mini-emulsion polymerization,40–43 self-assembly of polymer brushes,44 and dendrimer-based strategies.45 We shall focus our discussion on nanoparticles produced by the nano-precipitation method, which is a modification of the reprecipitation method developed by Nakanishi and co-workers for the preparation of micron- to submicron-sized organic particles,46–49and later applied to conjugated polymers by Masuhara and co-workers.34,35 Reprecipitation is essentially a solvent exchange method in which a small amount of the target compound dissolved in a “good” solvent rapidly mixes with excess of a “poor” solvent. Owing to the solubility discrepancy of the organic compound in two solvents and the miscibility of these two solvents, the sudden change of solvent quality induces the aggregation of the organic molecules, causing nanoparticle formation. We found that, for several conjugated polymers, a nano-precipitation method using dilute (<10 ppm) polymer solution in THF, followed by rapid addition to water under brief sonication to increase the mixing rate, results in the formation of very small nanoparticles (∼5 nm dia. in some cases, Fig. 1c) containing 1–3 polymer chains, with some fraction of the polymer forming larger aggregates.14,18,27 Despite the rigidity of the π-conjugated backbone, small (<30 nm dia.) hydrophobic conjugated polymer nanoparticles tend to exhibit roughly spherical particle morphology, since surface-tension effects typically predominate in this size range.43 The internal structure of the nanoparticles varies, with either amorphous or semicrystalline phases produced, depending on the polymer and preparation conditions.283. CPN fluorescence brightness, saturated emission rate, and photostability
It has become increasingly clear that nanoparticles vary significantly in terms of the signal provided for a given application. While there is no single metric for comparing fluorescence brightness in all applications, several photophysical parameters are useful for predicting relative signal level and suitability for a range of applications. The fluorescence cross-section, σfl, given by the product of the optical absorption cross-section and the fluorescence quantum yield, provides a measure of particle brightness under low to moderate excitation intensities for which saturation is not an issue. The ratio of fluorescence cross-section to the nanoparticle volume or mass provides a reasonable comparison of the relative brightness of nanoparticles of different sizes, or when characterizing the brightness of a type of nanoparticle that can be fabricated in a variety of sizes. Fig. 2a shows the fluorescence cross-section/volume ratios σflV−1 for several nanoparticles and other fluorophores, estimated from literature reports.50–60 The cross-section/volume ratios were compared for nanoparticles without biocompatibility layers, since the thickness of such layers is highly variable. The cross-section/volume ratios vary over several orders of magnitude for different types of nanoparticles, with CPNs based on poly(9,9-dioctylfluorene-co-benzothiadiazole) (PFBT) exhibiting the highest ratio of peak fluorescence cross-section to particle volume—nearly two orders of magnitude higher than those of CdSe quantum dots (at 488 nm) or dye-loaded silica spheres. These differences can be explained by taking into account the principal factors that contribute to the per-particle fluorescence cross-section, including the transition dipole moment, fluorescence quantum yield, and effective number of fluorophores. CdSe quantum dots possess a large transition dipole moment, resulting in typical fluorescence cross-sections roughly 10 times those of organic dyes such as Rhodamine 6G, for quantum dots with excitation and emission in the visible region of the spectrum.61 Dye-loaded silica particles derive their brightness from the large number of chromophores contained in a single nanoparticle, however, the chromophore density is relatively low, due to aggregation quenching effects that typically limit the optimum dye loading to a few percent, resulting in fluorescence cross-sections roughly 27 times those of a single dye molecule, for 30 nm diameter particles.54 In contrast, the effective chromophore density of CPNs is much higher, since they are almost entirely comprised of π-conjugated polymer. This results in fluorescence cross-sections as high as 1.1 × 10−13 cm2, for PFBT-based CPNs 15 nm in diameter, larger than the peak fluorescence cross-section of Rhodamine 6G by a factor of roughly 400.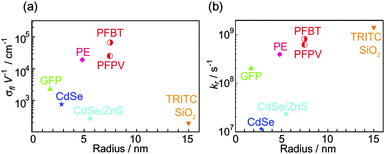 | ||
| Fig. 2 Fluorescence cross-section/volume ratio (a) and per-particle saturated radiative rate (b) vs. the radii of various fluorescent tags, including green fluorescent protein (GFP), phycoerythrin (PE), bare CdSe QDs, core–shell (CdSe)ZnCdS QDs, core–shell nanoparticles with multiple tetramethylrhodamine isothiocyanate (TRITC) as the core that is protected by silica shell, and π-conjugated polymer nanoparticles (PFBT and PFPV dots). The volumes of the fluorescent tags were calculated/estimated as following: GFP possesses a cylindrical structure with 4.2 nm long and 2.4 nm in diameter,50 PE protein exhibits a disc-shaped structure with a mean diameter of 10.1 nm and height of 5.4 nm,51 bare CdSe QDs have hydrodynamic diameter of 5.6 nm,52 Core–shell (CdSe)ZnCdS QDs have a hydrodynamic diameter of 11 nm,60 and TRITC silica nanoparticles have mean diameter of 30 nm with ∼8.7 incorporated TRITC molecules.54 For absorption cross-section values in unit of cm2, 6.5 × 10−17 for GFP,55 9.2 × 10−15 for PE protein,56 4.6 × 10−16 for the bare CdSe and the core–shell quantum dots,57 were used. For fluorescence quantum yields, 0.64 for GFP,58 0.98 for PE protein,59 0.15 for the bare CdSe QDs, 0.50 for the core–shell QDs,53 and 0.47 for TRITC SiO2 particles54 were used. For the fluorescence lifetimes in unit of ns: 3.2 for GFP,65 2.5 for PE protein,66 13.3 for the bare CdSe QDs,67 21.9 for the core–shell QDs,57,68 and 2.9 for TRITC SiO2 particles54 were used. All the parameters of the conjugated polymer nanoparticles involved in this figure (PFBT and PFPV dots) come from our group.15 | ||
For applications involving high excitation rates, such as high speed imaging and flow cytometry, the saturated per-particle photon emission rate is often a key factor in determining particle brightness. As shown in Fig. 2b, the saturated emission rates for several common nanoparticles and fluorophores vary by orders of magnitude. The nature of colloidal CdSe quantum dots as a single quantum system results in a much lower per-particle photon emission rate as compared to nanoparticles containing multiple fluorophores such as dye-loaded silica particles and CPNs—for a single quantum system, the maximum theoretical photon emission rate is equal to the radiative rate, while for a system containing multiple independent fluorophores, the theoretical maximum per-particle photon emission rate is given by the radiative rate of the fluorophore multiplied by the number of fluorophores. Other factors can also have a substantial detrimental impact on the per-particle emission rate, particularly the presence of long-lived dark states. For conventional organic fluorophores, such dark states are typically triplet excited states, charge-transfer states, or conformational states with weak fluorescence. In quantum dots, auger processes can result in the formation of dark states.62 In CPNs, an infrequent, reversible photoinduced electron transfer process leads to the formation of hole polarons (cations), which can act as efficient exciton quenchers,63,64 effectively reducing the observed saturated emission rate of single CPN well below the theoretical maximum.15 A more detailed discussion of quenching by hole polarons in CPNs and efforts towards limiting the effect of polaron-induced quenching on the emission rate are provided and discussed in Section 4.6.
Extended imaging and high resolution particle tracking studies often require fluorescent labels with high photostability. The most photostable organic dyes can emit millions of photons per dye molecule prior to photobleaching, under favorable conditions.69–71 A quantitative measure of photostability is the photon number or “death number”, which represents the average total number of photons emitted per particle prior to photobleaching, and is equal to the ratio of the fluorescence quantum yield to the photobleaching quantum yield.72 Quantitative photostability information for nanoparticles is rarely reported, in part due to the many factors that can affect measured photostability, including excitation intensity, pH, ionic strength, and the presence of reactive species (such as molecular oxygen). Furthermore, for nanoparticles, the presence of an encapsulating shell, as well as its quality and effectiveness in blocking access of reactive species to the nanoparticle core, is a key issue for photostability, resulting in highly variable photostability even within a given class of nanoparticle. Time-lapse microscopy images for biological samples labeled with different nanoparticles or comparing a nanoparticle-labeled sample to a dye-labeled sample are often employed as a qualitative measure of photostability. However, the results of such experiments can be highly misleading, favoring dim nanoparticles with low fluorescence cross-sections, which undergo fewer excitation–emission cycles over a given time interval. Despite these uncertainties, a preponderance of literature reports indicates that dye-loaded silica nanoparticles and quantum dots are more photostable than organic dyes.10,73 We determined the photon numbers of several CPNs by performing quantitative single nanoparticle photobleaching and bulk solution photobleaching measurements.15 PFBT-based CPNs were determined to exhibit extraordinarily high photostability, with photon numbers on the order of ∼109 for 10 nm diameter particles, roughly three orders of magnitude higher than those of typical fluorescent dyes,15 and preliminary results indicate that dye doping and encapsulation with a silica shell can further improve photostability. However, a quantitative direct comparison of the photostability of CPN and colloidal semiconductor quantum dots has not been performed.
4 Exciton diffusion and energy transfer in conjugated polymer nanoparticles
Single conjugated polymer chains and CPNs exhibit a number of complex and interesting photophysical phenomena at the single molecule or single particle level and in bulk suspensions, due to both their nanoscale size and their properties as organic semiconductor materials. Single conjugated polymer chains dispersed in a polymer matrix were observed to exhibit pronounced blinking behavior, with dark state lifetimes of ∼200 ms,74 which has been attributed to efficient quenching by reversibly photogenerated of hole polarons.25,75–77 The fluorescence of single conjugated polymer chain was also found to exhibit photon anti-bunching phenomena due to singlet–singlet exciton collisions.78 Singlet–triplet exciton collisions have also been explored in single conjugated polymer molecules.25 Electrochemical injection of holes into CPNs was observed.33 Efficient energy transfer in polymer blends18 and dye dopants19 has been observed, single CPN oxygen sensors have been demonstrated,20 and photoswitchable fluorescence in CPNs doped with photochromic dyes has been reported.21 Complex photobleaching kinetics have been observed in conjugated polymer films and in CPNs. These phenomena all involve highly efficient energy transfer resulting from a combination of exciton diffusion and Förster energy transfer, which results in much larger quenching volumes than Förster energy transfer alone. Exciton diffusion models have been developed that make predictions of energy transfer efficiency and exciton dynamics in doped molecular crystals79,80 and doped polymer films.81 We have developed a similar model that also takes into account the effects of nanoparticle size.19 In this section, these photophysical phenomena of CPNs are reviewed and the roles of exciton diffusion and energy transfer are discussed.4.1 Amplified energy transfer in blended conjugated polymer nanoparticles and dye-doped CPNs
Energy transfer in conjugated polymer blends has been demonstrated as a viable strategy for improving the quantum efficiency and tuning the emission color of light-emitting devices.82–85 Owing to the high absorption cross-section, high fluorescence quantum yield, and broad emission spectra resulting in excellent overlap with a large variety of energy acceptors, conjugated polymers are ideal photon “harvesting” materials. Furthermore, exciton diffusion (i.e., multiple energy transfer) occurring in conjugated polymers can increase the efficiency of energy transfer to acceptors.86In an effort to develop CPNs with improved optical properties, we recently investigated energy transfer phenomena in conjugated polymer blends and in dye-doped conjugated polymer nanoparticles, finding evidence of amplified energy transfer due to exciton diffusion.18,19 In the investigations of energy transfer in polymer blends, poly(9,9-dihexylfluorenyl-2,7-diyl) (PDHF), a blue-emitting conjugated polymer, was employed as the host material and energy donor, while three other green-to-red-emitting polymers, poly[{9,9-dioctyl-2,7-divinylenefluorenylene}-alt-co-{2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylene}] (PFPV), poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1′,3}-thiadiazole)] (PFBT), and poly[2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene] (MEH-PPV), were employed as the energy acceptor materials. Blended nanoparticles were prepared by nano-precipitation from a solution mixture of the two polymers dissolved in THF. AFM measurement results indicated that the blended polymer nanoparticles are approximately spherical and most particles possess diameters in the range of 25 ± 3 nm, corresponding to roughly 100 to 200 polymer molecules per nanoparticle. The blended polymer nanoparticles exhibit fluorescence excitation spectra very similar to those of the host polymer (polyfluorene), and fluorescence emission characteristic of the guest polymer, indicating energy transfer from the host polymer to the guest polymer (Fig. 3a and b). The large separation between the excitation peak and the emission peak, as much as 215 nm in the case of PF nanoparticles doped with MEH-PPV, is expected to facilitate reduction of the effects of autofluorescence and scattered excitation light in sensing and imaging experiments.
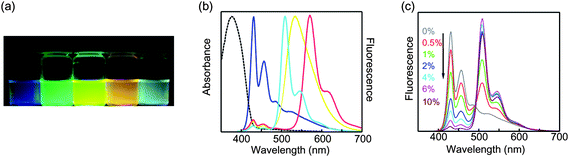 | ||
| Fig. 3 (a) Photograph of fluorescence emission from aqueous suspensions of the blend nanoparticles taken under a UV lamp (365 nm). (b) Normalized absorption (dashed) and fluorescence emission spectra (solid) of pure PDHF (blue), PDHF doped with 6% PFPV (cyan), 6% PFBT (yellow), and 6% MEH-PPV (red) polymer blend nanoparticles. (c) Concentration-dependent fluorescence spectra of PDHF/PFPV polymer blend nanoparticles under 375 nm excitation. Reproduced with permission from ref. 18. Copyright American Chemical Society, 2006. | ||
The high energy transfer efficiency observed (roughly 50% efficient at 2% acceptor doping and >90% efficient at 6% acceptor doping) is consistent with amplified energy transfer due to exciton diffusion, and also indicates negligible segregation at the nanoparticle level (few nanoparticles containing only one type of polymer) as well as negligible phase separation within the nanoparticles. In contrast, pronounced phase separation is often observed in cast thin films of conjugated polymer blends, due to the low entropy of mixing of polymers.87,88 It is likely that the lack of phase separation in the nanoparticles is at least in part due to the rapid particle formation process, which could lead to kinetic trapping of the blended state.
Amplified energy transfer was also observed in conjugated polymer nanoparticles doped with a variety of dyes.19 Polyfluorene (PDHF) nanoparticles were doped with several different organic-small-molecule fluorescent dyes: perylene, Nile Red, Coumarin 6, and tetraphenylporphyrin (TPP). The dyes were incorporated into the PDHF nanoparticles via the nano-precipitation process previously employed to prepare blended conjugated polymer nanoparticles. At a few percent doping fraction, the fluorescence from the host component (PDHF) is almost completely quenched and there is strong dopant fluorescence (Fig. 4a), indicating efficient energy transfer from the host polymer to the dopant dyes. Another noteworthy feature of these dye-doped CPNs is the highly red-shifted emission spectrum as compared to pure polymers and typical fluorescent dyes. In the case of TPP-doped PDHF CPNs, near UV (375 nm) excitation generates a vivid red (650 nm) fluorescence emission, with a remarkable separation (275 nm) between excitation and emission wavelength. The dye-doped CPNs also exhibit excellent photostability, hundreds or thousands of times higher than dyes in solution. Moreover, the combination of large per-particle absorptivity (from host polymers) and high fluorescence quantum yield (from guest fluorescent dyes) enables large improvements in fluorescence brightness. In the case of 30 nm PDHF CPNs doped with either perylene or Coumarin 6 (2 wt%), a fluorescence quantum yield of ∼40% was obtained. Furthermore, the highly red-shifted fluorescence emission of the dye-doped CPNs is advantageous for biological imaging, since a large red-shift facilitates rejection of undesired scattered light and autofluorescence.
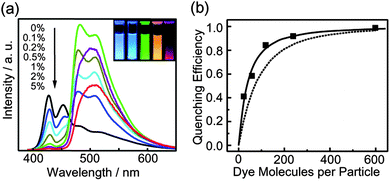 | ||
| Fig. 4 (a) Concentration-dependent fluorescence spectra of PDHF nanoparticles doped with Coumarin 6. Inset: photograph of fluorescence emission from aqueous suspensions of the dye-doped PDHF nanoparticles taken under UV lamp excitation (365 nm). From left to right: neat PDHF, PDHF doped with 2% perylene, PDHF doped with 2% Coumarin 6, PDHF doped with 5% Nile Red, and PDHF doped with 5% TPP. (b) Quenching efficiency as a function of the number of Coumarin 6 molecules per 15 nm dia. PDHF nanoparticle. The squares are experimental results, while the dotted curves represent the predicted energy transfer efficiency assuming only Förster energy transfer model, while the solid curves represent the results of the combined exciton diffusion and Förster transfer model. Reproduced with permission from ref. 19. Copyright American Chemical Society, 2008. | ||
4.2 Nanoparticle exciton diffusion and energy transfer model
In organic semiconductors such as conjugated polymers, the neutral excited state produced by photoexcitation is typically classified as a Frenkel-type exciton.89 Several energy transfer processes can result in highly mobile excitons whose motion can be described by an exciton diffusion length, which can range from a few nm in disordered polymers to hundreds of nanometres in molecular crystals. In order to gain an insight into how exciton diffusion enhances energy transfer in CPNs, we developed a straightforward model incorporating exciton diffusion, energy transfer, and particle size. The finite size of the nanoparticle has a significant effect on energy transfer efficiency, and also precludes a simple analytical solution, so the model was implemented as a stochastic simulation. The energy transfer efficiencies for the dye-doped PDHF nanoparticles determined at several dye loading fractions were compared to model calculations, treating the exciton diffusion length as a fit parameter. The model was implemented as a 3D random walk simulation, with excitons and acceptor dyes confined to the interior of a sphere. Details of the simulation are published elsewhere,20 but an outline of the key features is presented here. The system is represented as a set of randomly positioned excitons and randomly positioned acceptors confined to a sphere. At each time step of duration Δt, each exciton takes a step in a random direction, with the distance step length adjusted according to the exciton diffusion constant. In addition, at each time step, each exciton has a probability of decaying via radiative or non-radiative decay, given by p = (kr + knr)Δt, and a probability of a given exciton undergoing Förster energy transfer given by p = kET,iΔt, with the energy transfer rate constant given by,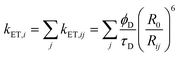 | (1) |
4.3 Energy transfer in mixed-phase conjugated polymer nanoparticles
Many conjugated polymers exhibit complex phase behavior, forming a variety of phases, depending on the structure of side chains, temperature, and solvent conditions.87,95–97 For instance, it is known that poly(9,9)-dioctylfluorene (PFO) exhibits two distinct phases—a disordered glassy phase and a crystalline β-phase containing planar polymer chains.98,99 The presence of β-phase in PFO films exerts significant influence on their photophysical properties, such as generation of polarons and triplet excitons, fluorescence spectrum, and fluorescence quantum yield.100–103 Recently, we investigated the influence of nanoparticle preparation conditions and solvent-induced swelling on the phase of PFO CPNs and the resulting changes in optical properties.28 PFO nanoparticles prepared via nano-precipitation initially exhibit the spectroscopic characteristics of the glassy phase, with no detectable β-phase. Upon adding organic solvent (either THF or toluene) to an aqueous suspension of PFO nanoparticles, solvent-induced swelling facilitated the formation of the thermodynamically favored β-phase, which persisted after removal of the organic solvent. The solvent-treated nanoparticles exhibit an additional narrow absorption peak at 435 nm, consistent with the presence of β-phase, while the fluorescence of the treated nanoparticles is red-shifted, with a pronounced vibronic progression (Fig. 5a).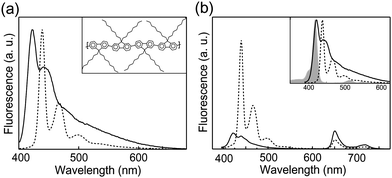 | ||
| Fig. 5 (a) Fluorescence spectra of glassy PFO nanoparticles (solid) and mixed-phase nanoparticles (dotted) obtained by toluene swelling. Inset: chemical structure and the β-phase conformation of the conjugated polymer PFO. (b) Fluorescence emission spectra of TPP-doped glassy PFO (solid) and β-phase nanoparticles (dotted). The inset displays spectral overlap between the TPP absorption (gray) and fluorescence emission of glassy PFO (solid) and β-phase nanoparticles (dotted). Reproduced with permission from ref. 28. Copyright American Chemical Society, 2008. | ||
Efficient energy transfer from the glassy-phase to the β-phase was observed. The red-shifted absorption features of the β-phase PFO provide good spectral overlap with the emission of the glassy phase, as shown in Fig. 5b. For nanoparticles containing a substantial fraction of β-phase, the emission of the glassy phase is completely quenched. The fluorescence quantum yield of PFO nanoparticles containing β-phase was ∼35%, as compared to ∼21% for the untreated (glassy-phase) nanoparticles. Fluorescence lifetime results indicate that the fluorescence quantum yield enhancement is attributable to a combination of efficient energy transfer to the β-phase and reduced non-radiative pathways in the β-phase relative to the glassy-phase. Another possibility is that the minority β-phase domains function as exciton traps, reducing exciton mobility and therefore the rate of energy transfer to quencher species such as hole polarons. In order to determine the effect of β-phase on exciton mobility and energy transfer efficiency, fluorescence measurements were performed on solvent-treated and untreated PFO nanoparticles doped with tetraphenylporphyrin (TPP). The TPP-doped glassy-phase PFO nanoparticles exhibit a strong TPP fluorescence (Fig. 5b), while solvent-treated TPP-doped PFO nanoparticles exhibit much less TPP fluorescence, indicating that the presence of β-phase significantly reduces the efficiency of energy transfer to acceptor dyes, likely due to competitive energy transfer to the β-phase as well as poor spectral overlap of the β-phase with TPP.
4.4 Superquenching of conjugated polymer nanoparticles by Au nanoparticles
In addition to the intra-particle energy transfer phenomena discussed above, inter-particle energy transfer from CPNs to Au nanoparticles was also investigated, and superquenching was observed.27 CPNs were prepared from PFO, PFPV, and MEH-PPV via modified nano-precipitation strategy as introduced previously, with diameter in the range of 3–14 nm. A silica encapsulation procedure was carried out via the condensation of octyltrimethoxysilane (OTMOS) on the surface of the nanoparticles, forming a silica shell around the CPNs. Subsequently, the silane coupling agent 3-aminopropyltrimethoxysilane (APTMS) was employed to functionalize the surface of silica-coated CPNs with amine groups, which are known to strongly bind to the surface of Au nanoparticles.104,105 Assembled nanostructures containing CPNs and Au nanoparticles (with diameter ∼15 nm) formed upon incubating amine-functionalized CPNs with Au nanoparticles, as shown in Fig. 6a.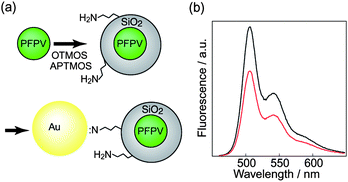 | ||
| Fig. 6 (a) Scheme demonstrates the functionalization of CPNs with amine groups and the subsequent assembly of CPNs with Au nanoparticles. (b) Fluorescence emission spectra of PFPV CPNs and CPN-Au assemblies. Reproduced with permission from ref. 27. Copyright American Chemical Society, 2006. | ||
Au nanoparticles are excellent fluorescence quenchers owing to their large absorption cross-section and broad absorption band.106 Fluorescence quenching of PFPV and MEH-PPV nanoparticles by Au nanoparticles was observed (Fig. 6b), and large Stern–Volmer constants (KSV) of ∼1 × 109 M−1 for PFPV CPNs and ∼2 × 109 M−1 for MEH-PPV CPNs were determined. This result is similar to the fluorescence superquenching effect reported for conjugated polymer electrolytes and Au nanoparticles.106 The observed superquenching effect is likely due to a combination of the large absorption cross-section of Au nanoparticles and exciton diffusion occurring in the CPN. This effect is promising for potential sensing applications or distance reporting using this nanoparticle dyad. Also, these results provided the first confirmation that CPNs can be encapsulated and functionalized, a significant step towards the broad application of CPNs as fluorescent labels for biological applications.
4.5 Fluorescence photoswitching in conjugated polymer nanoparticles by energy transfer to photochromic dyes
Photoswitchable fluorescence has attracted considerable attention because of the possible application of photoswitchable fluorescence to super-resolution biological imaging. Single-molecule-based super-resolution imaging, such as photoactivated localization microscopy (PALM)7 and stochastic optical reconstruction microscopy (STORM),107 has demonstrated resolution of as low as 10 nm. These techniques typically involve using light to activate or deactivate fluorophores, so that only a small number of fluorophores are active within the laser excitation spot. After many switching cycles, a super-resolution fluorescence image is constructed from the positions of thousands of fluorophores. An ideal photoswitchable fluorophore for this application would exhibit high brightness and photostability, single-step, reversible photoswitching, and a high ON/OFF contrast ratio. Further improvements in the spatial resolution and applicability of single-molecule-based super-resolution imaging will require the development of brighter, reversibly photoswitching fluorophores with a high contrast ratio. Therefore, there has been considerable interest in the development of photoswitchable nanoparticles, primarily based on energy transfer to photochromic dyes.13,108 While these nanoparticles are bright and exhibit reversible fluorescence photochromism, they exhibit more or less linear photoswitching behavior, not the single-step photoswitching required for STORM and PALM. However, recent results indicate that super-resolution imaging might also be possible using photoswitching nanoparticles lacking single-step photoswitching,109 and other super-resolution techniques such as stimulated emission depletion (STED) fluorescence microscopy110 do not require single-step photoswitching.CPNs exhibit the arguably highest fluorescence brightness to date in the 5–50 nm diameter size range, and are suitably small for super-resolution imaging. Also, as discussed above, CPNs exhibit highly efficient energy transfer to dye dopants, and should therefore be suitable for photoswitching by energy transfer to photochromic dyes dopants. Recently, Harbron et al. demonstrated that MEH-PPV nanoparticles doped with photochromic spirooxazine dye resulted in nanoparticles exhibiting reversible fluorescence photoswitching.21 In their investigation on the fluorescence photoswitching performance of the composite nanoparticle, 365 nm light was employed to convert spirooxazine to the merocyanine form, which slowly converts back to the spirooxazine form via a thermal process. While spirooxazine does not have significant absorbance in the 500–700 nm range, and thus does not act as a fluorescence quencher for MEH-PPV, UV-induced conversion from spirooxazine to merocyanine resulted in more than 90% quenching of the MEH-PPV fluorescence, corresponding to a photoswitching ratio larger than 10. Moreover, the photoswitching was reversibly cycled numerous times upon alternating UV irradiation and dark recovery, with no detectable fatigue effect. The efficient energy transfer observed is in good accordance with the exciton diffusion-mediated amplification of energy transfer efficiency as discussed above.
In order to test the application of fluorescence photoswitching in CPNs for imaging or possible nano-device applications requiring high single particle brightness, we evaluated single particle photoswitching using similar CPNs doped with photochromic dyes and evaluated the photostability and robustness of cycling. The conjugated polymer poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(1,4-benzo-{2,1′,3}-thiadiazole)] (PFBT) was used as the energy donor (fluorophores), and spironaphthoxazine was used as the photoresponsive energy acceptor. Composite nanoparticles were prepared via a reprecipitation–encapsulation method,27,111 yielding nanoparticles roughly 15 nm in diameter with a thin silica encapsulation layer, which was required to improve the colloidal stability of the nanoparticles. Fig. 7a demonstrates a representative FRET-based reversible fluorescence photoswitching system with PFBT functioning as the fluorophore and energy donor and spironaphthoxazine as the photochromic acceptor. The closed form of spironaphthoxazine (spiro form) has a weak absorption in the visible region, while the open form (merocyanine form) has a strong absorption band that overlaps with the PFBT emission band. Thus, photochemically converting the spironaphthoxazine from the closed form to the open form (merocyanine form), via UV light irradiation, activates energy transfer from PFBT to merocyanine form. As a result, PFBT fluorescence is effectively quenched, turning off nanoparticle fluorescence. The closed form is regenerated spontaneously over time, or can be driven by irradiation with visible light, resulting in the recovery of fluorescence. Fig. 7b displays the reversible photoswitching at the single particle level. Nanoparticles with a spironaphthoxazine derivative as photochromophore and PFBT as the fluorophore were employed, using a 405 nm diode laser acting both as the excitation source for fluorescence and for inducing the photochromic conversion. Single particle photoswitching measurements were obtained by acquiring a series of consecutive CCD frames (20 ms exposure time) upon switching on the laser. The first CCD frame, as shown in Fig. 7b, indicates clear detection of single nanoparticle fluorescence. The fluorescence from single particles was rapidly switched off in a few tens of milliseconds under continuous laser illumination. Subsequent imaging on the sample after switching off the laser for 30 seconds shows near-complete recovery of the fluorescence intensity. Such single nanoparticle reversible fluorescence ON/OFF conversion was performed for several switching cycles with no apparent photobleaching owing to the low laser intensity (∼30 W cm−2) employed. While these single particle results are encouraging, demonstration of super-resolution imaging based on photoswitching CPNs has not yet been achieved.
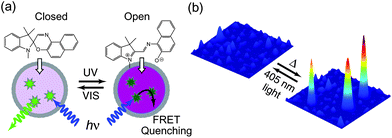 | ||
| Fig. 7 (a) Schematic representation demonstrating photoswitching and FRET-based fluorescence ON/OFF modulation of spironaphthoxazine-containing CPNs. (b) Single particle photoswitching of the spironaphthoxazine nanoparticles doped with the conjugated polymer PFBT. Excitation at 405 nm diode laser is used to both induce the switching to “OFF” state and to excite fluorescence. The fluorescence from single particles was rapidly switched off in a few tens of millisecond under continuous laser illumination. The “ON” state recovers after turning off the laser for a few seconds at room temperature. | ||
4.6 Reversible photobleaching due to fluorescence quenching by photogenerated polarons
It is hypothesized that the blinking phenomena observed in single conjugated polymer molecules and CPNs are due to reversible photogeneration of hole polarons,76,112 which efficiently quench the polymer fluorescence, with an effective quenching radius of 5–10 nm.64 It is also hypothesized that hole polarons are generated when molecular oxygen accepts an electron from the polymer excited state. In an effort to test and clarify aspects of this picture, we performed a series of fluorescence excitation modulation experiments on CPNs under various solution conditions using a conventional fluorimeter. For aqueous suspensions of MEH-PPV nanoparticles open to room atmosphere (∼20% oxygen), transient responses of the fluorescence intensity were observed upon modulation of the excitation intensity, indicating a photo-driven process (Fig. 8a). The fluorescence transients can be described as “reversible photobleaching”, in that the initial fluorescence intensity rapidly decays, with near complete recovery to the initial value if the excitation is blocked for a period of time. Additional experiments were performed using nitrogen purging, and also using anti-fade agents, causing partial or nearly total suppression of the excitation modulation transient, strongly indicating involvement of molecular oxygen. Polaron-induced quenching was also investigated by picosecond time-correlated single photon counting (TCSPC). Excitation modulation-induced transient fluorescence and picosecond lifetime results were compared to model calculations, yielding a detailed picture of the polaron generation, recombination, and quenching processes.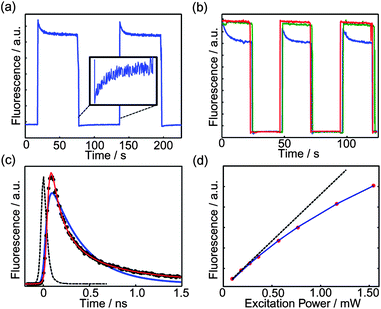 | ||
| Fig. 8 (a) Non-single exponential fluorescence photobleaching and single exponential fluorescence recovery of MEH-PPV CPNs bulk sample. (b) Alternation of high (∼400 µW) and low (∼40 µW) illumination powers in MEH-PPV CPNs aqueous dispersion in air without DABCO (blue, lowest curve), after bubbling with nitrogen (green, middle curve), and with the addition of 0.01 M DABCO (red, highest curve). (c) Fluorescence lifetime (black circles), IRF (black dotted line), and fits assuming exponential (blue) and KWW (red) kinetics. (d) A series of bulk fluorescence experiments on MEH-PPV CPdots indicating saturation. Fit parameters indicate emission saturation power of 3 mW. The dashed straight line represents the expected trend in a non-saturable system. | ||
The excitation modulation experiments were performed as follows. MEH-PPV nanoparticles were prepared via reprecipitation, with a diameter of ∼11 nm as determined by AFM. Nanoparticles suspended in deionized water were placed in a fluorimeter, and the emission intensity of the CPNs at 589 nm was recorded continuously over the course of the experiment, while the excitation intensity was modulated by periodically placing a neutral density filter in the excitation beam. It was found that the fluorescence intensity of MEH-PPV CPNs excited at a constant relatively high power (∼400 µW) exhibits complex (non-first-order) decay kinetics (Fig. 8a), in contrast to the typical first order, single exponential kinetics typically associated with the photodestruction of fluorophore via a low quantum yield process.72 After 20–30 seconds, the excitation was abruptly attenuated by a factor of 10 using a neutral density filter, which resulted in single exponential recovery of the fluorescence. After reverting to the higher intensity, the fluorescence intensity recovers to a value close to the previous initial fluorescence intensity, and similar complex kinetics are observed. Nearly identical behavior is observed over several cycles.
In order to test for the involvement of oxygen in the reversible photobleaching process, nitrogen bubbling was used to remove oxygen and the fluorescence excitation modulation transient was recorded as described above. Transient measurements were also performed using the anti-fade agent 1,4-diazabicyclo[2,2,2]octane (DABCO), which is often used in fluorescence imaging to prolong fluorescence through quenching of triplets and singlet oxygen, and it may also be involved in radical scavenging and reduction of oxidized species.113 As shown in Fig. 8b, under nitrogen or in the presence of DABCO, the excitation modulation-induced transient is nearly completely suppressed, indicating that oxygen is involved in the photodriven quenching process. Some slow, irreversible bleaching was observed, likely due to conventional, irreversible photooxidation of the polymer.
Quenching of nanoparticle fluorescence by hole polarons is believed to occur via efficient energy transfer.63,76 The absorption spectrum of a hole polaron is shifted to lower energy as compared to the neutral polymer, and the polaron has a low fluorescence quantum efficiency, thus the hole polaron has the characteristics required for efficient fluorescence quenching by Förster transfer. Exciton diffusion is also expected to increase quenching efficiency, as discussed above. A quenching radius of 4–10 nm has been estimated for polarons in MEH-PPV.64 We employed time-resolved fluorescence spectroscopy to investigate whether the transient reduction in fluorescence intensity was due to a dynamic quenching process or a static quenching process. If the decrease in fluorescence is primarily caused by photogenerated quenchers via an energy transfer process, then this is likely a dynamic quenching process that should lead to a decrease in the excited state lifetime. On the other hand, if the reduction in fluorescence is due primarily to depletion or photodestruction of polymer, this would constitute a static quenching process, which would not affect the lifetime of the excited state. The picosecond decay kinetics follow a KWW stretched-exponential functional form,93,94 with a β parameter of 0.4, indicating a wide range of exciton lifetimes (Fig. 8c). This is consistent with quenching by energy transfer to more or less randomly distributed quenchers, which results in a range of lifetimes,114 and is similar to the results of the exciton-diffusion-energy-transfer model described above. Also, the effect of bubbling nitrogen and lowering the excitation intensity on the fluorescence lifetime were determined. At low excitation power (62 µW) and under nitrogen protection, TCSPC results were obtained, and a biexponential fit yielded a weighted average lifetime of 480 ps, while the results obtained at a somewhat higher excitation power (281 µW) in air yielded a weighted average lifetime of 340 ps, consistent with the dynamic quenching by photogenerated hole polarons.
A continuum rate model was developed to gain an insight into the underlying mechanism of the experimental photobleaching results described above. This model does not explicitly include a microscopic description of the energy transfer processes resulting in quenching—such details (e.g. particle and quencher size, exciton mobility) are represented implicitly in the various rate constants. The detailed microscopic nature of energy transfer occurring within a CPdot is treated within the framework of a stochastic (random walk) formalism as discussed in previous sections.19 The model is described as follows: exciton dynamics within a CPN can be viewed as a continuous process of exciton creation (absorbance), exciton relaxation (fluorescence), non-radiative emission, quencher creation, quencher elimination, and energy transfer to quenchers. The combination of these processes can be described using a pair of coupled differential equations. The time-dependence of the exciton population is described by:
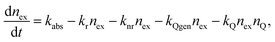 | (2) |
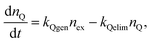 | (3) |
Another way to test the above picture of quenching by photogenerated hole polarons is measurement of fluorescence saturation. Fluorescence saturation is a well known phenomenon in many fluorescent systems, and is observed as a less-than-linear increase in fluorescence as the excitation power is increased.115 Using a fluorimeter, the emission intensities were acquired by stepping through the excitation powers and allowing the system to reach steady state fluorescence at each power. Based on a set of intensity-dependent fluorescence data, saturation was clearly observed, and a saturated excitation power of 3 mW was obtained via a numerical fit to the triplet-saturation equation,115 as shown in Fig. 8d. However, the observed saturation is also in good agreement with the steady-state emission predicted by the above coupled kinetic equations, indicating that the saturation may be due to polarons, though triplet-saturation may also occur. Preliminary single molecule fluorescence saturation results indicate that the presence of atmospheric oxygen reduces the saturation emission intensity of single MEH-PPV CPNs by a factor of 10 or more as compared to the oxygen-free sample, which favors the polaron-induced saturation hypothesis, since polaron generation is enhanced by oxygen, while triplet lifetimes are reduced by oxygen.
Taken together, the excitation modulation results, picosecond time-resolved fluorescence results, saturation results, and modeling results provide clear evidence that the complex photobleaching kinetics of MEH-PPV are not primarily due to polymer or NP heterogeneity, but can be ascribed to reversible photogeneration of hole polarons, which act as efficient fluorescence quenchers. The quencher population can be manipulated either by varying the excitation intensity, removal of molecular oxygen, or use of anti-fade compounds. This understanding of the processes that limit fluorescence brightness and cause fading of CPNs provides an excellent basis for the development of brighter, more fade-resistant nanoparticles.
4.7 Oxygen-sensing by dye-doped conjugated polymer nanoparticles
Molecular oxygen is a key metabolite in aerobic biological systems and tissue hypoxia is a critical parameter with respect to various tissue pathologies, such as retinal diseases, vascular abnormalities, and cancer. Measurement of oxygen consumption rate can help to elucidate alterations in metabolism caused by various stimuli or disease states. Accordingly, the development of local probes for high resolution mapping of tissue oxygen levels or oxygen consumption rate presents a challenging and important problem.116,117 We recently developed conjugated polymer nanoparticle-based oxygen sensor and demonstrated its application in biological oxygen sensing.20 Platinum(II) octaethylporphine (PtOEP) was employed as the oxygen sensitive dye, while nanoparticles consisting of polyfluorene derivatives poly(9,9-dihexylfluorene) (PDHF) and poly(9,9-dioctylfluorene) (PFO) were used as light-harvesting host materials. An energy transfer efficiency of 87% was determined for 10 wt% PtOEP-doped PFO nanoparticles. An additional indication of efficient energy transfer is the decrease in the donor excited state lifetime from 110 ps for the undoped CPNs to 18 ps for the doped CPNs, as determined by time-correlated single photon counting. Owing to the excellent light-gathering capacity of the PFO (absorption cross-section of 2 × 10−12 cm2 at 380 nm) and the efficient energy transfer to PtOEP, the doped nanoparticles are estimated to be at least 5 times brighter than similar-sized dye-doped silica particles.The oxygen sensing characteristics of the PtOEP-doped CPNs were experimentally determined. The nanoparticles exhibit moderate fluorescence (ca. 420 nm) from the PDHF host and oxygen-dependent phosphorescence (ca. 650 nm) from PtOEP dopant (Fig. 9a). It can be clearly seen that the nitrogen-purged suspension of CPNs exhibits intense red emission, while the air- and oxygen-saturated samples present weaker emission because of oxygen quenching. The blue residual host fluorescence is relatively insensitive to the presence of oxygen, while the red PtOEP fluorescence is sensitive to oxygen. Thus the nanoparticles are ratiometric,118 in that the blue conjugated polymer acts as a stable reference that is relatively insensitive to the presence of oxygen. An alternative strategy for oxygen mapping is to employ microsecond time-resolved imaging to measure local variations in the phosphorescence lifetime due to quenching by oxygen. This strategy is particularly useful if sample autofluorescence is high, since this can complicate ratiometric analysis, while measurements of the phosphorescence lifetime are almost completely unaffected by autofluorescence. The nanoparticles exhibited clear, oxygen-sensitive single exponential phosphorescence decays, with lifetimes of approximately 37, 10, and 5 µs for the nitrogen-, air-, and oxygen-saturated nanoparticle suspensions, respectively, as shown in Fig. 9b. Fig. 9c presents the oxygen sensing performance of the composite nanoparticles at single particle level. It can be seen that the phosphorescence of a single nanoparticle in the air atmosphere was quenched by oxygen to a great extent. In sharp contrast, subsequent imaging on the sample after resuming the nitrogen flow exhibits near-complete recovery of the phosphorescence intensity. Additionally, the phosphorescence intensity changes were observed over several nitrogen–air cycles and no apparent photobleaching was observed. The cellular uptake and oxygen sensing capabilities of the PtOEP-doped CPNs in cellular milieu were also experimentally evaluated. A comparison of the differential interference contrast (DIC) image and phosphorescence images of the nanoparticle-labeled macrophage-like murine cells clearly demonstrates uptake of the nanoparticles (Fig. 9d). No appreciable cytotoxicity or phototoxicity of the nanoparticle sensors employed in this study was found in course of incubation and imaging.
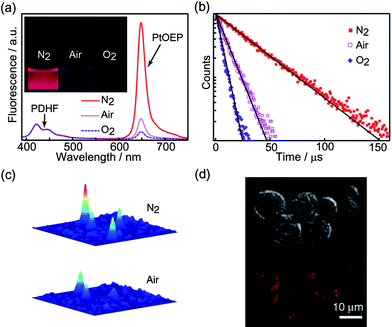 | ||
| Fig. 9 (a) Oxygen-dependent emission spectra of the 10% PtOEP doped PDHF dots. The inset shows doped PDHF dots in aqueous solutions saturated with nitrogen, air, and oxygen, respectively, under a UV lamp. (b) Phosphorescence decays of the 10% PtOEP-doped PDHF dots with different oxygen concentrations. (c) Single-particle phosphorescence images of the doped CPdots immobilized on a coverslip under nitrogen and air atmosphere, respectively. (d) DIC (upper) and phosphorescence (lower) images indicate that uptake of the nanoparticle sensor by macrophage cells. Reproduced with permission from ref. 20. Copyright Wiley-VCH, 2009. | ||
5. Conclusions
Conjugated polymer nanoparticles have emerged as a new type of fluorescent probe that possesses brightness orders of magnitude higher than those of typical fluorescent dyes and at least one order of magnitude higher than those of other fluorescent nanoparticles with diameters less than 20 nm. In recent particle tracking experiments, roughly 2 × 105 photons were detected per particle per 20 ms exposure, permitting video rate tracking with ∼1 nanometre resolution.16 CPNs also exhibit extraordinarily high two-photon-excited fluorescence cross-sections, as high as 2 × 105 GM for NIR (800 nm) excitation, which is currently the highest reported to date for a nanoparticle.17Another useful characteristic of CPNs is the ease with which a variety of dopants can be incorporated into the nanoparticle, and their high efficiency of energy transfer to dopants, even at relatively low doping ratios, due to amplified energy transfer caused by exciton diffusion. Potential applications of energy transfer to dye dopants have been demonstrated and were reviewed here, including highly red-shifted emission by doping with fluorescent dyes, oxygen sensor nanoparticles utilizing energy transfer to an oxygen-sensitive platinum porphyrin dye, and fluorescence photoswitching nanoparticles utilizing photochromic energy acceptors. By doping with dyes and other polymers, separations between excitation and emission of 275 nm were achieved, which facilitates rejection of unwanted scattering and autofluorescence while efficiently collecting the nanoparticle fluorescence. Also, by doping with dyes, the brightness, photostability, and saturated emission rate of the nanoparticles can be improved. Similarly, energy transfer was observed in mixed-phase polyfluorene CPNs, which caused trapping of excitons in the semicrystalline regions and improved fluorescence quantum yield relative to the glassy-phase nanoparticles. The demonstration of ratiometric oxygen sensing by single CPNs is promising for the possible development of nanoparticles for mapping other solution species, such as pH and metal ions. The observation of efficient fluorescence quenching by energy transfer to Au nanoparticles raises the possibility of other sensing schemes. Meanwhile, the high brightness of photoswitching CPNs could lead to improvements in super-resolution fluorescence imaging.
Studies of fluorescence quenching by photogenerated hole polarons in CPNs have led to improved understanding of seemingly unrelated phenomena including blinking in single CPNs, the complex photobleaching kinetics of conjugated polymers, and anomalous fluorescence saturation behavior. A simple rate model including photodriven quencher–polaron formation, second-order quenching, and first-order polaron recombination qualitatively reproduced the complex photobleaching kinetics and transients observed in excitation–modulation experiments. Picosecond time-resolved fluorescence experiments provided additional confirmation that a quenching process is involved. Fluorescence saturation behavior also agreed with the predictions of the rate model.
At the single particle level, energy transfer to photogenerated hole polarons results in blinking, complicating the use of smaller CPNs for particle tracking. Also, polaron-induced fluorescence saturation can limit particle brightness. On the basis of preliminary investigations and the work presented here, it appears that the addition of dye dopants could help to decrease blinking and increase the saturated intensity by competitive energy transfer to the dyes, which should lower the quenching efficiency of polarons. The facile preparation of doped CPNs could provide a rapid screening method for evaluating possible dye dopants to tune the emission wavelengths of light-emitting devices. Also, on the basis of the results reviewed here, we suggest that polaron-induced quenching, involving molecular oxygen or other electron-acceptor species, could be a significant factor limiting the brightness and longevity of polymer LEDs.
While this article is primarily focused on energy transfer phenomena, we will briefly address the challenges and opportunities for the future development of CPNs for biological imaging applications. The key challenge is the encapsulation, functionalization, and bioconjugation of CPNs, which are required in order to reduce nonspecific binding to biomolecules and cellular structures and to selectively bind to molecules of interest. While some progress has been made, including silica encapsulation and functionalization with amine groups,27 the development of reliable encapsulation and bioconjugation protocols remains an ongoing challenge. Cytotoxicity is also a concern, although the toxicity of conjugated polymers is expected to be lower than that of other nanoparticles containing heavy metals, and in any case, the concentration of nanoparticles used for applications such as single particle tracking is exceedingly low. Indeed, preliminary results indicate minimal cytotoxicity,15,20 though careful, detailed studies are in order. In addition to the applications that have been discussed, including conventional and two-photon fluorescence microscopy, ultraresolution fluorescence microscopy, and nanoscale single particle tracking, other applications requiring bright fluorophores could also benefit from the use of CPNs, such as flow cytometry. The development of conjugated polymer nanoparticles for biological imaging is still in its infancy. However, based on their extraordinary fluorescence brightness as well as the power and flexibility of extending the applications of CPNs by energy transfer to functional dopants acting as photoswitches and sensors, this type of probe is expected to play an important role in future imaging studies.
Acknowledgements
The authors acknowledge financial support from the NSF/EPSCoR under Grants No. 2001RII-EPS-0132573 and 2004RII-EPS-0447660, and NSF (CAREER) CHE-0547846. We also thank the ACS and Wiley-VCH for granting permission to reproduce figures.References
- W. Denk, J. H. Strickler and W. W. Webb, Science, 1990, 248, 73 CrossRef CAS.
- X. S. Xie and J. K. Trautman, Annu. Rev. Phys. Chem., 1998, 49, 441 CrossRef CAS.
- C. Joo, H. Balci, Y. Ishitsuka, C. Buranachai and T. Ha, Annu. Rev. Biochem., 2008, 77, 51 CrossRef CAS.
- S. J. Lord, H. D. Lee and W. E. Moerner, Anal. Chem., 2010, 82, 2192 CrossRef CAS.
- E. Toprak and P. R. Selvin, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 349 CrossRef CAS.
- X. S. Xie, P. J. Choi, G. W. Li, N. K. Lee and G. Lia, Annu. Rev. Biophys., 2008, 37, 417 Search PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642 CAS.
- B. Huang, M. Bates and X. W. Zhuang, Annu. Rev. Biochem., 2009, 78, 993 CrossRef CAS.
- W. Chen, J. Nanosci. Nanotechnol., 2008, 8, 1019 CrossRef CAS.
- A. P. Alivisatos, W. W. Gu and C. Larabell, Annu. Rev. Biomed. Eng., 2005, 7, 55 CrossRef CAS.
- S. Santra, P. Zhang, K. M. Wang, R. Tapec and W. H. Tan, Anal. Chem., 2001, 73, 4988 CrossRef CAS.
- H. Ow, D. R. Larson, M. Srivastava, B. A. Baird, W. W. Webb and U. Wiesner, Nano Lett., 2005, 5, 113 CrossRef CAS.
- Z. Y. Tian, W. W. Wu and A. D. Q. Li, ChemPhysChem, 2009, 10, 2577 CrossRef CAS.
- C. Szymanski, C. F. Wu, J. Hooper, M. A. Salazar, A. Perdomo, A. Dukes and J. McNeill, J. Phys. Chem. B, 2005, 109, 8543 CrossRef CAS.
- C. F. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415 CrossRef CAS.
- J. B. Yu, C. F. Wu, S. P. Sahu, L. P. Fernando, C. Szymanski and J. McNeill, J. Am. Chem. Soc., 2009, 131, 18410 CrossRef CAS.
- C. F. Wu, C. Szymanski, Z. Cain and J. McNeill, J. Am. Chem. Soc., 2007, 129, 12904 CrossRef CAS.
- C. F. Wu, H. S. Peng, Y. F. Jiang and J. McNeill, J. Phys. Chem. B, 2006, 110, 14148 CrossRef CAS.
- C. F. Wu, Y. L. Zheng, C. Szymanski and J. McNeill, J. Phys. Chem. C, 2008, 112, 1772 CrossRef CAS.
- C. F. Wu, B. Bull, K. Christensen and J. McNeill, Angew. Chem., Int. Ed., 2009, 48, 2741 CrossRef CAS.
- E. J. Harbron, C. M. Davis, J. K. Campbell, R. M. Allred, M. T. Kovary and N. J. Economou, J. Phys. Chem. C, 2009, 113, 13707 CrossRef CAS.
- P. J. Reid, D. A. Higgins and P. F. Barbara, J. Phys. Chem., 1996, 100, 3892 CrossRef CAS.
- T. Huser, M. Yan and L. J. Rothberg, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11187 CrossRef CAS.
- F. Schindler, J. M. Lupton, J. Feldmann and U. Scherf, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14695 CrossRef CAS.
- J. Yu, R. Lammi, A. J. Gesquiere and P. F. Barbara, J. Phys. Chem. B, 2005, 109, 10025 CrossRef CAS.
- A. J. Gesquiere, S. J. Park and P. F. Barbara, J. Am. Chem. Soc., 2005, 127, 9556 CrossRef CAS.
- C. F. Wu, C. Szymanski and J. McNeill, Langmuir, 2006, 22, 2956 CrossRef CAS.
- C. F. Wu and J. McNeill, Langmuir, 2008, 24, 5855 CrossRef CAS.
- A. J. Gesquiere, S. J. Park and P. F. Barbara, Eur. Polym. J., 2004, 40, 1013 CrossRef CAS.
- J. K. Grey, D. Y. Kim, B. C. Norris, W. L. Miller and P. F. Barbara, J. Phys. Chem. B, 2006, 110, 25568 CrossRef CAS.
- R. E. Palacios, F. R. F. Fan, J. K. Grey, J. Suk, A. J. Bard and P. F. Barbara, Nat. Mater., 2007, 6, 680 CrossRef CAS.
- Y. L. Chang, R. E. Palacios, F. R. F. Fan, A. J. Bard and P. F. Barbara, J. Am. Chem. Soc., 2008, 130, 8906 CrossRef CAS.
- R. E. Palacios, K. J. Lee, A. Rival, T. Adachi, J. C. Bolinger, L. Fradkin and P. F. Barbara, Chem. Phys., 2009, 357, 21 CrossRef CAS.
- V. V. Volkov, T. Asahi, H. Masuhara, A. Masuhara, H. Kasai, H. Oikawa and H. Nakanishi, J. Phys. Chem. B, 2004, 108, 7674 CrossRef CAS.
- N. Kurokawa, H. Yoshikawa, N. Hirota, K. Hyodo and H. Masuhara, ChemPhysChem, 2004, 5, 1609 CrossRef CAS.
- K. Landfester, R. Montenegro, U. Scherf, R. Guntner, U. Asawapirom, S. Patil, D. Neher and T. Kietzke, Adv. Mater., 2002, 14, 651 CrossRef CAS.
- T. Kietzke, D. Neher, K. Landfester, R. Montenegro, R. Guntner and U. Scherf, Nat. Mater., 2003, 2, 408 CrossRef CAS.
- T. Kietzke, B. Stiller, K. Landfester, R. Montenegro and D. Neher, Synth. Met., 2005, 152, 101 CrossRef CAS.
- K. Landfester, Angew. Chem., Int. Ed., 2009, 48, 4488 CrossRef CAS.
- M. C. Baier, J. Huber and S. Mecking, J. Am. Chem. Soc., 2009, 131, 14267 CrossRef CAS.
- K. Muller, M. Klapper and K. Mullen, Macromol. Rapid Commun., 2006, 27, 586 CrossRef.
- M. Green, P. Howes, C. Berry, O. Argyros and M. Thanou, Proc. R. Soc. London, Ser. A, 2009, 465, 2751 CrossRef CAS.
- Z. Q. Yang, W. T. S. Huck, S. M. Clarke, A. R. Tajbakhsh and E. M. Terentjev, Nat. Mater., 2005, 4, 486 CrossRef CAS.
- S. A. Jenekhe, M. M. Alam, Y. Zhu, S. Y. Jiang and A. V. Shevade, Adv. Mater., 2007, 19, 536 CrossRef CAS.
- P. Taranekar, J. Y. Park, D. Patton, T. Fulghum, G. J. Ramon and R. Advincula, Adv. Mater., 2006, 18, 2461 CrossRef CAS.
- H. Kasai, H. S. Nalwa, H. Oikawa, S. Okada, H. Matsuda, N. Minami, A. Kakuta, K. Ono, A. Mukoh and H. Nakanishi, Jpn. J. Appl. Phys., 1992, 31, L1132 CAS.
- H. S. Nalwa, H. Kasai, S. Okada, H. Oikawa, H. Matsuda, A. Kakuta, A. Mukoh and H. Nakanishi, Adv. Mater., 1993, 5, 758.
- H. Nakanishi and H. Katagi, Supramol. Sci., 1998, 5, 289 CrossRef CAS.
- H. O. Kasai and H. Nakanishi, in Organic Mesoscopic Chemistry, ed. H. Masuhara and F. C. Schryver, Blackwell Science, Oxford, 1999, p. 145 Search PubMed.
- M. Ormo, A. B. Cubitt, K. Kallio, L. A. Gross, R. Y. Tsien and S. J. Remington, Science, 1996, 273, 1392 CrossRef CAS.
- E. Gantt, Plant Physiol., 1969, 44, 1629 CrossRef CAS.
- A. M. Smith and S. Nie, J. Am. Chem. Soc., 2008, 130, 11278 CrossRef CAS.
- B. O. Dabbousi, J. RodriguezViejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463 CrossRef CAS.
- D. R. Larson, H. Ow, H. D. Vishwasrao, A. A. Heikal, U. Wiesner and W. W. Webb, Chem. Mater., 2008, 20, 2677 CrossRef CAS.
- M. F. Garcia-Parajo, G. M. J. Segers-Nolten, J. A. Veerman, J. Greve and N. F. van Hulst, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 7237 CrossRef CAS.
- A. C. Ley, Plant Physiol., 1984, 74, 451 CrossRef CAS.
- B. Lounis, H. A. Bechtel, D. Gerion, P. Alivisatos and W. E. Moerner, Chem. Phys. Lett., 2000, 329, 399 CrossRef CAS.
- G. H. Patterson, S. M. Knobel, W. D. Sharif, S. R. Kain and D. W. Piston, Biophys. J., 1997, 73, 2782 CrossRef CAS.
- M. Wu, P. M. Goodwin, W. P. Ambrose and R. A. Keller, J. Phys. Chem., 1996, 100, 17406 CrossRef CAS.
- M. Howarth, W. H. Liu, S. Puthenveetil, Y. Zheng, L. F. Marshall, M. M. Schmidt, K. D. Wittrup, M. G. Bawendi and A. Y. Ting, Nat. Methods, 2008, 5, 397 CrossRef CAS.
- C. A. Leatherdale, W. K. Woo, F. V. Mikulec and M. G. Bawendi, J. Phys. Chem. B, 2002, 106, 7619 CrossRef CAS.
- M. Nirmal, B. O. Dabbousi, M. G. Bawendi, J. J. Macklin, J. K. Trautman, T. D. Harris and L. E. Brus, Nature, 1996, 383, 802 CrossRef CAS.
- M. Deussen, P. H. Bolivar, G. Wegmann, H. Kurz and H. Bassler, Chem. Phys., 1996, 207, 147 CrossRef CAS.
- J. D. McNeill and P. F. Barbara, J. Phys. Chem. B, 2002, 106, 4632 CrossRef CAS.
- G. Jung, J. Wiehler and A. Zumbusch, Biophys. J., 2005, 88, 1932 CAS.
- R. A. Mathies, K. Peck and L. Stryer, Anal. Chem., 1990, 62, 1786 CrossRef CAS.
- G. Giraud, H. Schulze, T. T. Bachmann, C. J. Campbell, A. R. Mount, P. Ghazal, M. R. Khondoker, A. J. Ross, S. W. J. Ember, I. Ciani, C. Tlili, A. J. Walton, J. G. Terry and J. Crain, Int. J. Mol. Sci., 2009, 10, 1930 Search PubMed.
- V. Fomenko and D. J. Nesbitt, Nano Lett., 2008, 8, 287 CrossRef CAS.
- S. Wennmalm and R. Rigler, J. Phys. Chem. B, 1999, 103, 2516 CrossRef CAS.
- I. Rasnik, S. A. McKinney and T. Ha, Nat. Methods, 2006, 3, 891 CrossRef CAS.
- A. Yildiz, J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman and P. R. Selvin, Science, 2003, 300, 2061 CrossRef CAS.
- C. Eggeling, J. Widengren, R. Rigler and C. A. M. Seidel, Anal. Chem., 1998, 70, 2651 CrossRef CAS.
- A. H. Fu, W. W. Gu, C. Larabell and A. P. Alivisatos, Curr. Opin. Neurobiol., 2005, 15, 568 CrossRef CAS.
- D. A. VandenBout, W. T. Yip, D. H. Hu, D. K. Fu, T. M. Swager and P. F. Barbara, Science, 1997, 277, 1074 CrossRef.
- D. H. Hu, J. Yu, G. Padmanaban, S. Ramakrishnan and P. F. Barbara, Nano Lett., 2002, 2, 1121 CrossRef CAS.
- J. Yu, N. W. Song, J. D. McNeill and P. F. Barbara, Isr. J. Chem., 2004, 44, 127 CrossRef CAS.
- Y. J. Lee, S. J. Park, A. J. Gesquiere and P. F. Barbara, Appl. Phys. Lett., 2005, 87, 051906 CrossRef.
- P. Kumar, A. Mehta, M. D. Dadmun, J. Zheng, L. Peyser, A. P. Bartko, R. M. Dickson, T. Thundat, B. G. Sumpter, D. W. Noid and M. D. Barnes, J. Phys. Chem. B, 2003, 107, 6252 CrossRef CAS.
- R. C. Powell, Phys. Rev. B: Solid State, 1970, 2, 2090 CrossRef.
- R. C. Powell, Phys. Rev. B: Solid State, 1970, 2, 1207 CrossRef.
- M. Yokota and O. Tanimoto, J. Phys. Soc. Jpn., 1967, 22, 779 CrossRef CAS.
- L. C. Chen, L. S. Roman, D. M. Johansson, M. Svensson, M. R. Andersson, R. A. J. Janssen and O. Inganas, Adv. Mater., 2000, 12, 1110 CrossRef CAS.
- Q. F. Xu, H. M. Duong, F. Wudl and Y. Yang, Appl. Phys. Lett., 2004, 85, 3357 CrossRef CAS.
- X. Gong, S. Wang, D. Moses, G. C. Bazan and A. J. Heeger, Adv. Mater., 2005, 17, 2053 CrossRef CAS.
- X. Gong, J. C. Ostrowski, G. C. Bazan, D. Moses, A. J. Heeger, M. S. Liu and A. K. Y. Jen, Adv. Mater., 2003, 15, 45 CrossRef CAS.
- B. P. Lyons and A. P. Monkman, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 235201 CrossRef.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477 CrossRef CAS.
- X. H. Yang, F. Jaiser, D. Neher, P. V. Lawson, J. L. Bredas, E. Zojer, R. Guntner, P. S. de Freitas, M. Forster and U. Scherf, Adv. Funct. Mater., 2004, 14, 1097 CrossRef CAS.
- E. A. Silinsh, Organic Molecular Crystals: Their Electronic States, Springer-Verlag, Berlin, New York, 1980 Search PubMed.
- T. Förster, Ann. Phys. (Leipzig), 1948, 2, 55 Search PubMed.
- A. Haugeneder, M. Neges, C. Kallinger, W. Spirkl, U. Lemmer, J. Feldmann, U. Scherf, E. Harth, A. Gugel and K. Mullen, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 15346 CrossRef CAS.
- M. A. Stevens, C. Silva, D. M. Russell and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 165213 CrossRef.
- K. C. B. Lee, J. Siegel, S. E. D. Webb, S. Leveque-Fort, M. J. Cole, R. Jones, K. Dowling, M. J. Lever and P. M. W. French, Biophys. J., 2001, 81, 1265 CAS.
- M. N. Berberan-Santos, E. N. Bodunov and B. Valeur, Chem. Phys., 2005, 315, 171 CrossRef CAS.
- T. Q. Nguyen, V. Doan and B. J. Schwartz, J. Chem. Phys., 1999, 110, 4068 CrossRef.
- D. Perahia, R. Traiphol and U. H. F. Bunz, J. Chem. Phys., 2002, 117, 1827 CrossRef CAS.
- D. Perahia, X. S. Jiao and R. Traiphol, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 3165 CrossRef CAS.
- M. Grell, D. D. C. Bradley, X. Long, T. Chamberlain, M. Inbasekaran, E. P. Woo and M. Soliman, Acta Polym., 1998, 49, 439 CrossRef CAS.
- M. Grell, D. D. C. Bradley, M. Inbasekaran and E. P. Woo, Adv. Mater., 1997, 9, 798 CrossRef CAS.
- A. J. Cadby, P. A. Lane, H. Mellor, S. J. Martin, M. Grell, C. Giebeler, D. D. C. Bradley, M. Wohlgenannt, C. An and Z. V. Vardeny, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 15604 CrossRef CAS.
- E. J. W. List, C. H. Kim, A. K. Naik, U. Scherf, G. Leising, W. Graupner and J. Shinar, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 155204 CrossRef.
- A. Hayer, A. L. T. Khan, R. H. Friend and A. Kohler, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 241302 CrossRef.
- M. Ariu, D. G. Lidzey, M. Sims, A. J. Cadby, P. A. Lane and D. D. C. Bradley, J. Phys.: Condens. Matter, 2002, 14, 9975 CrossRef CAS.
- L. M. LizMarzan, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329 CrossRef CAS.
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243 CrossRef CAS.
- C. H. Fan, S. Wang, J. W. Hong, G. C. Bazan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 6297 CrossRef CAS.
- M. J. Rust, M. Bates and X. W. Zhuang, Nat. Methods, 2006, 3, 793 CrossRef CAS.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859 RSC.
- D. H. Hu, Z. Y. Tian, W. W. Wu, W. Wan and A. D. Q. Li, J. Am. Chem. Soc., 2008, 130, 15279 CrossRef CAS.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780 Search PubMed.
- H. S. Peng, C. F. Wu, Y. F. Jiang, S. H. Huang and J. McNeill, Langmuir, 2007, 23, 1591 CrossRef CAS.
- H. Z. Lin, S. R. Tabaei, D. Thomsson, O. Mirzov, P. O. Larsson and I. G. Scheblykin, J. Am. Chem. Soc., 2008, 130, 7042 CrossRef CAS.
- B. Herman, Fluorescence Microscopy, Bios Scientific Publishers Ltd, 1997 Search PubMed.
- B. P. Lyons, R. J. Jackson, K. S. Wong and A. P. Monkman, Synth. Met., 2003, 135, 367 CrossRef.
- X. Michalet, S. Weiss and M. Jager, Chem. Rev., 2006, 106, 1785 CrossRef CAS.
- Y. Amao, Microchim. Acta, 2003, 143, 1 CrossRef CAS.
- D. B. Papkovsky and T. C. O'Riordan, J. Fluoresc., 2005, 15, 569 CrossRef CAS.
- S. H. Im, G. E. Khalil, J. Callis, B. H. Ahn, M. Gouterman and Y. N. Xia, Talanta, 2005, 67, 492 CrossRef CAS.
Footnotes |
| † Current address: Department of Chemistry, University of Washington, Seattle, WA 98195, USA. |
| ‡ Current address: Department of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA 30332, USA. |
| This journal is © The Royal Society of Chemistry 2010 |