Surface plasmon resonance of gold nanoparticles assemblies at liquid | liquid interfaces†
Mohamad
Hojeij
,
Nathalie
Younan
,
Lydie
Ribeaucourt
and
Hubert H.
Girault
*
Laboratoire d'Electrochimie Physique et Analytique, Ecole Polytechnique Fédérale de Lausanne, Station 6, CH-1015, Lausanne, Switzerland. E-mail: hubert.girault@epfl.ch; Fax: +41 21 693 36 67; Tel: +41 21 693 31 45
First published on 8th July 2010
Abstract
Surface plasmon resonance (SPR) was observed when a planar close-packed assembly of gold nanoparticles (Au NPs) is adsorbed at the water|1,2-dichloroethane interface. Aqueous gold nanoparticles, 13 or 16 nm in diameter, are deposited at the interface by adding methanol to form a close-packed film with a visible gold mirror reflectance. By total internal reflection of a light beam on the interface, the angular dependence of the interfacial reflectivity was measured in a pseudo-Kretschmann configuration and compared to Fresnel simulations for a homogeneous gold film. The experimental angles for minimum reflectivity were found to match the simulated values. Then, the fluorescence of dye molecules co-adsorbed within 13 and 16 nm gold nanoparticles assemblies at the liquid|liquid interface was measured. The fluorescence intensity under SPR is revealed to be much greater than under total internal reflection conditions, yielding an enhancement factor of approximately 30 and 50 for 13 and 16 nm Au NPs assemblies, respectively. Also, the fluorescence lifetime was found to decrease under SPR conditions.
Introduction
Metallic nanoparticles (NPs), especially gold (Au) and silver (Ag), have been attracting growing interest over the last decade because of their unique optical and electronic properties. They became promising candidates in many applications such as biosensing and imaging, photo-diagnostics, selective photothermal therapy1 and surface-enhanced Raman scattering (SERS).2 Their intrinsic properties due to their localized surface plasmon resonance (LSPR) are strongly dependant on both size and shape.The liquid|liquid interface provides a defect-free platform for the study of optical properties of nanoparticles. Different approaches have already been considered to induce the formation of a gold film at a liquid|liquid interface. It has been shown that 16 nm Au citrate-stabilized nanoparticles can be assembled at the water|hexane interface.3,4 The self-assembly was carried out by addition of ethanol as a means to induce the adsorption of hydrophilic gold nanoparticles at the water|hexane interface. The role of ethanol is assumed to decrease the surface charge and bring the contact angle of the particles at the interface close to 90° such that they can adsorb at the interface to decrease their interfacial energy. This approach was further improved by the addition of a surfactant, dodecanethiol, in the organic phase with a controlled concentration. The dodecanethiol is believed to decrease the repulsive force between particles by forming organic coating layers around the particles when adsorbed at the interface.
Classical surface plasmon resonance (SPR) experiments are usually performed for Au films deposited on a glass slide optically coupled with a right-angled prism. A widely used configuration for these experiments is the Kretschmann configuration. Several spectroscopic measurements including fluorescence, Raman scattering and second harmonic generation (SHG), were also performed on such Au films. It was also reported that surface plasmons can be used to enhance the surface sensitivity of spectroscopic measurements. For example, Corn et al.5 showed that gold nanorods functionalized with single-stranded DNA can be used to enhance the sensitivity of surface plasmon resonance imaging (SPRI) measurements on DNA microarrays. Moreover, Lyon et al.6 reported that the sensitivity of the SPR biosensing can be greatly enhanced by immobilizing colloidal Au to an evaporated Au film. Enormous changes in the SPR curves were induced by simply modifying the immersion time in the Au colloidal solution. Furthermore, the modification of molecular fluorescence due to plasmon excitations has also been studied both experimentally and theoretically.7 During the past several years, the effects of silver and gold particles on the fluorescence of nearby fluorophores have been investigated. It was found that silver island films generally increase fluorescence intensity and decrease the lifetime.8–10 Stranik et al.11 reported the enhancement of fluorescence of ruthenium dye in the vicinity of spherical gold/silver alloy NPs. This plasmonic enhancement resulted from the localized surface plasmon resonance at the metal surface. The LSPR modifies the intensity of the electromagnetic field around the fluorophore, which can lead to an increase in the emitted fluorescence intensity. The effect is dependent on many parameters such as metal type, NP size and shape as well as the distance between the NP and the fluorophore. The enhancement takes place as an increase in the excitation of the dye and in its quantum efficiency.12 The increased quantum yields were also accompanied by decreased lifetimes. Cohanoschi et al.13 also reported a strong surface plasmon enhancement of multiphoton absorption of dyes in an activated gold colloidal solution. The 480-fold enhancement in two-photon absorption and 30-fold in three-photon absorption were attributed to the electric field enhancement via surface plasmon resonance. The surface plasmon enhancement of emission from quantum dots (QDs) was also investigated. 23-Fold enhanced photoluminescence intensity was reported for CdSe QDs located on evaporated gold films.14
On the other hand, SPR at liquid|liquid interfaces has not received yet a lot of attention. To the best of our knowledge, only one paper showed the fluorescence enhancement of Rose Bengal dye at p-xylene|water interface under SPR conditions using a pseudo-Kretschmann configuration.15 At a SPR angle of 78.1°, the fluorescence signal of Rose Bengal dye was 103 times higher than the signal without the gold film. The enhancement originates from two factors: the electric field enhancement; and the reduction of the fluorescence lifetime of dye molecules in the close vicinity of a metal surface. Herein, we display the SPR response for gold nanoparticles assemblies at liquid|liquid interfaces. The excitation wavelength used is 544 nm, the interface considered is water|1,2-dichloroethane (DCE). The self-assembly of both 13 and 16 nm citrate-stabilized Au NPs is induced by vigorous injection of methanol. Second, a 30-fold and 50-fold fluorescence intensity of Coumarin 343 co-adsorbed with the 13 and 16 nm Au NPs, respectively, is observed under surface plasmon resonance conditions, accompanied by a dramatic decrease in the fluorescence lifetime.
Experimental
The schematic of the experimental setup is shown in Fig. 1. A 5 mW Melles Griot laser pumping at 544 nm was used as the excitation source. An iris (I) was used to eliminate any scattered light coming from the laser source. With a polarizer P (Newport), p-polarization was obtained to ensure SP resonance. Different mirrors (protected silver mirrors M1 and M2, Thorlabs) were also used in order to focus the laser beam on the cell. The mirror (M1) is used to change the angle of incidence by moving on a translation stage (TS). The combination of mirrors and lenses enables the beam to remain focused at the center of the meniscus of the interface (in the focal plane of the lens L1), while changing the incidence angle. The TS was connected through a motion controller (Unidex 100, Aerotech) to a computer and thus, insures efficient control of the angle of incidence using a custom-made labview program. A lens (L2) was used to focus the reflected beam. A very sensitive (nW) power meter PM was used to detect the signal. Two mirrors (M3 and M4) followed by a lens (L3) were used to focus the reflected beam into the power meter. The system was programmed to show the reflected intensity as a function of the mirror position, which was related to the angle of incidence at the liquid|liquid interface, using optical geometry and Snell–Descartes law. All the measurements were done in the dark.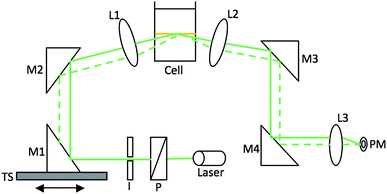 | ||
| Fig. 1 Schematic of the experimental setup of surface plasmon resonance at a liquid|liquid interface. | ||
Citrate-stabilized gold nanoparticles (average size of 13 and 16 nm) were synthesized due to the mild reduction of hydrogen tetrachloroaurate by sodium citrate. The high nucleation rate allows a narrow distribution of quasi-spherical shaped NPs. By modifying the stoïchiometric ratio of the reagents, the size of the synthesized NPs can be changed. For 16 nm Au NPs, the method used is the wet Turkevich method.16 Briefly, a solution of 190 ml of HAuCl4 aqueous solution, with a weight content of 10 mg of gold, was heated until boiling point under vigorous stirring. Then, 10 ml of a 1% sodium citrate aqueous solution was added. The solution was then stirred and kept at boiling conditions for another 45 min. At the end of the reaction, the mixture was showing a red-wine color, characteristic of the formation of Au nanoparticles. As for the 13 nm Au NPs, the procedure reported by Park et al. was followed.3 Briefly, 100 mL of a 1 mM aqueous HAuCl4·3H2O solution was added to 100 mL of triply deionized water (Millipore), which was then boiled. This step was followed by the addition of 10 mL of a 38.8 mM aqueous solution of sodium citrate and the mixture was further boiled for 20 min. A typical surface plasmon resonance band with maximum at 517 nm and 520 nm for 13 and 16 nm Au NPs solutions, respectively, was observed using a PerkinElmer Lambda XLS+ spectrophotometer. The nanoparticles’ size and distribution were determined by transmission electron microscopy (TEM) using a Philips CM20 operating at 200 kV. The NPs are found to be spherical, with an average size of 13 ± 2 nm and 16 ± 2 nm, respectively and with a narrow distribution (<15%) (Fig. S1†).
The procedure for forming Au NPs film at the interface between water and 1,2-dichloroethane consists of a vigorous injection of methanol. The use of alcohol in order to bring the particles to the interface was previously observed.17,18 An optic cell (20 × 40 × 50 mm) was used to prepare the film. To 20 mL of DCE was added 10 mL of the Au solution, a volume of 5 mL of methanol was then injected vigorously at the interface followed by stirring. The formed film showed yellow reflection and stayed stable for many days.
The time-resolved fluorescence measurements were performed using the time-correlated single photon counting (TCSPC) technique. The description of this setup is detailed elsewhere.19 The excitation laser beam was p-polarized in order to excite the surface plasmons and its excitation wavelength was set at 450 nm, corresponding to the absorption peak of the dye (Coumarin 343). The latter was dissolved in the aqueous phase with a concentration of 30 μM and at pH 10. These conditions were found to be the most appropriate in order to reach a full monolayer coverage of the dye at the interface.20
Results and discussion
Fig. 2 displays both calculated and experimental surface plasmon resonance responses for the gold films at water|DCE interface. Simulations of the SPR spectra were realized using the program developed by the research group of Robert M. Corn at Irvine Univeristy.21 In the case of a liquid|metal film|liquid system, the model is reduced to a three-layer model. The reflectivity is thus given by Fresnel's equations: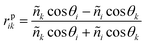 | (1) |
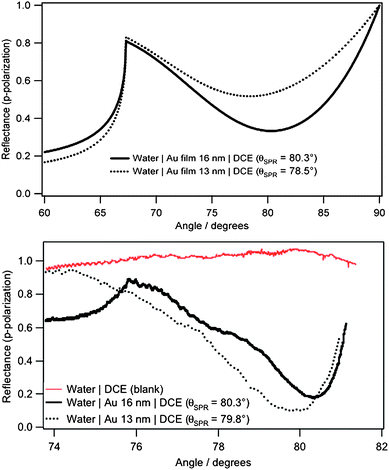 | ||
| Fig. 2 (Top) Calculated and (bottom) experimental SPR curves at water|DCE interface for an excitation wavelength of 544 nm (full line: 16 nm Au film, dotted line: 13 nm Au film). | ||
where ñ is the refractive index of the medium and θ the angle that the beam makes with the normal in the corresponding medium. The indexes i and k represents two adjacent media where the light passes. The total reflection becomes:
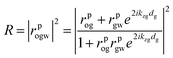 | (2) |
where o, g and w correspond to the organic, gold and aqueous media, respectively.
From eqn (1), one can calculate rpog as follows
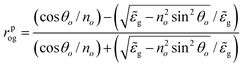 | (3) |
The phase factor kzgdg is the optical path length and is given by:
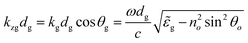 | (4) |
![[small epsilon, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0de.gif) g is the optical dielectric function of the gold film, ω the frequency of the incident light in vacuum, and c its velocity.
g is the optical dielectric function of the gold film, ω the frequency of the incident light in vacuum, and c its velocity.
The refractive index of gold is adjusted to the wavelength referring to the experimental values of Johnson and Christy.22 Two approximations are done using those values. First, the film of nanoparticles is considered as bulk gold. This approximation is partly based on the work of Stoller et al.23 They conclude that there is a good agreement between both real and imaginary parts of the dielectric constant measured in bulk gold and that on 10 to 15 nm gold nanoparticles in the range of the plasmon resonance, i.e. from 510 to 580 nm. Then, the values are linearly extrapolated from those given by Johnson and Christy (values given every 0.12 eV only). The refractive indexes of DCE and of water were taken as 1.445 and 1.333 respectively. For 16 nm gold nanoparticles at the water|DCE interface, the experimental resonance angle is found to be equal to 80.3°, corroborating the simulation. However, for 13 nm Au NPs, a slight shift of 1.3° is observed.
Obviously, the experimental and calculated SPR curves show a similar behaviour but still differ. First of all, the angle range for the experimental curves is narrower because of the limitations with the setup i.e. the total internal reflection of the system (∼ 68°) and the formation of the meniscus between the phases which can deviate the laser beam. Second, the behaviour and values of the reflectance are different as well: in the simulations, the reflectance is already decreasing at an angle of 72°. This is not the case for the experimental data where the reflectance fluctuates before reaching the minimum. All these deviations might be explained by the fact that the simulations take into account some approximations as previously mentioned. Furthermore, these fluctuations can also be due to the defects formed in the film like the presence of gaps between the particles network and the possible aggregation i.e. the thickness of the gold layer.
Fig. 3 displays the emission spectra of Coumarin 343 dissolved in the aqueous phase at a concentration of 30 μM, using total internal reflection (TIR) condition and at the obtained resonance angle of 80.3, in the presence of 16 nm and 13 nm Au NPs at the water|DCE interface. The excitation wavelength equals 450 nm. It was proved by simulation (not shown here) that at this wavelength, the SPR also occurs at the same angle.
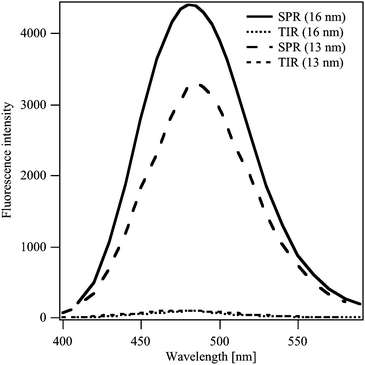 | ||
| Fig. 3 Emission spectra of the Coumarin 343 at water|Au film (16 nm or 13 nm)|DCE interface for TIR conditions and at the SPR. The excitation wavelength equals 450 nm. | ||
The fluorescence intensity under surface plasmon resonance is much greater than under TIR conditions, yielding an enhancement factor of approximately 50 in the case of 16 nm Au film and a bit lower for 13 nm Au film. Many authors24–27 previously reported fluorescence enhancement, especially for classic SPR with a gold film. The enhancement factor ranged from almost 10 to 90. Cohanoschi et al.15 reported a surface plasmon enhancement at liquid|liquid interface with an enhancement factor of approximately 103. In conclusion, when SPR occurs, the energy absorbed by the dye increases and as a consequence the fluorescence is enhanced. The influence of the plasmon excitation on the fluorescence lifetime of the Coumarin 343 co-adsorbed to nanoparticles film at the liquid|liquid interface was studied using the TCSPC technique.
Fig. 4 represents the lifetime decay of the co-adsorbed Coumarin 343, from tom to bottom, in bulk, under total internal reflection (TIR) conditions and under SPR conditions, respectively. The fit results of the obtained curves are depicted in Table 1. In bulk water, the fluorescence decay profile of Coumarin 343 fits nicely with a single exponential function with fluorescence lifetime of 4.5 ns. At the water|DCE interface (TIR condition), the fluorescence decay profile was better fit with a double exponential function with an average lifetime of 3.5 ns. The values of the shorter and longer lifetime components were found from the fitting equations to be 158 ps and 3.5 ns, respectively. This result shows that the Coumarin 343 molecules adsorbs in two different forms at the water|DCE interface as reported in a previous work.28 However, at the water|16 nm Au film|DCE interface at the SPR angle of 80.3°, the fluorescence decay profile was also fitted with a double exponential function, resulting in an average lifetime of 2.31 ns, where the values of the two lifetime components were equal to 1.4 and 3.99 ns, respectively. Similar results were obtained in the case of 13 nm Au film at SPR angle of 79.8°. However, a lower effect is observed which can be related to the decrease in the stability of the gold nanoparticles with decreasing their size due to their higher surface energy. As we notice in both 13 and 16 nm gold films, the longer lifetime component slightly increased but the shorter one became much more significant.29 These variations led to a dramatic decrease of the average fluorescence lifetime.
| τ 1/ns | τ 2/ns | τ/ns | A 1 | A 2 | χ2 | |
|---|---|---|---|---|---|---|
| Bulk | 4.50 | — | 4.5 | — | — | 1.04 |
| Water|DCE (TIR) | 0.15 | 3.5 | 3.5 | 0.04 | 0.96 | 1.03 |
| Au sol 13 nm|DCE (θSPR) | 1.65 | 3.5 | 3.0 | 0.22 | 0.78 | 1.09 |
| Au sol 16 nm|DCE (θSPR) | 1.40 | 4.0 | 2.3 | 0.65 | 0.35 | 1.18 |
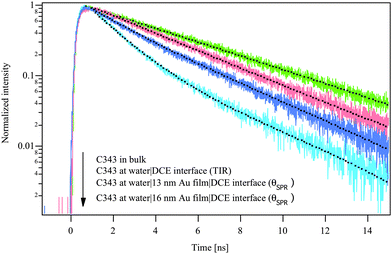 | ||
| Fig. 4 Fluorescence decay profiles, from top to bottom, of 30 μM of Coumarin 343 in bulk water, at water|DCE interface under TIR condition and at water|Au film (16 or 13 nm)|DCE interface at the resonance angle. The fitting curves are marked by dotted lines. | ||
The emission of a fluorophore is described in terms of quantum yield (Q0) and lifetime (τ0). The quantum yield represents the fraction of the excited molecules that relaxes by radiative decay (Γ) and is given by
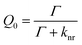 | (5) |
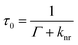 | (6) |
This means that when the localized plasmon resonance wavelength of a metal nanocrystal is close to the emission wavelength of the dye molecule, the emission will be enhanced due to the local electric field enhancement arising from the plasmon resonance. The emission enhancement takes effect as an increase in the radiative decay rate. When dye molecules are situated very close to the surface of metal nanocrystals, fluorescence quenching, taken into account by the nonradiative decay, can also occur. When significant local field enhancements are present, both enhancement and quenching can be observed for the same system by varying the distance between the dye molecule and the metal nanoparticle. The net result will be determined by the magnitude of the local field enhancement. In our case, the net fluorescence enhancement of the Coumarin 343 with an enhancement factor of almost 50 suggests that the quenching has been overcome by the fluorescence enhancement and thus, the reduction of the lifetime arises from the plasmon resonance phenomenon. The reduction of the fluorescence lifetime of dye molecules adsorbed at the interface suggests then that most molecules in the close proximity to the metal surface have their molecular transition dipole moment perpendicular to the surface. This effect has been theoretically proposed by Barnes30 and Chew,31 and experimentally demonstrated by many other authors.12,15,32 On the other hand, a fluorescence lifetime enhancement suggests that the molecular transition dipole is oriented parallel to the metal surface and was also reported in a previous work.33 According to Cohanoschi et al.,15 the reduction of the fluorescence lifetime of dye molecules in the close vicinity of the metal surface is one of the reasons that led to the fluorescence enhancement at the SPR conditions.
Conclusions
This work presents surface plasmon resonance (SPR) of gold nanoparticles assemblies at a liquid|liquid interface. The experiments reveal that surface plasmon resonance occurs for gold films at te liquid|liquid interface, and the obtained SPR angles corroborate those of the simulations. The ability of SPR to enhance the fluorescence of dye molecules in the vicinity of the metal surface is further demonstrated. Emission spectra show that when co-adsorbing with 16 nm Au NPs at the water|DCE interface, the Coumarin 343 molecules fluoresce at the SPR angle with an intensity that is approximately 50 times higher than for TIR conditions. In addition, the SPR leads to the reduction of the fluorescence lifetime of dye molecules suggesting that their molecular transition dipole moment is oriented perpendicular to the surface.Acknowledgements
The Swiss Science National Foundation is gratefully acknowledged. The authors are thankful to Dr S. Terretaz, Professors P.-F. Brevet, R. Corn, H. J. Lee and A. Kornyshev for useful discussions. Frédéric Gumy is also acknowledged for his technical assistance.Notes and references
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayad, Plasmonics, 2007, 2, 107–118 CrossRef CAS.
- S. Yamamoto and H. Watarai, Langmuir, 2006, 22, 6562–6569 CrossRef CAS.
- Y. K. Park and S. Park, Chem. Mater., 2008, 20, 2388–2393 CrossRef CAS.
- Y. K. Park, S. H. Yoo and S. Park, Langmuir, 2007, 23, 10505–10510 CrossRef CAS.
- I. E. Sendroiu, M. E. Warner and R. M. Corn, Langmuir, 2009, 25, 11282–11284 CrossRef CAS.
- L. A. Lyon, M. D. Musick and M. J. Natan, Anal. Chem., 1998, 70, 5177–5183 CrossRef CAS.
- S. K. Gray, Plasmonics, 2007, 2, 143–146 CrossRef CAS.
- J. R. Lakowicz, Y. B. Shen, S. D'Auria, J. Malicka, J. Y. Fang, Z. Gryczynski and I. Gryczynski, Anal. Biochem., 2002, 301, 261–277 CrossRef CAS.
- A. Parfenov, I. Gryczynski, J. Malicka, C. D. Geddes and J. R. Lakowicz, J. Phys. Chem. B, 2003, 107, 8829–8833 CrossRef CAS.
- C. D. Geddes, A. Parfenov, D. Roll, I. Gryczynski, J. Malicka and J. R. Lakowicz, J. Fluoresc., 2003, 13, 267–276 CrossRef CAS.
- O. Stranik, R. Nooney, C. McDonagh and B. D. MacCraith, Plasmonics, 2007, 2, 15–22 CrossRef CAS.
- I. Gryczynski, J. Malicka, Y. B. Shen, Z. Gryczynski and J. R. Lakowicz, J. Phys. Chem. B, 2002, 106, 2191–2195 CrossRef CAS.
- I. Cohanoschi and F. E. Hernandez, J. Phys. Chem. B, 2005, 109, 14506–14512 CrossRef CAS.
- K. Okamoto, S. Vyawahare and A. Scherer, J. Opt. Soc. Am. B, 2006, 23, 1674–1678 Search PubMed.
- I. Cohanoschi, A. Thibert, C. Toro, S. L. Zou and F. E. Hernandez, Plasmonics, 2007, 2, 89–94 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951 Search PubMed.
- F. Reincke, S. G. Hickey, W. K. Kegel and D. Vanmaekelbergh, Angew. Chem., Int. Ed., 2004, 43, 458–462 CrossRef CAS.
- N. Younan, M. Hojeij, L. Ribeaucourt and H. H. Girault, Electrochem. Commun., 2010, 12, 912 CrossRef CAS.
- M. Hojeij, EPFL Thesis, 2009.
- D. Pant, M. Le Guennec, B. Illien and H. H. Girault, Phys. Chem. Chem. Phys., 2004, 6, 3140–3146 RSC.
- R. M. Corn, Chemistry Department, University of California.
- P. B. Johnson and R. W. Christy, Phys. Rev. B: Solid State, 1972, 6, 4370–4379 CrossRef CAS.
- P. Stoller, V. Jacobsen and V. Sandoghdar, Opt. Lett., 2006, 31, 2474–2476 Search PubMed.
- A. Ishida and T. Majima, Analyst, 2000, 125, 535–540 RSC.
- H. Kano and S. Kawata, Opt. Lett., 1996, 21, 1848–1850 Search PubMed.
- S. C. Kitson, W. L. Barnes, J. R. Sambles and N. P. K. Cotter, J. Mod. Opt., 1996, 43, 573–582 CrossRef.
- B. H. Ong, X. C. Yuan, Y. Y. Tan, R. Irawan, X. Q. Fang, L. S. Zhang and S. C. Tjin, Lab Chip, 2007, 7, 506–512 RSC.
- D. Pant and H. H. Girault, Phys. Chem. Chem. Phys., 2005, 7, 3457–3463 RSC.
- C. F. Duan, H. Cui, Z. F. Zhang, B. Liu, J. Z. Guo and W. Wang, J. Phys. Chem. C, 2007, 111, 4561–4566 CrossRef CAS.
- W. L. Barnes, J. Mod. Opt., 1998, 45, 661–699 CrossRef CAS.
- H. Chew, J. Chem. Phys., 1987, 87, 1355–1360 CrossRef CAS.
- K. H. Drexhage, Prog. Opt., 1974, 12, 165 Search PubMed.
- F. E. Hernandez, S. J. Yu, M. Garcia and A. D. Campiglia, J. Phys. Chem. B, 2005, 109, 9499–9504 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM and photographic images; UV-vis spectra. See DOI: 10.1039/c0nr00241k |
| This journal is © The Royal Society of Chemistry 2010 |