Synthesis of continuous TiC nanofibers and/or nanoribbons through electrospinning followed by carbothermal reduction
Lifeng
Zhang
a,
Jianjun
Hu
*bc,
Andrey A.
Voevodin
b and
Hao
Fong
*a
aDepartment of Chemistry, South Dakota School of Mines and Technology, 501 East Saint Joseph Street, Rapid City, South Dakota 57701-3995. E-mail: Hao.Fong@sdsmt.edu; Fax: (+1)-605-394-1232; Tel: (+1)-605-394-1229
bMaterials and Manufacturing Directorate, Air Force Research Laboratory (AFRL/RXBT), 2941 Hobson Way, Wright-Patterson Air Force Base, Dayton, Ohio 45433-7750. E-mail: Jianjun.Hu@wpafb.af.mil; Fax: (+1)-937-255-2176; Tel: (+1)-937-255-9073
cUniversity of Dayton Research Institute, 300 College Park, Dayton, Ohio 45469-0050
First published on 8th July 2010
Abstract
Continuous titanium carbide (TiC) nanofibers that possess an intriguing nanoribbon morphology with a width and thickness of ∼300 nm and ∼40 nm, respectively, and containing TiC crystallites with sizes ranging from 5 nm to 30 nm were synthesized through electrospinning followed by carbothermal reduction.
Titanium carbide (TiC) possesses a high melting point, hardness, and wear resistance; conventionally, it has been widely used for the preparations of cermets, cutting tool tips, and wear resistant coatings.1,2 Recently, TiC has attracted growing attention as an important precursor for the development of porous carbonaceous materials with an average pore size of a few nanometres;3 and such materials are termed as carbide derived carbon (CDC). Unlike the syntheses of other porous carbonaceous materials, which usually involve the thermal decomposition of organic and/or polymeric materials, the synthesis of CDC is based upon selective removal of the metal from a metal carbide; for example, Ti can be removed from TiC using Cl2 at an elevated temperature.3,4 The exceptionally large pore volume and specific surface area make the CDC a promising material for applications such as catalyst support, gas storage, and super-capacitors;3,5–9 additionally, the unique combination of large pore volume, high surface-to-mass ratio, and superior thermal conductivity is also very useful to support and direct heat to phase change materials in thermal energy storage systems similar to currently used carbon foams.10
The nano-scaled TiC materials are mainly available in the forms of powder/particles and thin films. Several publications have reported the preparations of TiC nanowires and/or nanorods through “bottom-up” methods;11–13 to the best of our knowledge, few investigations have been carried out for making continuous TiC nanofibers.14 It is known that the “top-down” nanomaterials-processing technique of electrospinning is a straightforward method for the convenient fabrication of continuous polymeric, ceramic, and carbonaceous fibers with diameters ranging from tens to hundreds of nanometres.14–16 The electrospun TiC nanofibers in the form of overlaid fiber-mat are expected to be advantageous for the further development of CDC.
In general, TiC can be synthesized from precursors containing Ti and C. Although the chemical vapor deposition of precursors containing Ti–C bonds (such as tetraalkyltitanium) can be used to prepare TiC,17 the high-temperature reaction between two precursors containing Ti and C separately is the commonly adopted method. The Ti-containing precursors can be Ti metal, TiCl4, and TiO2. The metal of Ti can react with carbon at a high temperature to make TiC directly.18 The gas-phase reaction between TiCl4 and an appropriate compound such as C2H2, CCl4, CH4, and CaC2 is another method.19–22 Nonetheless, the carbothermal reduction of TiO2 is probably the most important method in industry for making TiC; TiO2 is used as the titanium source due to low cost, while various materials including carbon black,23,24 activated carbon,13 carbon film,25 and cellulose2,26 have been studied as carbon sources. It is noteworthy that the use of titanium alkoxide as the titanium source and organic/polymeric materials as the carbon source has also been investigated, particularly for the development of TiC nanomaterials.27–29 In this study, titanium(IV) n-butoxide (TiBO, Aldrich catalog number: 244112) and furfuryl alcohol (FA, Aldrich catalog number: 185930) were selected as the titanium and carbon sources, respectively.
Previous publications have demonstrated that the process of electrospinning followed by pyrolysis is a capable approach for the syntheses of ceramic materials in the form of continuous nanofibers.30–32 It is also known that TiBO can form the Ti–O–Ti three-dimensional network (gel) through the sol–gel process, and the gel can be converted into TiO2 through pyrolysis;30 and that FA can form a polymer through a condensation reaction, and the polymer can be turned into carbon through heat treatment in an inert atmosphere.27,31 The hypothesis for this study was that electrospinning of a spin dope containing TiBO and FA would result in the uniform dispersion of titanium and carbon sources in the nanofibers; and the carbothermal reduction of such nanofibers would result in continuous TiC nanofibers. To prepare the spin dope for electrospinning, polyvinylpyrrolidone (PVP, Aldrich catalog number: 437190, Mw = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300000) was selected as the carrying polymer,32N,N-dimethylformamide (DMF, Aldrich catalog number: 227056) was selected as the solvent, and acetic acid (HAc, Aldrich catalog number: 537020) was added into the spin dope as the catalyst for hydrolysis of TiBO and polymerization of FA.
300000) was selected as the carrying polymer,32N,N-dimethylformamide (DMF, Aldrich catalog number: 227056) was selected as the solvent, and acetic acid (HAc, Aldrich catalog number: 537020) was added into the spin dope as the catalyst for hydrolysis of TiBO and polymerization of FA.
Numerous spin dopes and different preparation procedures were investigated during this study, while the optimal ones based upon the morphologies and structures of electrospun precursor nanofibers and the resulting TiC nanofibers were prepared as follows: a spin dope containing 10 wt% TiBO, 10 wt% FA, 10 wt% PVP, and 2.5 wt% HAc was prepared using DMF as the solvent in a sealed glass bottle. The spin dope was then magnetically stirred for 48 h. Then, a plastic syringe was filled with the spin dope for electrospinning into precursor nanofibers; the syringe had an 18-gauge stainless-steel needle with 90° blunt tip. The electrospinning setup included an ES-30P high voltage power supply and a laboratory-made roller with diameter of 10 inches. The roller was electrically grounded and placed 10 inches below the tip of needle. During electrospinning, a positive high voltage of 15 kV was applied to the needle; and the feed rate of 1.0 ml h−1 was maintained using a KDS-200 syringe pump. The rotational speed of the roller was set at 100 rpm. This process of electrospinning was very stable, and the electrospinning jet ran continuously without breaking for many hours; the electrospun precursor nanofibers were collected as an overlaid nanofiber-mat on aluminium foil that covered the roller. The nanofiber-mat was then kept under ambient condition (with a temperature of ∼25 °C and relative humidity of ∼50%) for 4 days to allow the completion of chemical reactions inside the nanofibers. Subsequently, the nanofibers was carefully peeled off from the aluminium foil, transferred into a ceramic boat, and placed in a Lindberg 54453 Heavy Duty Tube Furnace for pyrolysis into final TiC nanofibers using the following procedure: (1) increasing the temperature at 5 °C min−1 from ∼25 °C to 325 °C, (2) holding the temperature at 325 °C for 6 h, (3) increasing the temperature at 5 °C min−1 to 1400 °C, (4) holding the temperature at 1400 °C for 12 h, and (5) naturally cooling off to room temperature. A constant flow of argon gas was maintained through the furnace during the heat treatment. Morphologies and structures of the electrospun precursor nanofibers and the final TiC nanofibers were examined by the Zeiss Supra 40VP field-emission SEM, FEI Titan 80–300 S/TEM, and Rigaku Ultima Plus XRD.
The SEM image in Fig. 1A indicated that the electrospun precursor nanofibers had a cylindrical morphology with diameters of 100–300 nm, and they were relatively uniform without microscopically identifiable beads and/or beaded-nanofibers.33 The nanofibers retained the fiber morphology after pyrolysis in argon at 325 °C, while it appeared that the surface of intermediate nanofibers became quite rough (Fig. 1B). The subsequent carbothermal reduction in argon at 1400 °C converted the intermediate nanofibers into the final TiC nanofibers, which possessed a ribbon morphology with the width and thickness being approximately 300 nm and 40 nm, respectively. However, if the intermediate nanofibers were prepared at 425 °C instead of 325 °C, the resulting TiC nanofibers would possess a cylindrical morphology instead of ribbon morphology (results not shown). It is noteworthy that PVP cannot completely decompose in argon until the temperature reaches ∼400 °C, as evidenced by thermal gravimetric analysis.30,32 The partial decomposition of PVP at 325 °C resulted in the increase of surface roughness for the intermediate nanofibers. In the meantime, the relatively fast heating rate of 5 °C min−1 during the following carbothermal reduction prevented the partially decomposed PVP to completely decompose before it became very rubbery. Such a situation resulted in flattening of the nanofibers and led to the formation of the ribbon morphology. With increasing the temperature to 1400 °C, the PVP in the nanofibers would eventually be decomposed/removed completely; and the final TiC nanofibers/nanoribbons would be obtained. As shown in Fig. 1D, the final TiC nanofibers/nanoribbons had tiny nanoparticles on the surface; and these nanoparticles were TiC crystallites (see the following results and discussion).
 | ||
| Fig. 1 SEM images showing the representative morphologies of electrospun precursor nanofibers (A), intermediate nanofibers (B), and final TiC nanofibers/nanoribbons with low (C) and high (D) magnifications. | ||
The TEM image (Fig. 2A) of intermediate nanofibers showed elongated bulges on the surface, which was consistent with the rough surface observed in the SEM image (Fig. 1B); additionally, no crystalline structure could be identified in the intermediate nanofibers as evidenced by the electron diffraction pattern (inset of Fig. 2A). In contrast, crystalline nanoparticles (i.e. crystallites) were observed all over the final TiC nanofibers/nanoribbons (Fig. 2B). The high-resolution TEM image (Fig. 3) confirmed that these crystallites had sizes typically in the range of 5–30 nm, and they were randomly distributed in an amorphous matrix; some crystallites (such as “1”, “2”, and “3” in Fig. 3) appeared to be single-crystalline and/or highly ordered, while others (such as “4”, “5”, and “6” in Fig. 3) appeared to contain defects and/or amorphous regions. The interplanar spacing “d” of the crystallite “2” in Fig. 3 was measured to be ∼2.6 Å, which was consistent with the reported value for the (111) crystallographic plane of TiC.13 The TEM energy dispersive spectroscopy (TEM-EDS) was employed to examine the chemical compositions of crystallites and their matrix in the TiC nanofibers/nanoribbons; and the acquired results indicated that both crystallites and the matrix contained carbon and titanium, yet the carbon component was enriched in the matrix.
 | ||
| Fig. 2 Representative TEM images showing the morphological structures of an intermediate nanofiber (A), and a final TiC nanofiber/nanoribbon (B). The insets are the corresponding electron diffraction patterns. | ||
 | ||
| Fig. 3 A high-resolution TEM image (left) showing that a representative electrospun TiC nanofiber/nanoribbon consisted of TiC crystallites (with sizes typically of ∼30 nm or less) embedded in an amorphous matrix (containing both C and Ti). The crystallites “1”, “2”, and “3” appeared to be single crystalline and/or highly ordered, while the crystallites “4”, “5”, and “6” appeared to contain defects and/or amorphous regions. The enlarged image (right) shows the lattice fringe in the crystallite “2” and indicates that the interplanar spacing was ∼2.6 Å. | ||
The crystalline structure of TiC nanofibers/nanoribbons was further investigated by XRD, and the results are shown in Fig. 4. It is noteworthy that TiC and TiO have similar crystalline structures of cubic unit cells with lattice constants of 4.17 Å and 4.33 Å, respectively.27 The analyses of the acquired XRD results revealed that the nanofibers/nanoribbons prepared at the carbothermal reduction temperature of 1400 °C had a predominantly TiC crystalline component (97% based upon calculations); nonetheless, if the nanofibers/nanoribbons were prepared at 1200 °C, it appeared that the crystalline component was mainly TiO (60% based upon calculation). With the combination of TEM and XRD results, it was conclusive that the crystallites observed in the final nanofibers/nanoribbons prepared at 1400 °C were TiC.
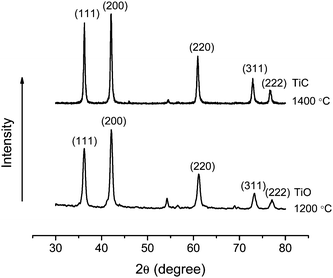 | ||
| Fig. 4 XRD curves of the final nanofibers/nanoribbons synthesized through the carbothermal reduction of electrospun precursor nanofibers in argon at 1200 °C (bottom) and 1400 °C (top). | ||
To understand the elemental distributions of Ti and C in the TiC nanofibers/nanoribbons, energy-filtered TEM images were acquired. The images “A” and “B” in Fig. 5 indicated the presence of C and Ti in a representative TiC nanofiber/nanoribbon, respectively. It was evident that both Ti and C were distributed throughout the nanofiber/nanoribbon; therefore, after selective removal of Ti through the Cl2 treatment at an elevated temperature,3,4 the extremely porous CDC with nanofiber/nanoribbon morphology would be developed.
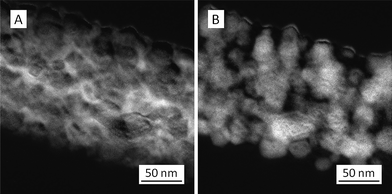 | ||
| Fig. 5 Energy-filtered TEM images showing the presence of (A) carbon and (B) titanium in representative electrospun TiC nanofibers/nanoribbons (note: the brightness corresponds to the abundance of elements; i.e. brighter areas represent higher abundance of elements). | ||
The following are the explanations about structural evolution from the electrospun precursor nanofibers to the final TiC nanofibers/nanoribbons: in the electrospun precursor nanofibers with the presence of trace amounts of HAc (most of the HAc, as well as the solvent DMF, had evaporated during electrospinning16), TiBO was converted into a Ti–O–Ti network (gel) through the sol–gel process,34 while FA was converted into poly(furfuryl alcohol) through condensation and/or dehydration.27 An electrospun precursor nanofiber containing the Ti–O–Ti gel, poly(furfuryl alcohol), and PVP was likely to possess the structure similar to multi-component interpenetration network (IPN), except that the interpenetration might occur on the nanometre scale instead of molecular scale. After the pyrolysis in argon at 325 °C, the intermediate nanofibers were formed; and they consisted of partially decomposed PVP, partially pyrolyzed Ti–O–Ti gel, and partially carbonized poly(furfuryl alcohol). The subsequent carbothermal reduction in argon at 1400 °C had several consequences including (1) the PVP was completely decomposed/removed,32 (2) the poly(furfuryl alcohol) was converted into carbon,31 (3) the Ti–O–Ti gel was turned into TiO2, which might exist merely as an intermediate, because (4) the freshly generated carbon and TiO2 reacted almost instantly and formed TiC. It was speculated that the initially formed TiC was likely in the amorphous form; subsequently, homogeneous nucleation would occur, leading to the formation of TiC crystallites. Presumably, if the total amounts of Ti and C atoms in the final TiC nanofibers could be precisely controlled to be the same, and the optimal heat-treatment conditions could be applied, the resulting electrospun TiC nanofibers/nanoribbons could be completely polycrystalline without an amorphous component. Nonetheless, for the further development of CDC, the synthesized electrospun TiC nanofibers/nanoribbons (with TiC crystallites embedded in the amorphous matrix containing both Ti and C) was probably preferred, because the presence of amorphous matrix would facilitate the Cl2 treatment at an elevated temperature to selectively remove titanium.3,4
In summary, continuous TiC nanofibers that could be designed to possess the intriguing nanoribbon morphology were synthesized through electrospinning of a spin dope containing TiBO and FA followed by heat treatments in argon first at 325 °C and then at 1400 °C. The electrospun TiC nanoribbons were morphologically uniform and had width and thickness of ∼300 nm and ∼40 nm, respectively; these nanoribbons contained TiC crystallites embedded in the amorphous matrix. The TiC crystallites had sizes ranging from 5 nm to 30 nm; the amorphous matrix also contained both titanium and carbon, yet the carbon component was enriched. The electrospun TiC nanofibers/nanoribbons in the form of overlaid fiber-mat could be an innovative precursor for the development of CDC with exceptionally large pore volume and specific surface area, which would be particularly useful for applications such as catalyst support, gas storage, super-capacitor, and phase change material support in thermal management systems.
Acknowledgements
This research was supported by the US Air Force Research Laboratory (AFRL) under the Cooperative Agreement Number (CAN) FA9453-06-C-0366 and by the Department of Energy under the Grant Number DE-FG02-08ER64624.References
- S. Zhang, Mater. Sci. Eng., A, 1993, 163, 141–148 CrossRef.
- Y. Gotoh, K. Fujimura, M. Koike, Y. Ohkoshi, M. Nagura, K. Akamatsu and S. Deki, Mater. Res. Bull., 2001, 36, 2263–2275 CrossRef CAS.
- R. Dash, J. Chmiola, G. Yushin, Y. Gototsi, G. Laudisio, J. Singer, J. Fisher and S. Kucheyev, Carbon, 2006, 44, 2489–2497 CrossRef CAS.
- J. Zheng, T. C. Ekstrom, S. K. Gordeev and M. J. Jacob, J. Mater. Chem., 2000, 10, 1039–1041 RSC.
- Y. Gogotsi, R. K. Dash, G. Yushin, T. Yildirim, G. Laudisio and J. E. Fischer, J. Am. Chem. Soc., 2005, 127, 16006–16007 CrossRef CAS.
- G. Yushin, R. Dash, J. Jagiello, J. E. Fischer and Y. Gogotsi, Adv. Funct. Mater., 2006, 16, 2288–2293 CrossRef CAS.
- S. H. Yeon, S. Osswald, Y. Gogotsi, J. P. Singer, J. M. Simmons, J. E. Fischer, M. A. Lillo-Rodenas and A. Linares-Solano, J. Power Sources, 2009, 191, 560–567 CrossRef CAS.
- J. Chmiola, G. Yushin, R. K. Dash, E. N. Hoffman, J. E. Fischer, M. W. Barsoum and Y. Gogotsi, Electrochem. Solid-State Lett., 2005, 8, A357–A360 CrossRef CAS.
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765–772 CrossRef CAS.
- N. C. Gallego and J. W. Klett, Carbon, 2003, 41, 1461–1466 CrossRef CAS.
- Y. Choi, Mod. Phys. Lett. B, 2006, 20, 1777–1780 CrossRef CAS.
- S. R. Qi, X. T. Huang, Z. W. Gan, X. X. Ding and Y. Cheng, J. Cryst. Growth, 2000, 219, 485–488 CrossRef CAS.
- K. Huo, Y. Hu, Y. Ma, Y. Lu, Z. Hu and Y. Chen, Nanotechnology, 2007, 18, 145615 CrossRef.
- P. Zhu, Y. Hong, B. Liu and G. Zou, Nanotechnology, 2009, 20, 255603 CrossRef.
- Y. Dzenis, Science, 2004, 304, 1917–1919 CrossRef CAS.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS.
- G. S. Girolami, J. A. Jensen and D. M. Pollina, J. Am. Chem. Soc., 1987, 109, 1579–1580 CrossRef CAS.
- D. C. Halverson, K. H. Ewald and Z. A. Munir, J. Mater. Sci., 1993, 28, 4583–4594 CrossRef CAS.
- R. Alexandrescu, E. Borsella, S. Botti, M. C. Cesile, S. Martelli, R. Gigogi, S. Turtu and G. Zappa, J. Mater. Sci., 1997, 32, 5629–5635 CrossRef CAS.
- M. S. Noel and D. Kovar, J. Mater. Sci., 2002, 37, 689–697 CrossRef CAS.
- D. W. Lee and B. K. Kim, Scr. Mater., 2003, 48, 1513–1518 CrossRef CAS.
- X. Feng, Y. J. Bai, B. Lu, C. G. Wang, Y. X. Liu, G.-L. Geng and L. Li, J. Cryst. Growth, 2004, 264, 316–319 CrossRef CAS.
- R. Koc and J. S. Folmer, J. Mater. Sci., 1997, 32, 3101–3111 CrossRef CAS.
- D. Sarkar, M. C. Chu, S. J. Cho, Y. I. Kim and B. Basu, J. Am. Ceram. Soc., 2009, 92, 2877–2882 CrossRef CAS.
- G. A. Swift and R. Koc, J. Mater. Sci., 1999, 34, 3083–3093 CrossRef CAS.
- Y. Shin, X. S. Li, C. Wang, J. R. Coleman and G. J. Exarhos, Adv. Mater., 2004, 16, 1212–1215 CrossRef CAS.
- Z. Jiang and W. E. Rhine, Chem. Mater., 1991, 3, 1132–1137 CrossRef CAS.
- D. R. Stanley, J. D. Birchall, J. N. Kenneth Hayland, L. Thomas and K. Hodgetts, J. Mater. Chem., 1992, 2, 149–156 RSC.
- S. Dutremez, P. Gerbier, C. Guerin, B. Henner and P. Merle, Adv. Mater., 1998, 10, 465–470 CrossRef CAS.
- R. Chandrasekar, L. Zhang, J. Y. Howe, N. E. Hedin, Y. Zhang and H. Fong, J. Mater. Sci., 2009, 44, 1198–1205 CrossRef CAS.
- E. Fitzer, W. Schaefer and S. Yamada, Carbon, 1969, 7, 643–648 CAS.
- L. Zhang, J. Y. Howe, Y. Zhang and H. Fong, Cryst. Growth Des., 2009, 9, 667–670 CrossRef CAS.
- H. Fong, I. Chun and D. H. Reneker, Polymer, 1999, 40, 4585–4592 CrossRef CAS.
- N. Y. Turova, E. P. Turevskaya, V. G. Kessler and M. I. Yanovskaya, The Chemistry of Metal Alkoxides, Kluwer Academic Publishers, Norwell, 2002, pp. 107–126 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |