Formation of periodic nanoring arrays on self-assembled PS-b-PMMA film under rapid solvent-annealing
Yuanjun
Liu
a,
Yanchun
Gong
b,
Longbin
He
a,
Bo
Xie
a,
Xi
Chen
a,
Min
Han
*a and
Guanghou
Wang
c
aNational Laboratory of Solid State Microstructures and Department of Materials Science and Engineering, Nanjing University, Nanjing, 210093, China. E-mail: sjhanmin@nju.edu.cn; Fax: +86-25-83686248; Tel: +86-25-83686248
bInstitute of Sciences, PLA University of Science and Technology, Nanjing, 211101, China
cNational Laboratory of Solid State Microstructures and Department of Physics, Nanjing University, Nanjing, 210093, China
First published on 27th August 2010
Abstract
Periodic nanoring arrays are prepared by self-assembled poly styrene-block-polymethylmethacrylate (PS-b-PMMA) diblock copolymer under rapid solvent-annealing. The dimension of the nanorings can be modified by controlling the solvent-annealing time.
Nanoring arrays have recently attracted considerable attention, motivated by potential applications due to their unique magnetic, optical or electrical properties.1–8 A variety of fabrication methods have been explored to construct nanorings. Conventional optical lithography is limited in spatial resolution. Focused ion beam (FIB) milling or electron beam lithography can offer high resolution and versatility, but they lack the efficiency that is essential for nanostructure fabrication over large area. Alternatively, template-based synthesis techniques have been demonstrated in recent years9–12 to fabricate nanoring arrays over a large scale and at low cost.
The self-assembly processes of block copolymers offer interesting strategies to create high-resolution patterns on the nanometre scale as templates to generate functional nanostructures. Block copolymer nanopatterns with lamellar, cylinder, or spherical morphologies have been well exploited and used to prepare nanodot and nanowire arrays.13–17 Recently, there have been a number of works to prepare ring-shaped patterns using block copolymers. The fabrication of nanoring arrays over a large area has been demonstrated by self-assembling of block copolymers in dilute solution18 or mixed solution,19,20 as well as using very slow solvent-annealing.21 They provide simple and efficient ways for nanoring array generation but the generated patterns lack the long-range ordering that is essential for certain applications. Concentric ring patterns with tunable dimensions have been formed by templated self-assembly of a polystyrene-block-polydimethylsiloxane diblock copolymer under confinement in shallow circular pits.22 Compared with untemplated self-assembly, it is more complicated. Formation of ordered nanoring arrays upon selective swelling via a polymer-analogous hydrolysis reaction has been reported in thin films of ABC triblock terpolymer poly(styrene)-block-poly(2-vinylpyridine)-block-poly(tert-butyl methacrylate).23 Other triblock copolymers can form nanoring structures by self-assembling with the solvent-annealing method.24–26 For example, by adjusting the solvent-annealing conditions and the film thickness, a thin film with PMMA-core/PS-shell cylinders in a PB matrix was formed in polybutadiene-block-polystyrene-block-polymethyl-methacrylate triblock terpolymer. An array of PS rings was then generated after removing the PMMA cylinder cores and the PB matrix with selective etching.24
In this paper, we report on a novel approach to fabricate nano-sized rings made of PS-b-PMMA diblock copolymer with high reproducibility over a large area. It is shown that nanoring arrays with hexagonal close packing structures can be formed with the symmetric AB diblock copolymer, a compositionally much simpler system as compared to the previously explored ABC triblock terpolymers, by rapid solvent-annealing of nanopatterns with nanoscale depressions morphology in toluene vapor, and the dimension of the nanorings can be modified by controlling the solvent-annealing time.
The symmetric diblock copolymer, PS-b-PMMA (MPS = 130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, MPMMA = 133000, polydispersity index = 1.10) purchased from Polymer Source, Inc. was dissolved in toluene with a typical concentration of 0.5 wt%. About 20 μL of the solution was spin-coated onto a freshly cleaned silica glass substrate at 2500 rpm for 40 s to form a film. The film was placed in a glass vessel of 27 cm3 volume sealed with a stopper for 5.5 h with 100 μL of acetone added as annealing solvent. The temperature of the acetone vapor was kept at 25 °C. After solvent annealing, the morphologies of the as-prepared PS-b-PMMA thin films were measured with an atomic force microscopy (AFM, NT-MDT, Russia). Commercial silicon cantilevers with force constants ranging from 2.7 to 7.7 N m−1 was used. To obtain non-artifact images, the height and phase images were recorded simultaneously under a moderate tapping force levels of rsp = 0.85 (the set-point amplitude ratio rsp is defined as Asp/A0, where A0 is the free oscillation amplitude and Asp is the set point amplitude) with a free amplitude of 0.2–0.8 V. As shown in Fig. 1, the film has a porous morphology with long range ordering, which is also confirmed by the corresponding fast Fourier transformation shown as an inset. It has been reported that with such moderate tapping conditions the magnitude of the phase shift is directly related to the elastic modulus of the sample when the amplitude of the oscillating cantilever is only slightly reduced.27–30 When tapping a region of higher modulus, the force interaction leads to a more positive phase shift and hence appears brighter in the phase image. PMMA has higher modulus than PS at room temperature (bulk elastic modulus of PS and PMMA are 3.0 and 3.3 GPa, respectively),31 therefore, the brighter and the darker areas in Fig. 1(b) represent the PMMA and the PS domains respectively, while in the AFM height image [Fig. 1(a)] their brightness was reversed, indicating that the PMMA domains appear as depressions. The PS-b-PMMA thin film contains highly ordered two-dimensional arrays of PMMA nanoscale depressions with hexagonal close packing structures, distributing in the continuous PS matrix. The diameter of the nanoscale depressions is ∼55 nm on average, the center-to-center distance between adjacent PMMA domains is ∼100 nm, and the depth of the pits is around 2 nm, as can be measured from the AFM images. The film thickness is about 25 nm, judged from the concentration of the solution as well as confirmed by an AFM profile across the edge of the film.
000, MPMMA = 133000, polydispersity index = 1.10) purchased from Polymer Source, Inc. was dissolved in toluene with a typical concentration of 0.5 wt%. About 20 μL of the solution was spin-coated onto a freshly cleaned silica glass substrate at 2500 rpm for 40 s to form a film. The film was placed in a glass vessel of 27 cm3 volume sealed with a stopper for 5.5 h with 100 μL of acetone added as annealing solvent. The temperature of the acetone vapor was kept at 25 °C. After solvent annealing, the morphologies of the as-prepared PS-b-PMMA thin films were measured with an atomic force microscopy (AFM, NT-MDT, Russia). Commercial silicon cantilevers with force constants ranging from 2.7 to 7.7 N m−1 was used. To obtain non-artifact images, the height and phase images were recorded simultaneously under a moderate tapping force levels of rsp = 0.85 (the set-point amplitude ratio rsp is defined as Asp/A0, where A0 is the free oscillation amplitude and Asp is the set point amplitude) with a free amplitude of 0.2–0.8 V. As shown in Fig. 1, the film has a porous morphology with long range ordering, which is also confirmed by the corresponding fast Fourier transformation shown as an inset. It has been reported that with such moderate tapping conditions the magnitude of the phase shift is directly related to the elastic modulus of the sample when the amplitude of the oscillating cantilever is only slightly reduced.27–30 When tapping a region of higher modulus, the force interaction leads to a more positive phase shift and hence appears brighter in the phase image. PMMA has higher modulus than PS at room temperature (bulk elastic modulus of PS and PMMA are 3.0 and 3.3 GPa, respectively),31 therefore, the brighter and the darker areas in Fig. 1(b) represent the PMMA and the PS domains respectively, while in the AFM height image [Fig. 1(a)] their brightness was reversed, indicating that the PMMA domains appear as depressions. The PS-b-PMMA thin film contains highly ordered two-dimensional arrays of PMMA nanoscale depressions with hexagonal close packing structures, distributing in the continuous PS matrix. The diameter of the nanoscale depressions is ∼55 nm on average, the center-to-center distance between adjacent PMMA domains is ∼100 nm, and the depth of the pits is around 2 nm, as can be measured from the AFM images. The film thickness is about 25 nm, judged from the concentration of the solution as well as confirmed by an AFM profile across the edge of the film.
 | ||
| Fig. 1 AFM height (a) and phase (b) images of PS-b-PMMA thin film after being annealed in acetone for 5.5 h. The scanning size of both images is 1 × 1 μm2. (c) Cross-section profile along the line in (a). Inset of (a) is corresponding fast Fourier transformation. | ||
The morphology of the as-prepared porous films was further modified through solvent annealing in saturated toluene vapor. To explore the solvent vapor induced morphology reconfiguration, the PS-b-PMMA thin films were annealed in a carefully sealed vessel of 27 cm3 volume for different times: 60 s, 90 s, 120 s, 160 s, and 180 s, respectively, with 100 μL of toluene dripped in. After solvent annealing, the samples were quickly removed to ambient atmosphere and dried. Fig. 2 illustrates the representative AFM images scanned from these samples. In the earlier stage of solvent annealing, the modification of the morphology of the polymer film is indistinct, the original hexagonal nanoscale depressions arrays can still be clearly observed in the AFM height image, as shown in Fig. 2(a). However, a few areas of the PS matrix start to be replaced by PMMA phases [indicated by arrows in Fig. 2(a)]. They grow with annealing time and become more and more visible. On the AFM images scanned over the surface of PS-b-PMMA thin film annealed in toluene vapor for 90 s, as shown in Fig. 2(d) and Fig. 2(e), full-grown nanoring arrays of PS domains can be clearly observed all around. The original hexagonal arrays of nanoscale depressions have completely transformed into hexagonal arrays of nanorings. Measuring from Fig. 2(d), it can be found that the original hexagonal lattices remain unchanged, while nanoscale depressions are replaced with nanorings at each lattice point. The outer diameter of the nanorings is about 100 nm on average, being approximately the same as the center-to-center distance between adjacent depressions in the original nanoscale depressions arrays. Therefore, the nanorings are closely contacted, as can be seen in Fig. 2(d). The inner diameter of the nanorings is about 38 nm, as measured from Fig. 2(f). Enclosed by the PS nanorings are PMMA domains, as can be seen clearly from the AFM phase image shown in Fig. 2(e). They appear as depressed nanopits, corresponding to those depressions in Fig. 1(a).
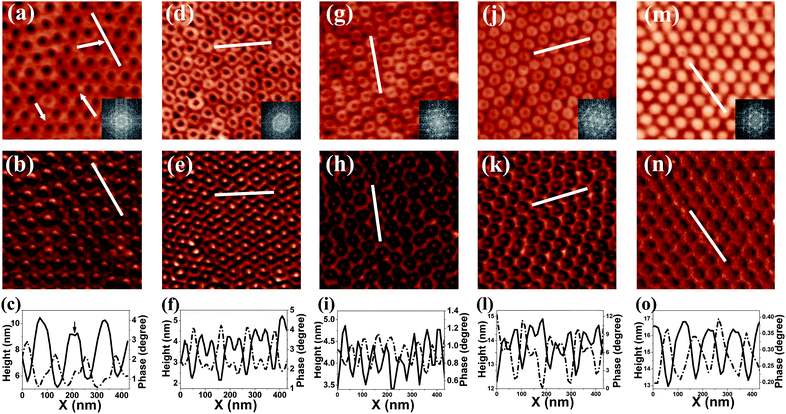 | ||
| Fig. 2 AFM micro-images of the morphologies of the PS-b-PMMA thin film annealed in toluene for (a) 60 s, (d) 90 s, (g) 120 s, (j) 160 s, and (m) 180 s. The original morphology is hexagonal nanopore arrays which shown in Fig. 1a. The scanning size of all images is 1 × 1 μm2. (b), (e), (h), (k), and (n) are phase images corresponding to (a), (d), (g), (j), and (m). Cross-section profiles along the lines present in the corresponding height (solid line) and phase (dash line) images are shown in (c), (f), (i), (l), and (o), respectively. Insets of (a), (d), (g), (j), and (m) are corresponding fast Fourier transformations. | ||
With the increase of the annealing time, the nanorings shrink both on the outer diameter and inner diameter. The nanorings shown in Fig. 2(g) are scanned from the surface of the PS-b-PMMA thin film with 120 s annealing. The outer diameter of the nanorings has reduced to about 93 nm, while their inner diameter changes from 38 nm to 27 nm. The reduction of the inner diameter, or the shrinking of the PMMA phase surrounded by the PS ring, is more rapid than the reduction of the outer diameter. Comparing the phase images, from Fig. 2(e) to Fig. 2(h), changes on the morphology of the polymer film are remarkable. The PMMA phase has become the matrix of the self-assembled nanopattern instead of the PS phase. In the PMMA matrix, the hexagonal arrays of PS nanorings embed, as can be seen from Fig. 2(g). A further increase of the annealing time only induces a minor change on the external diameter of the nanorings, however, the inner diameter of the nanorings keeps on decreasing continuously. As shown in Fig. 2(j), with 160 s annealing, the external diameter of the nanorings is still around 90 nm, while the inner diameter of the nanorings is reduced to 18 nm. Finally, after 180 s annealing, the nanorings grow into complete nanocylinders. Self-assembled nanocylinder arrays, corresponding to PS cylindrical cores surrounded by PMMA layers32 which are stable against toluene vapor, are formed, as shown in Fig. 2(m).
The evolution of the morphology from the original nanoscale depression arrays to the final nanocylinder arrays along with the solvent-annealing process is summarized schematically in Fig. 3. Depression arrays are first formed upon solvent-annealing. With further annealing, they gradually transform into nanoring arrays. The nanoring arrays become the dominant morphology of the PS-b-PMMA thin film after 90 s annealing. When annealing proceeds, the external diameter shrinks from about 100 nm to about 90 nm and then becomes unchanged. Meanwhile the internal diameter of the nanorings decreases at a much more rapid rate, and after 160 s annealing, the morphology is transformed completely to nanocylinder arrays. Throughout the solvent annealing process of the PS-b-PMMA thin films, the hexagonal ordering can be perfectly retained, as can be confirmed by the corresponding fast Fourier transformations of the AFM images, which are shown as insets of Fig. 2(a), (d), (g), (j), and (m). In Fig. 4, a large scale AFM image of PS-b-PMMA nanoring arrays is shown. Ordered domains as large as 2 × 2 μm2 can be found all over the film area.
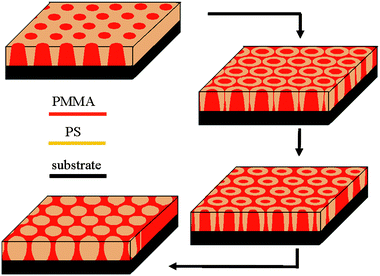 | ||
| Fig. 3 Schematic illustration of the morphology evolution of PS-b-PMMA thin film during solvent-annealing. | ||
 | ||
| Fig. 4 A large area AFM height-image of the morphologies of the PS-b-PMMA thin film annealed in toluene for 120 s. The scanning size of the images is 5.0 × 5.0 μm2. Inset of the image is the fast Fourier transform (FFT) pattern. | ||
The formation of the nanorings can be attributed to the different behaviors of PS and PMMA chains in a given solvent, which have been determined to be the reason for solvent-induced microphase separation in block copolymer.33 In the light of the Flory–Huggins criterion,34,35 the miscibility between a polymer and a solvent is governed by the interaction parameter χPS (P is the polymer, S is the solvent). This value determines the affinity between solvent and polymer through an inverse relation which can be quantitatively written as: χPS = Vs[(δdS − δdP)2 + (δpS − δpP)2]/RT. Where Vs is the molar volume of the solvent, R is the gas constant, δd is the dispersion solubility parameter, and δP is the polar solubility parameter. From this relation we get χTol-PMMA = 0.45, χTol-PS = 0.34, χAce-PMMA = 0.18, χAce-PS = 1.1.32 Thus acetone is a selective solvent for PMMA, and toluene is a slightly selective solvent for PS. When the porous film is exposed to toluene, both PS and PMMA blocks are mobile and can reconstruct themselves easily. Considering the selective dissolving of PS in toluene, there is a trend for the polymer film to have a morphology that is composed of close packed spherical PS cores aggregated within a PMMA matrix so as to maintain a largest contact area between PS phase and the solvent, and minimize the contact area between PMMA phase and the solvent. On the transition from the morphology of PMMA nanopits surrounded by PS matrix to the morphology of PS cylindrical cores surrounded by PMMA matrix, arrays of PS nanorings are formed as an intermediate morphology. Furthermore, the dimension of the nanorings can be controlled by properly choosing the solvent annealing time to terminate the morphology evolution at a certain point in the transition stage.
The lattice parameters of the ordered self-assembled patterns of diblock copolymer, such as the size of the phase separated domains and the center-to-center distance between adjacent domains are mainly influenced by the molecular weights of the two blocks.36 There is no change in the molecular weights of the blocks in the self-assembling process, therefore the period of the ordered patterns is not changed throughout the morphology transition process, as can be seen from the AFM images in Fig. 2. On the other hand, the formation of the nanorings and the change of their diameters should be related to the reorganization of the spatial distribution of the two blocks, which induces local fluctuations in the film height, rather than a change in the film area or the number of phase separated domains. From this point of view, the self-assembling process of the ring-shaped morphology may be affected not only by the molecular weights of the PS and PMMA blocks but also by the thickness of the film. Their detailed relations remain an interesting issue for further investigation.
In summary, with a proper control of the annealing time for porous PS-b-PMMA diblock copolymer thin film in toluene solvent, ordered arrays of polymer nanorings can be obtained, and the dimension of the nanorings can be finely adjusted. This provides a facile way for the large scale fabrication of well-ordered nanoring arrays on block copolymer film. Such high-resolution patterns may be used as templates to generate nanoring arrays of functional nanostructures through hierarchical self-assembling.
We thank the financial support from NSFC (Grant Nos. 10674063, 10974092, and 90606002), the Hi-tech Research and Development Program of China under contract NO. 2006AA03Z316, the National Basic Research Program of China (973 Program, contract NO. 2009CB930501), as well as the Provincial Hi-tech Research Program of Jiangsu in China (BG2007041).
Notes and references
- R. J. Warburton, C. Schäflein, D. Haft, F. Bickel, A. Lorke, K. Karrai, J. M. Garcia, W. Schoenfeld and P. M. Petroff, Nature, 2000, 405, 926 CrossRef CAS.
- T. Mano, T. Kuroda, S. Sanguinetti, T. Ochiai, T. Tateno, J. Kim, T. Noda, M. Kawabe, K. Sakoda, G. Kido and N. Koguchi, Nano Lett., 2005, 5, 425 CrossRef CAS.
- A. Matos-Abiague and J. Berakdar, Phys. Rev. Lett., 2005, 94, 166801 CrossRef CAS.
- J. Podbielski, F. Giesen and D. Grundler, Phys. Rev. Lett., 2006, 96, 167207 CrossRef CAS.
- F. J. Castaño, D. Morecroft, W. Jung and C. A. Ross, Phys. Rev. Lett., 2005, 95, 137201 CrossRef CAS.
- M. M. Miller, G. A. Prinz, S. F. Cheng and S. Bounnak, Appl. Phys. Lett., 2002, 81, 2211 CrossRef CAS.
- P. Földi, O. Kálmán, M. G. Benedict and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 155325 CrossRef.
- L. W. Yu, K. J. Chen, J. Song, J. Xu, W. Li, X. F. Li, J. M. Wang and X. F. Huang, Phys. Rev. Lett., 2007, 98, 166102 CrossRef CAS.
- F. Q. Zhu, D. L. Fan, X. X. Zhu, J. G. Zhu, R. C. Cammarata and C. L. Chien, Adv. Mater., 2004, 16, 2155 CrossRef CAS.
- D. Jia and A. Goonewardene, Appl. Phys. Lett., 2006, 88, 053105 CrossRef.
- D. K. Singh, R. V. Krotkov, H. Xiang, T. Xu, T. P. Russell and M. T. Tuominen, Nanotechnology, 2008, 19, 245305 CrossRef.
- K. L. Hobbs, P. R. Larson, G. D. Lian, J. C. Keay and M. B. Johnson, Nano Lett., 2004, 4, 167 CrossRef CAS.
- S. B. Darling, Prog. Polym. Sci., 2007, 32, 1152 CrossRef CAS.
- W. A. Lopes and H. M. Jaeger, Nature, 2001, 414, 735 CrossRef CAS.
- M. Aizawa and J. M. Buriak, J. Am. Chem. Soc., 2006, 128, 5877 CrossRef CAS.
- Z. T. Shi, M. Han, F. Q. Song, J. F. Zhou, J. G. Wan and G. H. Wang, J. Phys. Chem. B, 2006, 110, 18154 CrossRef CAS.
- J. N. Chai, D. Wang, X. N. Fan and J. M. Buriak, Nat. Nanotechnol., 2007, 2, 500 CrossRef.
- J. T. Zhu, Y. G. Liao and W. Jiang, Langmuir, 2004, 20, 3809 CrossRef CAS.
- D. J. Pochan, Z. Y. Chen, H. G. Cui, K. Hales, K. Qi and K. L. Wooley, Science, 2004, 306, 94 CrossRef CAS.
- T. Pietsch, N. Gindy and A. Fahmi, Polymer, 2008, 49, 914 CrossRef CAS.
- Y. M. Gong, Z. J. Hu, Y. Z. Chen, H. Y. Huang and T. B. He, Langmuir, 2005, 21, 11870 CrossRef CAS.
- Y. S. Jung, W. Jung and C. A. Ross, Nano Lett., 2008, 8, 2975 CrossRef CAS.
- S. Ludwigs, K. Schmidt and G. Krausch, Macromolecules, 2005, 38, 2376 CrossRef CAS.
- V. P. Chuang, C. A. Ross, P. Bilalis and N. Hadjichristidis, ACS Nano, 2008, 2, 2007 CrossRef CAS.
- V. P. Chuang, C. A. Ross, J. Gwyther and I. Manners, Adv. Mater., 2009, 21, 3789 CrossRef CAS.
- A. Sperschneider, F. Schacher, M. Gawenda, L. Tsarkova, A. H. E. Müller, M. Ulbricht, G. Krausch and J. Köhler, Small, 2007, 3, 1056 CrossRef CAS.
- Ph. Leclère, G. Moineau, M. Minet, Ph. Dubois, R. Jérôme, J. L. Brédas and R. Lazzaroni, Langmuir, 1999, 15, 3915 CrossRef CAS.
- S. N. Magonov, V. Elings and M. H. Whangbo, Surf. Sci., 1997, 375, L385 CrossRef CAS.
- J. P. Cleveland, B. Anczykowski, A. E. Schmidt and V. B. Elings, Appl. Phys. Lett., 1998, 72, 2613 CrossRef CAS.
- Y. Wang, R. Song, Y. S. Li and J. S. Shen, Surf. Sci., 2003, 530, 136 CrossRef CAS.
- J. Brandrup, E. H. Immergut, E. A. Grulke, A. Abe and D. R. Bloch, Polymer Handbook, 4th edn, Wiley, New York, 1999 Search PubMed.
- J. Peng, D. H. Kim, W. Knoll, Y. Xuan, B. Y. Li and Y. C. Han, J. Chem. Phys., 2006, 125, 064702 CrossRef.
- J. Peng, Y. Xuan, H. F. Wang, Y. M. Yang, B. Y. Li and Y. C. Han, J. Chem. Phys., 2004, 120, 11163 CrossRef CAS.
- S. A. Chen, J. Appl. Polym. Sci., 1971, 15, 1247 CrossRef CAS.
- Y. Xuan, J. Peng, L. Cui, H. F. Wang, Y. M. Yang, B. Y. Li and Y. C. Han, Macromolecules, 2004, 37, 7301 CrossRef CAS.
- T. Xu, H. C. Kim, J. DeRouchey, C. Seney, C. Levesque, P. Martin, C. M. Stafford and T. P. Russell, Polymer, 2001, 42, 9091 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |