Towards multifunctional, targeted drug delivery systems using mesoporous silica nanoparticles – opportunities & challenges
Jessica M.
Rosenholm
ab,
Cecilia
Sahlgren
*cd and
Mika
Lindén
*a
aCenter for Functional Materials, Department of Physical Chemistry, Åbo Akademi University, Porthansgatan 3–5, FI-20500, Turku, Finland. E-mail: Cecilia.Sahlgren@btk.fi; Mika.Linden@abo.fi
bNano Biomedical Research Center, Med-X Institute, Shanghai Jiao Tong University, 200030, Shanghai, P.R. China
cDepartment of Biology, Åbo Akademi University, Artillerigatan 6A, FI-20520, Turku, Finland
dTurku Centre for Biotechnology, University of Turku and Åbo Akademi University, P.O. Box 123, FI-20521, Turku, Finland
First published on 23rd August 2010
Abstract
One of the big challenges of medicine today is to deliver drugs specifically to defected cells. Nanoparticulate drug carriers have the potential to answer to this call, as nanoparticles can cross physiological barriers and access different tissues, and also be provided in a targetable form aimed at enhancing cell specificity of the carrier. Recent developments within material science and strong collaborative efforts crossing disciplinary borders have highlighted the potential of mesoporous silica nanoparticles (MSNs) for such targeted drug delivery. Here we outline recent advances which in this sense push MSNs to the forefront of drug delivery development. Relatively straightforward inside-out tuning of the vehicles, high flexibility, and potential for sophisticated release mechanisms make these nanostructures promising candidates for targeted drug delivery such as ‘smart’ cancer therapies. Moreover, due to the large surface area and the controllable surface functionality of MSNs, they can be controllably loaded with large amounts of drugs and coupled to homing molecules to facilitate active targeting, simultaneously carrying traceable (fluorescent or magnetically active) modalities, also making them highly interesting as theragnostic agents. However, the increased relative surface area and small size, and flexible surface functionalization which is beneficially exploited in nanomedicine, consequently also includes potential risks in their interactions with biological systems. Therefore, we also discuss some safety issues regarding MSNs and highlight how different features of the drug delivery platform influence their behaviour in a biological setting. Addressing these burning questions will facilitate the application of MSNs in nanomedicine.
 Jessica M. Rosenholm | Jessica Rosenholm obtained her Doctor of Science in Technology degree in Physical Chemistry in 2008 under the supervision of Prof. Mika Lindén at Åbo Akademi University, funded by the national Biomaterials and Tissue Engineering Graduate School. She recently returned from a post-doc visit at the Nano Biomedical Research Center, headed by Prof. Hongchen Gu, at the Med-X Institute of Shanghai Jiao Tong University, focusing on magnetic nanoparticles for biomedicine. Her research interests thus include nanomaterial synthesis and functionalization for biomedical applications such as drug delivery and bioimaging. |
 Cecilia Sahlgren | Cecilia Sahlgren recieved her PhD in Cell Biology December 2002 at Åbo Akademi University. Subsequently she was a post doctoral fellow at the Karolinska Institute. Currently she is an Academy research fellow at the Academy of Finland and leads a research group situated at the Turku Centre for Biotechnology. The focus of her research group is to understand the role of cellular interactions in cancer and development using cellular and mouse disease models. The research team has a special interest in developing systems to target specific cell populations in vivo for various applications including drug delivery and bioimaging. |
 Mika Lindén | Mika Lindén is working as a group leader at the department of physical chemistry, Åbo Akademi University. He obtained his doctorate degree in 1996 under the supervision of Prof. Jouko Peltonen, followed by a post-doc in Prof. Ferdi Schüth's group at the University of Frankfurt. He has also worked as a Humboldt fellow in the group of Prof. Schüth at the MPI, Mülheim/Ruhr, and as visiting senior scientist in the groups of Profs. Clément Sanchez and David Grosso, Paris, and Jean Rouquerol, Marseille. His current research interests include biomaterials, materials for solar cell applications, and proteomics. |
Introduction
Targeted drug delivery is of special importance in cancer therapy as current cancer treatments based on chemotherapy are typically associated with serious side-effects due to non-specific uptake of the drug by healthy cells. Furthermore, many cancer drug candidates are hydrophobic, which makes direct administration a challenge. Therefore there is a clear need for the development of cancer cell-specific drug carrier systems capable of delivering poorly water-soluble drugs, which is why a lot of nanomedical research is focused on cancer therapies. Among the different types of particles being pushed forward in nanomedicine, ceramic (inorganic) particles have been highlighted to provide increased mechanical strength, chemical stability, biocompatibility, and resistance to microbial attack as compared to their organic (polymeric) counterparts.1–3 Moreover, the ceramic matrix effectively protects immobilized guest molecules against enzymatic degradation or denaturation induced by external pH and temperature as no swelling or porosity changes occur as a response to environmental changes such as pH. After the introduction of mesoporous materials to the drug delivery scene in the early 2000s,4 mesoporous silica nanoparticles (MSNs) have recently also shown to be promising candidates for cell-specific delivery, which will be outlined in the following. One of the strengths of the MSN delivery platforms is that they are also especially suitable for the delivery of hydrophobic substances, as the cargo is incorporated via post-synthesis processes unlike many predecessor sol–gel silica-derived particles, and both their size and surface chemistry can be modified within a wide range. Moreover, the high specific pore volume and surface area of MSNs (typical values range from 0.6–1 cm3 g−1 and 700–1000 m2 g−1, respectively) make it possible, depending on the drug, to reach drug loadings exceeding 30 wt%, and allow tuning of the drug loading to that of the particle concentration in order to minimize particle-induced toxicity. Furthermore, the particles can readily be made bio-imageable, for example by covalent attachment of fluorophores for optical microscopy studies or by incorporation of superparamagnetic iron oxide nanoparticles (SPIONs)5–9 or paramagnetic Gd-chelate complexes10–13 for magnetic resonance imaging, which makes it possible to study the cell specificity and biodistribution of the MSNs both under in vitro and in vivo conditions. The current development curve related to the use of MSNs for drug delivery and bioimaging is very steep, thanks to close collaborations between material chemists, biologists, and physicians, and the current drivers for development are based on solving real biological problems. This development is naturally also reflected in the experimental designs, and increasingly more complex biological evaluations are reported in addition to detailed material characterizations. In this minireview we critically discuss recent developments related to the use of MSNs for drug delivery, with special focus on actively targeted drug delivery, i.e. when a cell specific recognition moiety is linked to the outer surface of the MSN in order to promote carrier uptake by the target cells. As this kind of research are generally carried out on cancerous vs. healthy cells due to the urgent need of specific delivery devices especially in cancer therapy, our focus will be from a cancer point of view even though the trends discussed are of a general nature for any nanomedical approach. We also give our view on some of the open challenges that need to be addressed in order to bring the highly promising MSNs to practical use as drug delivery vehicles.Synthesis and functionalization of MSNs
Nanoparticle synthesis
The particle size range of interest for targeted intracellular delivery applications in the case of MSNs is about 50–200 nm, as larger particles cannot easily bypass physical membranes in the body, and smaller MSN particles are difficult to synthesize as a consequence of their inherent mesoporosity. A narrow particle size distribution is also preferred due to size being an important complementary strategy for spatial control of nanoparticle distribution and behavior in a living system.14–19 Therefore, wet-chemical synthesis methods are generally more beneficial as compared to other physical methods like spray-drying, which typically lead to a relatively wide particle size distribution with non-negligible fractions of fine and larger particles. Some reported syntheses for the production of MSNs are summarized in Table 1. In most reported syntheses, tetraethoxysilane, TEOS, has been used as the precursor for silica, and cationic quaternary ammonium salts, the most common being cetyltrimethyl ammonium bromide (CTAB), have been used as structure directing agents under alkaline conditions. This typically leads to materials with mesopore dimensions in the range 3–4 nm, often with cylindrical mesopores arranged in a two-dimensional hexagonal fashion, characteristic of MCM-41 type materials.20,21 In addition, functional silanes can be used together with TEOS as a precursor for silica, typically at a concentration of 10 mol-% of the TEOS, and allows the introduction of different functional groups in situ onto the silica surface already during synthesis. Currently, much focus is made on attempts to increase the pore sizes of MSNs. The driver for this development is that while the size of smaller mesopores is sufficient to host small-molecule drugs, larger pores are needed in order to efficiently encapsulate most macromolecular substances such as proteins (enzymes, antibodies) and nucleic acids (DNA, RNA). Pore size-engineering is rather straightforward for larger particle sizes, and can be achieved by addition of auxiliary organics such as trimethylbenzene, TMB, as pore swelling agent to the synthesis. However, pore size-expansion in nanoparticulate syntheses is more challenging, as the dilute conditions often also involve co-solvents such as alcohols. Therefore, the swelling agent may preferentially remain dissolved in the solvent rather than being solubilized by the surfactant micelles. Successful pore-swelling of MSNs under alkaline conditions has been accomplished, however, by addition of an almost 20-fold amount of TMB as compared to CTAB, yielding pore sizes of 5.4 nm22 and by hydrothermal treatment of the dried silica/surfactant composite in the presence of either TMB23 or surfactants with longer alkyl chains,24,25 yielding pore diameters reaching 6.5 nm. The reverse miniemulsion technique has been used to prepare MSNs using a dual surfactant system, CTAB and amphiphilic block copolymer PE/B-b-PEO, giving rise to pore sizes as large as 13 nm.26 The largest mesopore diameters reported for MSNs to date is 20 nm. Here, the MSNs were synthesized under acidic conditions using polymeric surfactants of the ABA-type as structure directing agents.27 However, generally the particle size distributions of these MSNs are clearly broader as compared to MSNs synthesized under alkaline conditions using quaternary ammonium salts as structure directing agents.| Main precursor | SDA | Particle size/nm | Pore diameter/nm | pH condition | Means of particle size control | Authors |
|---|---|---|---|---|---|---|
| TEOS | CTAB | 15 | 3 | Alkaline | Dilution + pH quenching | Mann et al.28,29 |
| TEOS | CTAC | 20–50 | 3 | Alkaline | Quenching by P123 | Suzuki et al.42 |
| TEOS/APTS | Fatty acid | 270 | 3 | Alkaline | Quenching by Pluronics | Garcia-Bennett et al.43 |
| TEOS | CTAB, dodecylamine | 65–740 | 3 | Alkaline | Dilute conditions, water–EtOH cosolvent | Nooney et al.30 |
| TEOS/organosilanes | CTAB | 50–200 | 3 | Alkaline | Complexation by triethanol amine | Bein et al.45,46,47 |
| TEOS/APTS | CTAB | 70–100 | 3 | Alkaline | Ethylene glycol as co-solvent | Okubo et al.31 |
| Waterglass | P123 | 50–300 | 5–7 | Acidic | Addition of salts, varying amount of water | Berggren and Palmqvist32 |
| TEOS/APTMS | CTAB | 30–280 | 3 | Alkaline | pH-adjustment | Mou et al.33 |
| TEOS | CTAB | 40–550 | 3 | Neutral | Propanetriol as co-solvent + quenching by Brij-56 | Shi et al.34 |
| TMOS | CTAB | <20 | 3 | Alkaline | High ratio of C16TMABr/Si | Urata et al.35,36 |
| TEOS | F127 | 50–300 | 17–20 | Acidic | Quenching using a cationic fluorocarbon surfactant | Gao et al.27 |
| TEOS/thiol-silane | CTAB | 200–250 | 2.3–2.7 | Alkaline | Dilute conditions | Lin et al.37,38 |
| TEOS | CnTAB (n = 14, 16 and 18), CnTAC (n = 10, 16) | 70–150 | 1.6–3 | Close to neutral | Combined acid–base and liquid evaporation | Kapoor et al.39 |
| TMOS | CnTAC (n = 14, 16 and 18) | 150–870 | 2–2.3 | Alkaline | Methanol-to-water ratio | Yano and Fukushima40 |
| TEOS | F127 | 120 | about 6 nm | Acidic | — | Thomas et al.41 |
A key property of any MSN synthesis is to avoid secondary aggregation of particles after nucleation and growth of the primary MSN particles. Two main approaches have been used to prevent secondary aggregation during the synthesis: 1) performing the synthesis under highly dilute conditions, and 2) the use of particle growth quenchers. Performing the synthesis under dilute conditions is a very reproducible method for producing monodisperse MSNs in the laboratory, but up-scaling of the synthesis for the production of larger quantities may become difficult. In this respect, the use of particle growth quenchers, like non-ionic surfactants or polymers,25,42–44 fluorocarbon-based cationic surfactants,27 triethanolamine,45–47 propanetriol,48etc., is an interesting alternative, as much higher dry contents can be achieved without secondary aggregation. Even though many papers report these type of suspensions are stable for extensive amounts of time, this typically applies to as-synthesized MSNs in the mother liquid. Most dynamic light scattering (DLS) results reported are measured on the mother liquid and specific data on particle aggregation/redispersion after extraction, and even more seldom, subsequent drying, are rarely reported. Moreover, removal of the growth quenchers by leaching may again lead to problems with subsequent re-dispersion of the particles after drying. We also note that the use of organosilanes in the synthesis together with TEOS often lead to smaller particle diameters than in the case of corresponding synthesis where TEOS is the only source for silica, which indicates that the functional silanes also can serve as growth quenchers, alternatively influence the particle nucleation process so that more nuclei are formed leading to a larger number of particles and therefore to smaller final particle sizes.
The most critical step in obtaining well-dispersed, discrete nanoparticles is the template-removal process. Removal of the structure directing agent is normally performed by solvent extraction procedures, as thermal decomposition (calcination) can easily lead to inter-particle condensation leading to redispersability problems. Thermal decomposition could also easily lead to decomposition of organic functions originating from the co-condensation reactions, which normally is undesired. Dialysis has also been shown to be an attractive means for surfactant removal in the case of very small MSNs (<20 nm), which otherwise tend to aggregate irreversibly upon repeated centrifugation and re-dispersion cycles.36 Surfactant removal can also be accomplished using H2O2-mediated oxidation,49 but in this approach also oxidation of the organic functional groups occurs resulting in purely siliceous MSNs. Another critical aspect that has to be considered in relation to template removal is the efficiency of the template removal process, as the most employed porogen, CTAB, is cytotoxic and incomplete surfactant removal may thus limit the applicability of such particles in biological systems. Whereas syntheses using more biocompatible nonionic structure-directing agents for the synthesis of macroscopic mesoporous materials can readily be found in the literature, the reports on the production of nanoscaled particles using these routes are scarce (see Table 1).
Surface functionalization
Surface functionalization of the pore walls of MSNs is typically carried out using co-condensation approaches, which enables the surface functions to be introduced already during the synthesis step. This also minimizes the need for particle separation, drying and re-dispersion that otherwise would be needed if post-synthesis grafting methods are to be used. A wide range of functional groups have successfully been introduced to MSNs using the co-condensation approach, and covalent attachment of both fluorophores and biologically active molecules onto these functional groups have been demonstrated. Often the fluorophore is pre-reacted with the aminosilane that is subsequently used in the co-condensation synthesis, yielding inherently fluorescent MSNs. We note that this approach may also contribute positively on the redispersion of the nanoparticles after surfactant extraction (see below). However, when aiming at targeted delivery applications, additional functionalities, such as targeting groups, have to be introduced to the outer particle surface. Furthermore, it may also be beneficial to introduce a surface layer or pore capping agents (e.g. ultrasmall nanoparticles consisting of some other material, “supermolecules” or mechanically interlocked molecules, MIMs),50 onto the outer particle surface to create gate-keeping properties, i.e. being able to keep the pores closed to prevent guest molecules from being released and then “open” upon an external stimulus, for example irradiation, application of a magnetic field or changes in temperature. On this topic, the interested reader is recommended to consult some of the excellent recent reviews covering different means for introducing gate-keeping properties to MSNs.50 A schematic structure comprising a stimuli-responsive polymer layer is illustrated in Fig. 1 and a more general illustration of the biofunctionalization and cargo-loading possibilities is shown in Fig. 2.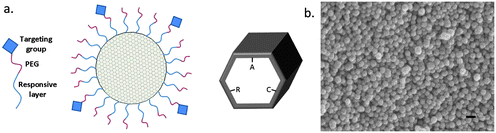 | ||
| Fig. 1 (a) Schematic representation of a targetable MSN. The high pore volume of MSNs makes it possible to achieve a high loading degree of active cargo. Selective functionalization of the outer surface of the particles with molecular or polymeric layers changing their structure upon changes in temperature or pH allows the release of the active components to be controlled by external passive or active triggering. Furthermore, introducing an outer PEG layer to which a cell-specific targeting group is attached can be introduced in order to minimize protein adsorption, increasing the circulation time in vivo, and also increases the accessibility to the targeting group. Furthermore, different functional groups can be introduced onto the pore walls, such as cationic (–C), anionic (–A), and non-ionic (–R) groups, making it possible to independently optimize the interactions between the cargo and the carrier. (b) Scanning electron microscopy image of MSNs synthesized according to the synthesis procedure described in ref. 31. Scale bar 100 nm. | ||
Some interesting hybrid structures relying on encaspulation of MSNs into lipid shells have also been described. Such ‘protocells’ was recently reported by the Brinker group, where they encapsulated mesoporous silica nanoparticles as cores into liposomal bilayer shells.51 The thus formed nanostructure takes advantage of the low toxicity and immunogenicity of liposomes whereas the protocell is more stable due to the silica core, and simultaneously also utilizes the mesoporous core to control both drug loading and release. On a related structure, Nordlund et al.52 recently reported on mesoporous silica particles surrounded by a proteolipid membrane incorporating a multisubunit redox-driven proton pump (cytochrome c oxidase). The enzyme was found to be functional both with respect to catalysis of O2 reduction to water and transmembrane charge separation. As the inner, water-filled pore structure thus may mimic a cytoskeleton, the structure yields a solid-supported biofunctional cellular surface, which could provide a basis for functional studies of membrane-bound transport proteins and also for applications within pharmaceutical drug delivery.52
Drug loading
The loading of drugs into MSNs is typically carried out as the final step. Due to the high specific areas and pore volumes of MSNs, large quantities of drug can be incorporated into the porous matrix by adsorption to the pore walls. MSNs are especially suitable for the delivery of hydrophobic drugs, which may be difficult with many other types of carrier particles. This is due to the fact that the MSN's structural integrity is kept intact in organic solvents, thanks to the ceramic matrix. Thus, the drug loading process can be performed in non-aqueous media, and repulsive forces between the carrier material and the drug that might be an issue under aqueous conditions due to electrostatic interactions (charges) and pH-dependencies can be neglected.53,54 Moreover, the optimal solvent can be chosen in terms of drug solubility, in order to facilitate the drug interactions with the pore wall over solvent–drug interactions. Consequently, the drug is most often absorbed from an organic solvent, and typically a monolayer adsorption is observed, and can be modeled using a Langmuir adsorption isotherm.55 Monolayer adsorption onto the pore walls has also been observed for proteins from aqueous solutions.56 This makes it possible to form a rational basis for the loading process,57 and to fine-tune the drug loading degree to the needs of the specific application. For more hydrophilic drugs, “pH-matching” in aqueous solvent can be utilized to reach higher drug loading levels than possible from organic media.53 Many studies have highlighted the possibility of using these kinds of specific interactions between the drug and functional groups present on the pore wall also for controlling the drug release process. In most cases, the vast majority of the groups present on the silica surface even functionalized MSNs are still silanol groups, why the influence of the silanol groups on the overall surface chemistry of the pore surface should not be underestimated.53,54 As the literature is ample with such reports of (macroscopic) mesoporous materials studied under sink conditions, we will limit ourselves to drug release demonstrated in actual combination with a biological system (to date almost exclusively demonstrated with cell cultures under in vitro conditions, see the section on controlled release). Another possibility for drug incorporation into MSNs is to covalently link the drug to functional groups present on the pore wall,58–60 and this is especially attractive if premature drug release otherwise could be a problem. However, in this case the drug activity should be retained after decoupling from the pore wall, and thus preferably the original functional group used to attach the drug molecule should be re-formed upon detachment in order to preserve the original drug molecular structure.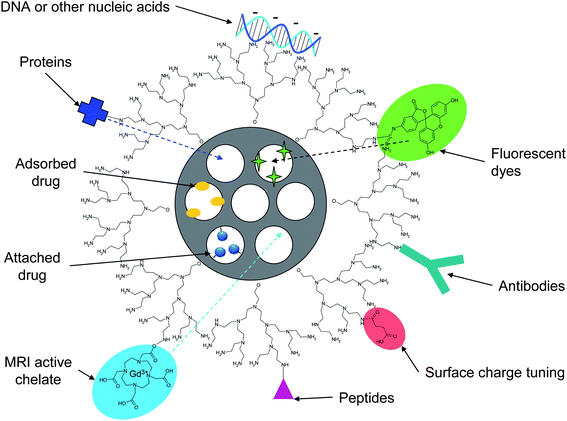 | ||
| Fig. 2 Cargo loading possibilities of MSNs. Small-molecule drugs are most often loaded into the mesopores of MSNs by adsorption from solution, but can also be attached to pending functional groups on the pore walls. Biomolecules such as proteins (enzymes, antibodies) or nucleic acids can be either adsorbed or attached to the particle surface; and potentially loaded into the porous matrix in case pore-expanded materials are used. The MSN system can furthermore be made bio-imageable for instance by attachment of fluorescent dyes or MRI-active complexes to either the particle surface and/or the pore walls for concurrent imaging of the drug delivery site/process. | ||
Dispersability
In addition to gate keeping properties, surface layers are highly beneficial in terms of particle re-dispersability (as well as suspension stability) after drying, as this is one often encountered problem with MSNs, increasingly so with decreasing particle size. Native MSNs typically have a high surface concentration of silanol groups, especially when not thermally treated, and drying of the particles can easily lead to formation of siloxane bridges between the particles making re-dispersability a challenge. As mentioned above, very often re-dispersability after drying is not demonstrated in the literature, and particle size analyses based on light-scattering measurements are often reported for as-synthesized particles. Our experience is that co-condensed particles are easier to re-disperse, but the nature of the organic function also seems to play an important role. Amino-functions seem to be beneficial for re-dispersability as compared to thiol functions, for example. This could be due to the more hydrophilic nature of amino groups in comparison to thiol groups, probably due to the charged nature of the amino group under aqueous conditions. Also the surface concentration of the organic groups plays a key role. For example, MSNs where the outer surface has been functionalized by surface hyperbranching polymerization of poly(ethylene imine) are easier to re-disperse than corresponding MSNs where the amino groups solely originate from the aminosilane used together with TEOS in the co-condensation synthesis (see Fig. 3). This is due to the fact that the surface concentration of chargeable organic groups is much higher in the former case, 61–63 and thus also due to the fact that the surface charge, as measured by the zeta-potential of the particles, is much higher in the case of poly(ethylene imine) functionalized particles.64,115 Adsorption or attachment of polymers to the surface of the MSNs may also result in a better dispersability of the MSNs due to steric stabilization. Here, biocompatibility of the used polymer has to be ensured when aiming at biological applications.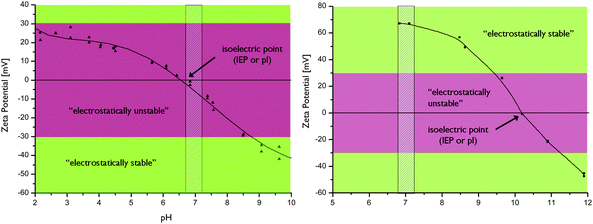 | ||
| Fig. 3 Electrokinetic titrations of by PEI-functionalization electrostatically stabilized MSNs (right) as compared to amino-co-condensed MSNs which are not electrokinetically stable (left) at physiological pH (shaded area).64,65 | ||
Further functionalization affecting targeting and ‘stealth’ properties
It is important that the surface layer contains functional groups to which additional functions can be linked, as the targeting moieties, e.g. peptides, antibodies, or simple molecules such as folic acid, have to be readily accessible on the particle surface. In this respect, it is also important to ensure that extensive protein adsorption to the particles does not occur under physiological conditions, as this could decrease the targetability of the particles as well as increase the recognition of the particles as foreign by the body defense mechanisms (the reticuloendothelial system, RES), which would lead to rapid removal from the blood circulation. Protein adsorption onto the MSNs may also influence the toxicity of the MSNs, as discussed in more detail below. An often used means for reducing protein adsorption to nanoparticles is to cover them with a layer of poly(ethylene glycol), PEG, as this creates a hydrophilic “molecular cushion” around the particles that ‘shields’ them from the blood serum proteins (see Fig. 1). In a recent study, Mou et al.66 reported a synthesis of PEG-functionalized MSNs that could be targeted to breast cancer cells overexpressing the HER2/neu receptor under in vitro conditions. Here, the targeting monoclonal antibody was first linked to PEG (molecular weight 5000) before covalent attachment to thiol-functionalized MSNs. The non-selective uptake of the MSNs was clearly reduced when the particles were covered by PEG. The surface concentration of PEG as well as the PEG molecular weight both affect the extent to which the particles exhibit ‘stealth’ properties, i.e. minimum protein adsorption (opsonization), as both parameters influence the polymer conformation on the surface. Many studies have concluded that PEG molecular weights ranging from 2000–5000 and PEG chain surface densities of about 1–2 chains nm−2 are optimum for minimizing protein adsorption on non-porous nanoparticles (see for example the study of Fang et al.67). Such a high packing density of PEG chains may be difficult to achieve on MSNs if all mesopores are open on the particle surface, which has been shown to be the case for MCM-41 silica which mainly is discussed here.68 In a recent study, He et al.69 studied the influence of PEG molecular weight and packing density on the adsorption of serum proteins to all-silica MSNs with a mean particle diameter of 150 nm. Here, the PEG chains were linked to the MSNs using silane coupling chemistry. They concluded that PEG molecular weights of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 were optimum for minimizing non-specific protein adsorption. One may speculate that the higher PEG molecular weights needed to minimize protein adsorption onto MSNs can be correlated with the porous nature of the MSNs, reducing the achievable PEG surface packing density as compared to non-porous counterparts. However, the efficient targetability observed by Mou et al. already at lower PEG molecular weights may also indicate that the optimum PEG molecular weights may be a function of the used linking chemistry. Clearly, this highly important topic requires more detailed future studies.
000–20000 were optimum for minimizing non-specific protein adsorption. One may speculate that the higher PEG molecular weights needed to minimize protein adsorption onto MSNs can be correlated with the porous nature of the MSNs, reducing the achievable PEG surface packing density as compared to non-porous counterparts. However, the efficient targetability observed by Mou et al. already at lower PEG molecular weights may also indicate that the optimum PEG molecular weights may be a function of the used linking chemistry. Clearly, this highly important topic requires more detailed future studies.
Hydrolytic stability
Another key property of MSNs in relation to drug delivery applications is the hydrolytic stability of the particles. Also in this case, surprisingly very few systematic studies devoted to understanding the dissolution behavior of MSNs in biological media have been carried out. However, the bulk dissolution kinetics of MSNs is important from several points of view. Firstly, the MSNs have to remain stable (intact) in the body on time scales corresponding to the circulation times of the particles. Secondly, the hydrolytic stability of the particles is important for the drug-particle formulation and also determines its shelf-life if stored as an aqueous dispersion. The targeting and imaging groups, as well as the drug, have to remain associated with the MSN until it reaches its target site in order for the targeting/delivery to be efficient. Furthermore, once the MSN has reached its target, it is important to know the kinetics for intra-cellular degradation in relation to the kinetics of which the particles are expelled from the cells. It is clear that the extracellular dissolution kinetics is different, probably much faster, than that of the intracellular dissolution kinetics due to the limited liquid volume inside the cell, but this topic remains the focus of future studies. In an interesting study, Etienne and Walcarius70 studied the dissolution of (micron sized) mesoporous silica in water as a function of pH, both in its native form and as amino-functionalized, and could show that amino-functionalized silica did dissolve at a faster rate than all-silica particles under biologically relevant pH conditions. For the amino-silane functionalized particles as much as 2/3 of the amino-groups were already found in the supernatant 4 h into the experiment, after which the concentration of amino species in solution reached a plateau. Thus, not all amino-functions were liberated in solution, but clearly factors such as particle surface charge could be expected to be dramatically influenced during the process. Furthermore, as fluorophores are frequently linked to amino-groups, care must be taken in ensuring that the fluorescence signal is indeed originating from fluorescent MSNs rather than from liberated free fluorophore in in vitro and in vivo experiments. In a recent study, Onida and co-workers studied the influence of SBF (simulated body fluid) immersion on the structural parameters of MSNs with particle sizes ranging from 200–800 nm.71 They observed a rapid decrease in the mesopore volume during the first hours after immersion, while signs of mesopore ordering were still seen in XRD. The authors thus concluded that the dissolved silica is re-desposited close to the mesopore openings gradually closing the pores. Further support for this conclusion was obtained from drug release kinetic measurements, where the drug release rate reached a plateau at times corresponding to the time at which the mesopore volume was strongly diminished. Decelerated drug release rates connected to silica dissolution behavior have also been observed for micron sized mesoporous silica.55 Similar observations were made by He et al.72 who reported on a three-stage degradation behavior of MSNs in SBF (see Fig. 4). Also this study revealed rapid initial bulk degradation, followed by the deposition of a calcium/magnesium silicate layer which drastically decreased (or counteracted) the degradation rate, similar to the observations of Onida et al. (see above).71 The third stage involved a maintained slow diffusion of dissolved silica species detected as free Si, whereas the whole sample was degraded after 15 days when immersed at a concentration of 0.5 mg ml−1. Importantly, the particle morphology remained intact after 24 h even though hollowing out and enlargement of the mesopores was clearly observed. The particle disintegration behavior becomes of paramount importance under in vivo conditions as smaller particles (or in this case, particle fragments) have different clearance mechanisms and kinetics as compared to larger (in this case, intact) particles. Again, the extrapolation from sink to actual in vivo conditions may be challenging. These results thus clearly highlight the need for detailed particle stability/degradation/disintegration studies in relation to drug delivery applications of MSNs.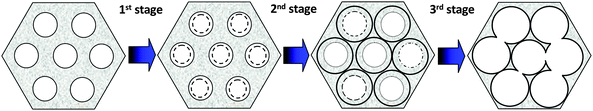 | ||
| Fig. 4 Schematic illustration of the three-stage degradation process of MSNs in SBF proposed by He et al. The initial fast bulk degradation (stage 1) is retarded by the formation of Ca/Mg-silicate layers (stage 2) followed by a slow diffusion-controlled degradation behavior spanning over several days (stage 3). Adapted from ref. 72. | ||
Toxicity and safety of MSNs
Also in vivo biodistribution of MSNs will be influenced by surface charge and size. To date, only a few studies have addressed the influence of these parameters on in vivo behavior of these potential drug carriers. Mesoporous nanoparticles in the size range of 50–100 nm possessing a relative positive charge have been shown to mainly be targeted to the liver after intravenous injections,84 while 50–200 nm non-porous silica particles were shown to be cleared to urine and bile over time in a size dependent manner.85 Smaller particles were preferably cleared by the urine and bile, whereas larger particles were trapped by macrophages and accumulated in the liver and spleen where they remained up to 4 weeks after injection. 130–180 nm mesoporous silicon particles were shown to be cleared from the body within 4 weeks after intravenous injections.86 This was contributed to disintegration of the particles followed by renal clearance. He et al. showed that differently modified non-porous silica nanoparticles (OH-SiNPs, COOH-SiNPs and PEG-SiNPs) with a size of approximately 45 nm were all partly excreted through the urine.87
Detailed analyses of biodistribution of differently charged MSNs are still lacking. In general, negatively charged surfaces should be less bio-reactive and thus mitigate cellular interactions that can give rise to unspecific toxicity. More positively charged particles on the other hand are expected to be more prone to interact with the RES and mobilize an immune response. High absolute charge, either positive or negative, is prone to decay the stealth properties of any particle,88,89 by increasing the opsonization (protein adsorption) of the particles, a property well-known to be induced also by hydrophobicity. Critical evaluation of how these physicochemical parameters of MSNs affect biodistribution is needed to avoid accumulation in healthy organs, disruption of biological membranes and unwanted activation of immune response.
Biological test conditions and particle dispersion media should thus also be taken into account during safety testing and particles should preferably be dissolved and tested in medium containing serum proteins to mimic physiological conditions. Recent studies indicate that serum protein adsorption might influence particle toxicity,92,93,94 although other studies suggest that serum protein opsonization do not influence particle uptake and toxicity.95
At the time of writing, proper assessment of in vivo biocompatilibility for MSNs is lacking and will be an important focus of future studies. A recent study showed that mesoporous silica materials of different size and porosity were lethal when injected intravenously or intraperitoneally, whereas no adverse responses were found for subcutaneously injected particles.96 However, these materials were all pristine (non-functionalized) silica, which may account for this observation, in accordance with the in vitro cytotoxicity observations discussed above. Importantly, the study also confirmed that no toxicity originated from the degradation products, in line with what is known for the degradation/dissolution behavior of amorphous silica. Thus, it is important to keep in mind that all components used to create the nanostructure should be biocompatible; on this topic we note that in the case of gate-keeping MSN systems, it could be beneficiary for instance to use USPIO nanoparticles instead of for instance CdS, as the latter is known to be cytotoxic while the former is frequently used as T2 contrast agents in MRI and should hence be more tolerable. Still regarding in vivo biocompatibility, a recent report investigating the biodistribution of MSNs did not reveal any adverse reactions within 6 h after intravenous injections, although toxicity were not the main focus addressed in this study.84 Likewise, other in vivo studies regarding MSNs have not revealed any signs of (acute) toxicity in the animals,7–12 thus suggesting that MSNs in their surface functionalized form exhibit sufficient biocompatibility to be suitable also for in vivo biomedical applications.
Although MSNs are promising therapeutic tools several issues regarding their safe use need to be addressed. Surface properties, size and other physicochemical properties affect circulation times, biodistribution and unspecific interactions with non-targeted cells and may influence the integrity of biological membranes. Thus careful analyses of in vivo toxicity and particle behavior are needed to understand how to fine tune particle characteristics for biomedical applications in humans.
MSNs in targeted therapeutics for cancer
Successful application of nanoparticles in cancer therapy will require detailed characterization of particle “biobehaviour”. As discussed above, size and surface modifications are critical determinants of particle pharmacokinetics and biodistribution. These parameters will affect particle stability, circulation time, tumour accumulation, cellular uptake and therapeutic efficacy. The complex interaction between particle characteristics and behaviour in vivo is summarized in Fig. 5.
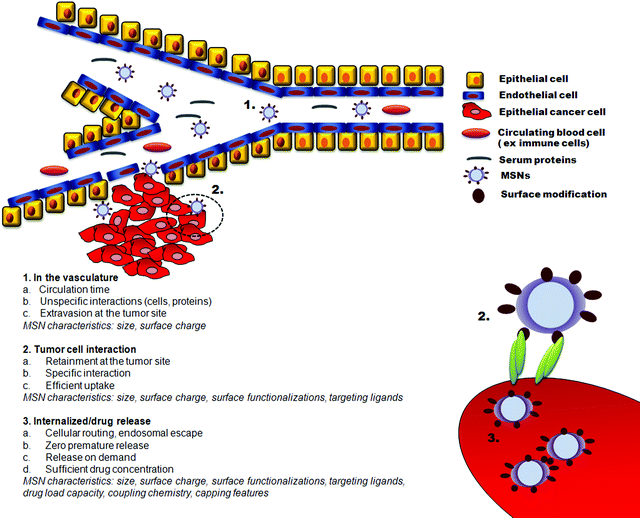 | ||
| Fig. 5 Critical parameters to be accounted for in the design of MSNs for targeted cancer therapy. | ||
Due to the leaky vasculature and poorly operational lymph system of tumors, nanoparticles can exit the blood vessels and accumulate at the tumor site by passive targeting via the enhanced permeability and retention (EPR) effect (Fig. 5). The diffusion rate in the extracellular spaces of the tumor is determined by the size and the surface charge of the particles. Small particles easily diffuse out and in of the tumor vasculature and within the tumor institium and the active concentration of the drug carrier at the tumor site might be low. Free movement is also affected by surface charge and negatively charged or weakly positively charged particles in the 50–150 nm range easily pass through tumor tissue.88
Although different surface modifications, the most common being PEGylation, can reduce unspecific interactions with proteins and thus have a positive influence on circulation times, active cellular uptake at the tumor site and enhanced tumor retention times can be further facilitated by active targeting. The attachment of targeting ligands to the particles provides specific particle–cell interactions and enhance cellular uptake. The ligand-mediated binding of the particles to the cell is regulated by the availability, affinity and the density of the targeting moiety (see Fig. 5). Surface functionalizations should be regulated for optimized ligand coupling and prevention of protein adsorption that might block active targeting and lead to rapid clearance of the particles from the body by the RES.
As compared to monoclonal antibodies – drug conjugates, the first “magic bullet” products for treating cancer that entered the market, particles are large enough to contain multiple targeting ligands and thus allow for more efficient binding to cell surface receptors (Fig. 5). This may broaden the repertoire of potential targeting ligands, as it allows for the use of low affinity ligands. Although the density-affinity relationship is of interest and the influence of these parameters on cellular uptake can easily be determined in vitro, the rate of cellular internalization, and hence the biological effect in vivo, will be related to the receptor surface density and availability on the targeted cell population. We are not aware of any studies on biodistribution of targeted vs. non-targeted MSNs in vivo and there are only a few studies showing cell specific targeting in vitro. MSNs have been conjugated with ligands such as sugar moieties,31,110,111 and folic acid (FA),31,59,112–116 as well as monoclonal antibody anti-HER2/neu66 and DNA aptamers123 for active targeting purposes. The efficiency of the active targeting is related to the ability of the binding of the particle-bound ligand with cell-surface receptors to trigger receptor-mediated endocytosis; the effectiveness of which thus can be quite easily pinpointed by competition experiments with free ligand.59,66,110–112,115,109 Given the complex influence of the physicochemical properties of particles on cellular uptake,14–19,73,88,89 uptake of ligand-linked particles should be compared to the uptake of particles linked to an binding-deficient mock or scrambled ligand. Competition experiments indicate that even in the presence of specific ligand–receptor interactions a certain degree of unspecific uptake also occurs,59,115 emphasizing that a more detailed understanding of the mechanisms of cellular internalization is needed. Moreover, the cellular system should mimic the complexity of the in vivo situation, and targeted delivery should preferably be verified in co-culture conditions,115 under 3-dimensional growth conditions and on several different cell types including primary cells. Critical evaluation of targetability of MSNs in intelligently designed biological settings in vitro will yield important information speeding up the transit from bench to bed side while keeping tedious in vivo testing at reasonable levels.
The primary role of targeting ligands in vivo is to enhance cellular uptake into cancer cells and to minimize accumulation in normal tissue. Specific particle–cell interactions will also serve to immobilize the particles within the tumor site. The colloidal properties of the particles will determine their biodistribution in vivo, an assumption supported by biodistribution studies on other carrier systems.88,117 A proper balance of particle functionalizations that influence circulation times, passive accumulation and retention at the site of action on one hand, and active cellular uptake on the other hand, will offer the optimal therapeutic outcome in vivo. Understanding how particle functionalizations and further ligand coupling influence biodistribution and tumor accumulation in vivo will be a highly interesting and important endeavor.
For the drug delivery process, the drug should not leak out (desorb) before cellular internalization, but once internalized, the drug should be able to reach the right intracellular target, depending on its mode of action. Thus, intracellular trafficking of both the cargo and the particles are of central interest (see Fig. 5). These can be preferentially studied by labeling the particles and the cargo with different dyes, or using specific dyes as cargo,116,122 as labeling of a cargo molecule will significantly alter its properties, especially for a small-molecule drug. Some drug molecules, such as camptothecin and doxorubicin, also exhibit fluorescent properties, and the uptake and routing can thus be studied by optical methods on time scales when the drug has not affected the cellular integrity. Due to the difficulties in quantification of intracellularly released amounts of drug, the intracellular drug release efficiency is often studied in terms of cell viability as a function of treatment with cytostatic drug-loaded MSNs; although targeted delivery has also been demonstrated with fluorescing dyes, whereas the intracellular amount delivered could be (semi)quantified by flow cytometry.116 Generally, the biocompatibility, as well as the effect of cytostatic drugs on MSNs is determined by cell viability assays such as the MTT assay. This technique has many advantages when compared to other toxicity assays because it requires minimal physical manipulation of the model cells and yields quick, reproducible results requiring simple optical density acquisition.128,129 However, a proper assessment of the influence of particles on both targeted and non-targeted cell populations requires more in-depth and long-term analyses of cellular function including effects on proliferation, apoptotic and necrotic cell death, synthesis of biomolecules, and cell metabolism.
The ultimate goal is, naturally, to obtain (targeted) cell death in cancerous cell lines whereas non-cancerous cell lines should be minimally affected – the core problem of current anticancer drugs used in chemotherapy. On this topic, Zink and co-workers reported cell growth inhibition by folate-functionalized MSNs containing the anticancer drug camptothecin adsorbed inside the pores. They observed enhanced cytotoxicity, about 60% cell death after 24 h, for PANC-1 cells, whereas the non-specific cytotoxicity of the HFF cell line used as the reference was in the range of 30% after 24 h.113 Similar numbers were reported by Zhu et al.123 who investigated the effect of doxorubicin-loaded MSN-PEM-aptamer conjugates on cell viability in (cancerous) HeLa and (healthy) QGY7703 cells and observed about 40% and 60% viability after 24 h, respectively. In a recent study, Rosenholm et al.59 conjugated the anticancer drug MTX (methotrexate), structurally similar to folate, mainly to the outside of MSNs. This way MTX was able to function dually as a targeting ligand and a cytostatic drug. The MTX-MSN-induced apoptotic cell death reached bout 33% after 72 h in the cancerous HeLa cell line as compared to non-cancerous HEK293 cells, where no apoptosis over the control was observed at concentrations where the biological effect of free MTX was equal on both cell lines. Moreover, the level of apoptosis induced by MTX-conjugated nanoparticles on the cancer cells was significantly higher than that of corresponding amounts of free MTX, while the opposite was, consequently, true for the non-cancerous cells. The above-mentioned results demonstrate that the drug delivery ability can be successfully combined with cell-specificity in a synergistic fashion over free drug therapy, thus advancing the applicability of MSNs in targeted cancer therapy.
For highly cytotoxic drugs, as naturally is the case for most cancer drugs, the most important feature is to prevent premature release and optimize the particles for efficient release after cellular uptake at the tumor site. Capping mechanisms and drug release kinetics must thus be carefully tested in physiological conditions mimicking in vivo situations (physiological buffer, serum proteins, pH, appropriate volume concentrations etc.). Whereas ibuprofen is the by far most used model drug in test tube release assays, (fluorescing) dyes are beneficially used as model drugs in combination with cellular studies as the intracellular release process can be followed microscopically (see Fig. 6) or quantified by flow cytometry. When cytotoxic drugs are used as the cargo the actual biological effect, as a function of time, induced by the drug is the most interesting parameter to pinpoint rather than drug concentration vs. time, i.e. experimental release curve. The release curves measured under test-tube conditions hardly correspond to intracellular conditions, though such experiments give important indications on the functionality of the gate-keeping mechanisms.
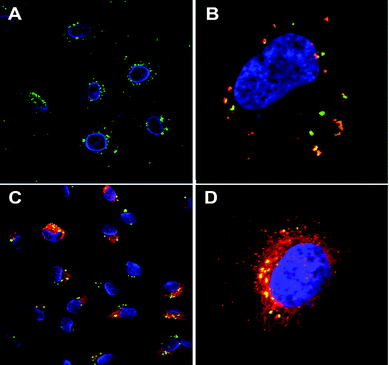 | ||
| Fig. 6 MSN-mediated delivery of hydrophobic dye DiI (red) into HeLa cells. FITC-labeled particles (green) without cargo, cell nucleus DAPI stained (blue) (A), DiI (red) loaded MSNs compartmentalized inside the cell, prior to payload release (B), DiI released into the cytoplasm while MSNs remain compartmentalized (C) and close-up of a cell (3D image) after 24 h incubation (D). Addition of free DiI to the cell medium at corresponding concentrations and conditions exhibits no detectable fluorescence. (For details, see ref. 116.) | ||
If carriers are optimally targeted to the tumor site, the release mechanism could include mechanisms that utilize the changes in redox states, and pH occurring as particles exit the extracellular space and enters the cell. Related to this, in a recent study by Lin and co-workers a cysteine residue was covalently attached to thiol-functionalized MSNs, which were effectively internalized by HeLa cells.60 Cysteine itself is toxic in high doses, membrane-impermeable and unstable in extracellular media. Covalent attachment of the cysteine by S–S-linkage to the silica nanoparticle prevented premature release in the extracellular media, but cysteine was released inside the cells due to reduction of the S–S bond. Due to high intracellular concentrations of gluthatione, the intracellular redox-potential is much higher compared to the outside medium, thereby providing an inbuilt differential release mechanism.
Intelligent systems may utilize changes in the functional states of the cells to provide a release-on-demand system, such as the self-regulated insulin double-drug delivery device reported by Lin et al. capable of releasing both G-Ins and cAMP for blood glucose regulation and insulin secretion as a response to saccharide triggers (i.e. glucose levels).130 Such systems naturally require detailed knowledge of the function of the targeted cell population, in combination with knowledge of the applied chemistry. An interesting angle would be to utilize some cancer-specific cellular feature or specific conditions in the tumor microenvironment to induce the release, some accordingly sheddable bond alternatives of which have been recently reviewed.131 In addition to cell intrinsic mechanisms external forces such as light, ultrasound, or magnetic field, can be applied to trigger the release of the drug.50 To our knowledge there are no demonstrations of controlled release of drugs at the tumor site in vivo. In vivo the situation is more complex and modifications for capping mechanisms/controlled release need to be considered in the background of the time spent in the extracellular environment i.e. circulation times, rate of unspecific uptake, type of drug, site of unspecific uptake and variable cellular uptake rates.
Conclusions
Recent developments within material science and strong collaborative efforts crossing disciplinary borders have highlighted the potential of MSNs for targeted drug delivery, the ultimate goal in cancer therapy. Relatively straightforward inside-out tuning of the vehicles, high flexibility, large drug load potential and potential for ‘smart’ release mechanisms make these nanostructures very interesting candidates for intelligent cancer medicine or other nanomedical applications. Despite recent successful demonstrations in cellular systems, there are critical issues that need to be addressed to facilitate the development of MSNs for biomedicine. The success rely on developments on both the material and the biological side in order to understand the cellular functions of the population to be targeted, the physiological restrictions that need to be overcome for successful delivery and the behavior of the drug carriers in an biological environment. Protocols for reproducible synthesis and functionalizations and adequate systems for in vitro testing of toxicity, targetability and drug delivery are critical components for this process. Detailed understanding on how “biobehaviour” is influenced by surface modifications, size, charge, roughness (porosity), protein adsorption and conjugated molecules will be a critical aim. Further, biocompatibility and biodistribution and biodegradability in vivo are burning topics. These issues have been discussed above and are summarized in the form of bullet points in Table 2. Future interdisciplinary research efforts addressing these topics will spur the quest to develop ‘smart’ nanomedicines.| MSNs in targeted drug delivery |
|---|
| Qualifications |
| • Highly flexible |
| • Tunable surface chemistry |
| • High drug load, potential for multidrug delivery |
| • Suitable for medical imaging |
| • Potential for intelligent drug release mechanisms |
| Requirements |
| • Reproducible synthesis and control over surface properties. |
| • Intelligent biological design for in vitro tests of toxicity, targetability and drug release |
| • Standardized protocols and quantification methods of uptake, cell function and drug release |
| • Animal models for in vivo on biodistribution, pharmacokinetics and biocompatibility. |
| Burning Questions |
| • Detailed understanding of the interactions at the nano-bio interface |
| • Uptake mechanisms and intracellular routing |
| • Biodistribution and biodegradation in vivo |
| • Biomarkers for efficient active targeting |
| • Discrete nanoparticle morphology retained throughout all processing steps |
| • Intactness of nanostructure matched with time needed to reach target |
Acknowledgements
Regarding Fig. 1b, Alain Duchanoy is acknowledged for the synthesis and Mikael Järn is thanked for recording the image. Boris Ufer and Alain Duchanoy are acknowledged for assembling Fig. 2 & 3. Emilia Peuhu is acknowledged for recording Fig. 6.References
- C. Barbé, J. Bartlett, L. Kong, K. Finnie, H. Q. Lin, M. Larkin, S. Calleja, A. Bush and G. Calleja, Adv. Mater., 2004, 16, 1959 CrossRef CAS.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013 RSC.
- W. Tan, K. Wang, X. He, X. J. Zhao, T. Drake, L. Wang and R. P. Bagwe, Med. Res. Rev., 2004, 24, 621 CrossRef.
- M. Vallet-Regi, A. Ramila, R. P. del Real and J. Perez-Pariente, Chem. Mater., 2001, 13, 308 CrossRef CAS.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C.-H. Shin, J.-G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688 CrossRef CAS.
- Y.-S. Lin, S.-H. Wu, Y. Hung, Y.-H. Chou, C. Chang, M.-L. Lin, C.-P. Tsai and C.-Y. Mou, Chem. Mater., 2006, 18, 5170 CrossRef CAS.
- H.-M. Liu, S.-H. Wu, C.-W. Lu, M. Yao, J.-K. Hsiao, Y. Hung, Y.-S. Lin, C.-Y. Mou, C.-S. Yang, D.-M. Huang and Y.-C. Chen, Small, 2008, 4, 619 CrossRef CAS.
- S.-H. Wu, Y.-S. Lin, Y. Hung, Y.-H. Chou, Y.-H. Hsu, C. Chang and C.-Y. Mou, Chem. Bio. Chem., 2008, 9, 53 CrossRef CAS.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS.
- Y.-S. Lin, Y. Hung, J.-K. Su, R. Lee, C. Chang, M.-L. Lin and C.-Y. Mou, J. Phys. Chem. B, 2004, 108, 15608 CrossRef CAS.
- K. M. Taylor, J. S. Kim, W. J. Reiter, H. An, W. Lin and W. Lin, J. Am. Chem. Soc., 2008, 130, 2154 CrossRef CAS.
- J. K. Hsiao, C. P. Tsai, T. H. Chung, Y. Hung, M. Yao, H. M. Liu, C. Y. Mou, C. S. Yang, Y. C. Chen and D. M. Huang, Small, 2008, 4, 1445 CrossRef CAS.
- F. Carniato, L. Tei, W. Dastrù, L. Marchese and M. Botta, Chem. Commun., 2009, 1246 RSC.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16 CrossRef CAS.
- L. Hu, Z. Mao and C. Gao, J. Mater. Chem., 2009, 19, 3108 RSC.
- H. Hess and Y. Tseng, ACS Nano, 2007, 1, 390 CrossRef CAS.
- M. De, P. S. Ghosh and V. M. Rotello, Adv. Mater., 2008, 20, 4225 CrossRef CAS.
- J. Gao and B. Xu, Nano Today, 2009, 4, 37 CrossRef.
- W. H. Suh, Y.-H. Suh and G. D. Stucky, Nano Today, 2009, 4, 27 CrossRef.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth and J. C. Vartuli, Nature, 1992, 359, 710 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2007, 129, 8845–8849 CrossRef CAS.
- A. Ahmed, R. Clowes, E. Willneff, H. Ritchie, P. Myers and H. Zhang, Ind. Eng. Chem. Res., 2010, 49, 602 CrossRef CAS.
- M. Mizutani, Y. Yamada and K. Yano, Chem. Commun., 2007, 1172 RSC.
- M. Mizutani, Y. Yamada, T. Nakamura and K. Yano, Chem. Mater., 2008, 20, 4777 CrossRef CAS.
- R. Schiller, C. K. Weiss, J. Geserick, N. Hüsing and K. Landfester, Chem. Mater., 2009, 21, 5088 CrossRef CAS.
- F. Gao, P. Botella, A. Corma, J. Blesa and L. Dong, J. Phys. Chem. B, 2009, 113, 1796 CrossRef CAS.
- C. E. Fowler, D. Khushalani, B. Lebeau and S. Mann, Adv. Mater., 2001, 13, 649 CrossRef CAS.
- S. Sadasivan, C. E. Fowler, D. Khushalani and S. Mann, Angew. Chem., Int. Ed., 2002, 41, 2151 CrossRef CAS.
- R. I. Nooney, D. Thirunavukkarasu, Y. Chen, R. Josephs and A. E. Ostafin, Chem. Mater., 2002, 14, 4721 CrossRef CAS.
- J. Gu, W. Fan, A. Shimojima and T. Okubo, Small, 2007, 3, 1740 CrossRef CAS.
- A. Berggren and A. E. C. Palmqvist, J. Phys. Chem. C, 2008, 112, 732 CrossRef CAS.
- F. Lu, S.-H. Wu, Y. Hung and C.-Y. Mou, Small, 2009, 5, 1408 CrossRef CAS.
- Q. He, X. Cui, F. Cui, L. Guo and J. Shi, Microporous Mesoporous Mater., 2009, 117, 609 CrossRef CAS.
- C. Urata, Y. Yamauchi, Y. Aoyama, J. Imasu, S. Todoroki, Y. Sakka, S. Inoue and K. Kuroda, J. Nanosci. Nanotechnol., 2008, 8, 3101 CrossRef CAS.
- C. Urata, Y. Aoyama, A. Tonegawa, Y. Yamauchia and K. Kuroda, Chem. Commun., 2009, 5094 RSC.
- V. S.-Y. Lin, C.-Y. Lai, J. Huang, S.-A. Song and S. Xu, J. Am. Chem. Soc., 2001, 123, 11510 CrossRef CAS.
- C.-Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and Victor S.-Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451 CrossRef CAS.
- M. P. Kapoor, W. Fujii, M. Yanagi, Y. Kasama, T. Kimura, H. Nanbu and L. R. Juneja, Microporous Mesoporous Mater., 2008, 116, 370 CrossRef CAS.
- K. Yano and Y. Fukushima, J. Mater. Chem., 2004, 14, 1579 RSC.
- M. J. K. Thomas, I. Slipper, A. Walunj, A. Jain, M. E. Favretto, P. Kallinteri and D. Douroumisa, Int. J. Pharm., 2010, 387, 272 CrossRef CAS.
- K. Suzuki, K. Ikari and H. Imai, J. Am. Chem. Soc., 2004, 126, 462 CrossRef CAS.
- H. Vallhov, S. Gabrielsson, M. Strømme, A. Scheynius and A. E. Garcia-Bennett, Nano Lett., 2007, 7, 3576 CrossRef CAS.
- K. Lund, Structural and morphological studies of mesoporous materials, doctoral dissertation in structural chemistry, Stockholm University, 2007 Search PubMed.
- K. Möller, J. Kobler and T. Bein, Adv. Funct. Mater., 2007, 17, 605 CrossRef.
- K. Möller, J. Kobler and T. Bein, J. Mater. Chem., 2007, 17, 624 RSC.
- K. Möller, J. Kobler and T. Bein, ACS Nano, 2008, 2, 791 CrossRef CAS.
- Q. He, X. Cui, F. Cui, L. Guo and J. Shi, Microporous Mesoporous Mater., 2009, 117, 609 CrossRef CAS.
- J. Kecht and T. Bein, Microporous Mesoporous Mater., 2008, 116, 123 CrossRef CAS.
- K. K. Cotí, M. E. Belowich, M. Liong, M. W. Ambrogio, Y. A. Lau, H. A. Khatib, J. I. Zink, N. M. Khashab and J. F. Stoddart, Nanoscale, 2009, 1, 16–39 RSC.
- J. Liu, A. Stace-Naughton, X. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2009, 131, 1354 CrossRef CAS.
- G. Nordlund, J. B. S. Ng, L. Bergström and Peter Brzezinski, ACS Nano, 2009, 3, 2639 CrossRef CAS.
- J. M. Rosenholm and M. Lindén, J. Controlled Release, 2008, 128, 157 CrossRef CAS.
- J. Rosenholm, Modular design of mesoporous silica materials: towards multifunctional drug delivery systems, doctoral dissertation in physical chemistry, Faculty of Technology, Åbo Akademi University, 2008 Search PubMed.
- J. Andersson, J. Rosenholm, S. Areva and M. Lindén, Chem. Mater., 2004, 16, 4160 CrossRef.
- A. Vinu, V. Murugesan, O. Tangermann and M. Haartman, Chem. Mater., 2004, 16, 3056 CrossRef CAS.
- J. Rosenholm, Synthesis and characterization of ordered micro- and mesoporous silica materials prepared by supramolecular templating and their application as carrier matrixes for controlled drug release, M. Sc. (Tech.) thesis in physical chemistry, Åbo Akademi University, 2002 Search PubMed.
- C. Tourné-Péteilh, D. Brunel, S. Bégu, B. Ciche, F. Fajula, D. A. Lerner and J.-M. Devoisselle, New J. Chem., 2003, 27, 1415 RSC.
- J. M. Rosenholm, E. Peuhu, L. T. Bate-Eya, J. E. Eriksson, C. Sahlgren and M. Lindén, Cancer-cell specific induction of apoptosis using mesoporous silica nanoparticles as drug delivery vectors, Small, 2010, 6, 1234 CAS.
- R. Mortera, J. Vivero-Escoto, I. I. Slowing, E. Garrone, B. Onida and V. S.-Y. Lin, Chem. Commun., 2009, 3219 RSC.
- J. M. Rosenholm, A. Penninkangas and M. Lindén, Chem. Commun., 2006, 3909 RSC.
- J. M. Rosenholm and M. Lindén, Chem. Mater., 2007, 19, 5023 CrossRef CAS.
- J. M. Rosenholm, A. Duchanoy and M. Lindén, Chem. Mater., 2008, 20, 1126 CrossRef CAS.
- L. Bergman, J. Rosenholm, A.-B. Öst, A. Duchanoy, P. Kankaanpää, J. Heino and M. Lindén, J. Nanomater., 2008, 2008, 1, DOI:10.1155/2008/712514.
- B. Ufer, J. M. Rosenholm, A. Duchanoy, L. Bergman and M. Lindén, Stud. Surf. Sci. Catal., 2008, 174, 353.
- C.-P. Tsai, C.-Y. Chen, Y. Hung, F.-H. Chang and C.-Y. Mou, J. Mater. Chem., 2009, 19, 5737 RSC.
- C. Fang, B. Shi, Y.-Y. Pei, M.-H. Hong, J. Wu and H.-Z. Chen, Eur. J. Pharm. Sci., 2006, 27, 27 CrossRef CAS.
- H. Tuysuz, C. W. Lehmann, H. Bongard, B. Tesche, R. Schmidt and F. Schüth, J. Am. Chem. Soc., 2008, 130, 11510 CrossRef CAS.
- Q. He, J. Zhang, J. Shi, Z. Zhu, L. Zhang, W. Bu, L. Guo and Y. Chen, Biomaterials, 2010, 31, 1085 CrossRef CAS.
- M. Etienne and A. Walcarius, Talanta, 2003, 59, 1173 CrossRef CAS.
- R. Mortera, S. Fiorilli, E. Garrone, E. Verné and B. Onida, Chem. Eng. J., 2010, 156, 184 CrossRef CAS.
- Q. He, J. Shi and F. Chen, Microporous Mesoporous Mater., 2010, 131, 314 CrossRef CAS.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS.
- F. Lu, S.-H. Wu, Y. Hung and C.-Y. Mou, Small, 2009, 5, 1408 CrossRef CAS.
- T.-H. Chung, S.-H. Wu, M. Yao, C.-W. Lu, Y.-S. Lin, Y. Hung, C.-Y. Mou, Y.-C. Chen and D.-M. Huang, Biomaterials, 2007, 28, 2959 CrossRef CAS.
- M. Fisichella, H. Dabboue, S. Bhattacharyya, M.-L. Saboungi, J.-P. Salvetat, T. Hevor and M. Guerin, Toxicol. in Vitro, 2009, 23, 697 CrossRef CAS.
- B. G. Trewyn, I. I. Slowing, S. Giri, H.-T. Chen and V. S.-Y. Lin, Acc. Chem. Res., 2007, 40, 846 CrossRef CAS.
- A. J. Di Pasqua, K. K. Sharma, Y.-L. Shi, B. B. Toms, W. Ouellette, J. C. Dabrowiak and T. Asefa, J. Inorg. Biochem., 2008, 102, 1416–1423 CrossRef CAS.
- D. Napierska, L. C. J. Thomassen, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, J. A. Martens and P. H. Hoet, Small, 2009, 5, 846 CrossRef CAS.
- D. R. Radu, C.-Y. Lai, K. Jeftinija, E. W. Rowe, S. Jeftinija and V. S.-Y. Lin, J. Am. Chem. Soc., 2004, 126, 13216 CrossRef CAS.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133 Search PubMed.
- Z. Tao, B. B. Toms, J. Goodisman and T. Asefa, Chem. Res. Toxicol., 2009, 22, 1869 CrossRef CAS.
- A. J. Di Pasqua, K. K. Sharma, Y.-L. Shi, B. B. Toms, W. Ouellette, J. C. Dabrowiak and T. Asefa, J. Inorg. Biochem., 2008, 102, 1416 CrossRef CAS.
- C.-H. Lee, S.-H. Cheng, Y.-J. Wang, Y.-C. Chen, N.-T. Chen, J. Souris, C.-T. Chen, C.-Y. Mou, C.-S. Yang and L.-W. Lo, Adv. Funct. Mater., 2009, 19, 215 CrossRef CAS.
- M. Cho, W. S. Cho, M. Choi, S. J. Kim, B. S. Han, S. H. Kim, H. O. Kim, Y. Y. Sheen and J. Jeong, Toxicol. Lett., 2009, 189, 177 CrossRef CAS.
- J.-H. Park, L. Gu, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331 CrossRef CAS.
- X. He, H. Nie, K. Wang, W. Tan, X. Wu and P. Zhang, Anal. Chem., 2008, 80, 9597 CrossRef CAS.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12 CrossRef CAS.
- Y.-S. Lin and C. L. Haynes, Chem. Mater., 2009, 21, 3979 CrossRef CAS.
- I. I. Slowing, C.-W. Wu, J. L. Vivero-Escoto and V. S.-Y. Lin, Small, 2009, 5, 57 CrossRef CAS.
- M. S. Lord, B. G. Cousins, P. J. Doherty, J. M. Whitelock, A. Simmons, R. L. Williams and B. K. Milthorpe, Biomaterials, 2006, 27, 4856 CrossRef CAS.
- D. Dutta, S. K. Sundaram, J. G. Teeguarden, B. J. Riley, L. S. Fifield, J. M. Jacobs, S. R. Addleman, G. A. Kaysen, B. M. Moudgil and T. J. Weber, Toxicol. Sci., 2007, 100, 303 CrossRef CAS.
- M. S. Ehrenberg, A. E. Friedman, J. N. Finkelstein, G. Oberdörster and J. L. McGrath, Biomaterials, 2009, 30, 603 CrossRef CAS.
- E. Witasp, N. Kupferschmidt, L. Bengtsson, K. Hultenby, C. Smedman, S. Paulie, A. E. Garcia-Bennett and B. Fadeel, Toxicol. Appl. Pharmacol., 2009, 239, 306 CrossRef CAS.
- S. P. Hudson, R. F. Padera, R. Langer and D. S. Kohane, Biomaterials, 2008, 29, 4045 CrossRef CAS.
- M. Hartmann, Chem. Mater., 2005, 17, 4577 CrossRef CAS.
- M. Vallet-Regı', F. Balas and D. Arcos, Angew. Chem., Int. Ed., 2007, 46, 7548 CrossRef CAS.
- D. Avnir, T. Coradin, O. Lev and J. Livage, J. Mater. Chem., 2006, 16, 1013 RSC.
- B. G. Trewyn, S. Giri, I. I. Slowing and V. S.-Y. Lin, Chem. Commun., 2007, 3236 RSC.
- S. Giri, B. G. Trewyn and V. S.-Y. Lin, Nanomedicine, 2007, 2, 99 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn, S. Giri and V. S.-Y. Lin, Adv. Funct. Mater., 2007, 17, 1225 CrossRef CAS.
- B. G. Trewyn, I. I. Slowing, S. Giri, H.-T. Chen and V. S.-Y. Lin, Acc. Chem. Res., 2007, 40, 846 CrossRef CAS.
- I. I. Slowing, L. Vivero-Escoto, C.-W. Wu and V. S.-Y. Lin, Adv. Drug Delivery Rev., 2008, 60, 1278 CrossRef CAS.
- S. Angelos, M. Liong, E. Choi and J. I. Zink, Chem. Eng. J., 2008, 137, 4 CrossRef CAS.
- M. Liong, S. Angelos, E. Choi, K. Patel, J. F. Stoddart and J. I. Zink, J. Mater. Chem., 2009, 19, 6251 RSC.
- L. Pasqua, S. Cundari, C. Ceresa and G. Cavaletti, Curr. Med. Chem., 2009, 16, 3054 CrossRef CAS.
- S. Wang, Microporous Mesoporous Mater., 2009, 117, 1 CrossRef CAS.
- J. Rosenholm, C. Sahlgren and M. Lindén, J. Mater. Chem., 2010, 20, 2707 RSC.
- I. Y. Park, I. Y. Kim, M. K. Yoo, Y. J. Choi, M.-H. Cho and C. S. Cho, Int. J. Pharm., 2008, 359, 280 CrossRef CAS.
- D. Brevet, M. Gary-Bobo, L. Raehm, S. Richeter, O. Hocine, K. Amro, B. Loock, P. Couleaud, C. Frochot, A. Morere, P. Maillard, M. Garcia and J. O. Durand, Chem. Commun., 2009, 1475 RSC.
- I. Slowing, B. G. Trewyn and V. S.-Y. Lin, J. Am. Chem. Soc., 2006, 128, 14792 CrossRef CAS.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889 CrossRef CAS.
- V. Lebret, L. Raehm, J. O. Durand, M. Smaihi, M. H. V. Werts, M. Blanchard-Desce, D. Methy-Gonnod and C. Dubernet, J. Sol-Gel Sci. Technol., 2008, 48, 32 CrossRef CAS.
- J. M. Rosenholm, A. Meinander, E. Peuhu, R. Niemi, J. E. Eriksson, C. Sahlgren and M. Lindén, ACS Nano, 2009, 3, 197 CrossRef CAS.
- J. M. Rosenholm, E. Peuhu, J. E. Eriksson, C. Sahlgren and M. Lindén, Nano Lett., 2009, 9, 3308 CrossRef CAS.
- K. F. Pirollo and E. H. Chang, Trends Biotechnol., 2008, 26, 552 CrossRef CAS.
- J. Lu, M. Liong, J. I. Zink and F. Tamanoi, Small, 2007, 3, 89 CAS.
- J. Lu, M. Liong, S. Sherman, T. Xia, M. Kovochich, A. E. Nel, J. I. Zink and F. Tamanoi, NanoBiotechnology, 2007, 3, 89 CrossRef CAS.
- T. Xia, M. Kovochich, M. Liong, H. Meng, S. Kabehie, S. George, J. I. Zink and A. E. Nel, ACS Nano, 2009, 3, 3273 CrossRef CAS.
- J. L. Vivero-Escoto, I. I. Slowing, C.-W. Wu and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 3462 CrossRef CAS.
- A. M. Chen, M. Zhang, D. Wei, D. Stueber, O. Taratula, T. Minko and H. He, Small, 2009, 5, 2673 CrossRef CAS.
- C.-L. Zhu, X.-Y. Song, W.-H. Zhou, H.-H. Yang, Y.-H. Wen and X.-R. Wang, J. Mater. Chem., 2009, 19, 7765 RSC.
- J. Lu, E. Choi, F. Tamanoi and J. I. Zink, Small, 2008, 4, 421 CrossRef CAS.
- S. C. De Smedt, J. Demeester and W. E. Hennink, Pharm. Res., 2000, 17, 113 CrossRef.
- F. Torney, B. G. Trewyn, V. S.-Y. Lin and K. Wang, Nat. Nanotechnol., 2007, 2, 295 CrossRef CAS.
- C.-L. Zhu, X.-Y. Song, W.-H. Zhou, H.-H. Yang, Y.-H. Wen and X.-R. Wang, J. Mater. Chem., 2009, 19, 7765 RSC.
- N. J. Marshall, C. J. Goodwin and S. J. Holt, Growth Regul., 1995, 5, 69 Search PubMed.
- B. J. Marquis, S. A. Love, K. L. Braun and C. L. Haynes, Analyst, 2009, 134, 425 RSC.
- Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S.-Y. Lin, J. Am. Chem. Soc., 2009, 131, 8398 CrossRef CAS.
- B. Romberg, W. E. Hennink and G. Storm, Pharm. Res., 2008, 25, 55 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |