Monodispersity control in the synthesis of monometallic and bimetallic quasi-spherical gold and silver nanoparticles
Qingbo
Zhang†
,
Jianping
Xie†
,
Yue
Yu
and
Jim Yang
Lee
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, 10 Kent Ridge Crescent, Singapore, 119260. E-mail: cheleejy@nus.edu.sg; Fax: +65 6779 1936; Tel: +65 6516 2899
First published on 11th August 2010
Abstract
In order for the nanoparticles of Au and Ag to be useful for a wide range of applications, the tailorability of the nanoparticle properties through controlled changes of composition, size, shape, and crystal structure is absolutely essential. Furthermore, these nanoparticle attributes must be delivered in high monodispersity to assure a consistent application performance. High monodispersity is also a requirement for the assembly of nanoparticles into extended nanostructures or mesoscale devices which may be needed for some applications. This article describes a variety of colloidal synthesis methods in use today for the preparation of monodisperse Au, Ag, and bimetallic Au–Ag nanoparticles. The emphasis is on the analysis of the efficacy of each method for tailoring the nanoparticle size and crystal structure. The scope of work surveyed is confined to quasi-spherical nanoparticles in the size range from sub-nanometre to several tens of nanometres.
 Qingbo Zhang | Dr Qingbo Zhang received his BS and MS from Tianjin University of China, and PhD from the National University of Singapore under the supervision of Prof. Jim Yang Lee. He is currently a postdoctoral research fellow in the Department of Chemical & Biomolecular Engineering of NUS. His research interests include precision synthesis and hierarchical assembly of monodisperse metal nanoparticles, exploration of their unique physiochemical properties and potential applications of individual nanoparticles and nanoparticle assemblies. |
 Jianping Xie | Dr Jianping Xie received his BS and MS from Tsinghua University of China. He graduated with a PhD from the Singapore–MIT Alliance (SMA) program under the co-supervision of Professors Jim Yang Lee (NUS) and Daniel I. C. Wang (MIT). He did postdoctoral work in the groups of Professors Jackie Y. Ying of the Institute of Bioengineering and Nanotechnology (IBN) in Singapore and Francesco Stellacci (MIT). He is currently an Assistant Professor in the Department of Chemical & Biomolecular Engineering of NUS. His current research interests include interfacing nanotechnology and biotechnology for biomedical applications and the green chemistry synthesis of metal nanomaterials. |
 Yue Yu | Ms Yue Yu received her BS in the School of Materials Science and Engineering from Nanyang Technological University of Singapore. She is currently a PhD candidate in the Department of Chemical & Biomolecular Engineering of NUS under the supervision of Professor Jim Yang Lee. Her research interests include shape-controlled synthesis and self-assembly of monometallic and bimetallic nanoparticles, and exploration of their optical and catalytic properties in practical applications. |
 Jim Yang Lee | Dr Jim Yang Lee received his BS from National University of Singapore and PhD from the University of Michigan. He is currently a professor of the Department of Chemical & Bimolecular Engineering at NUS. He also serves as a program co-chair of the Singapore–MIT Alliance and a senior fellow of the Energy Studies Institute. His main research interest is the synthesis of nanomaterials for energy related applications such as batteries, fuel cells and photoelectrochemical water splitting. |
1. Introduction
Nowadays it is common knowledge that materials at the nanoscale can display properties vastly different from the properties of the bulk and the properties of the constituent atoms or molecules.1 Among the different nanoparticles, those consisting of Au, Ag, and their composites have drawn the greatest interest because their properties can be useful for many applications.2,3 Indeed the use of Au nanoparticles as a pigment started as early as 2000 years ago. However, extensive investigations of Au/Ag nanoparticles targeting the tunability of their properties only occurred in the last three decades. Perhaps the most interesting feature of Au/Ag nanoparticles is their optical properties due to surface plasmon resonance (SPR). When the size of the particles is reduced to a value comparable to the electron mean free path (∼50 nm for Au and Ag), a nanoparticle in a beam of light can strongly scatter the incident light and the electromagnetic field around the particle can be enhanced.4Fig. 1 shows some of the colloidal Au/Ag nanoparticles synthesized in our laboratory. Their brilliant colors are the result of the tunable SPR band,5 which make the Au/Ag nanoparticles valuable as sensors, biolabels, and the building blocks of optical devices.6,7 When the size of the nanoparticles is further reduced to a value comparable to the Fermi wavelength of the conduction electrons (∼1 nm for Au and Ag), the strong quantum confinement of free electrons in the particle will result in discrete and size-tunable electronic transitions, thus leading to more interesting molecule-like properties such as fluorescence,8–11 magnetism,12–14 and quantized charging.15–20 The transition from quantized to quasi-continuous electronic energy is however not distinct, as quantized charging was also observed for nanoparticles of ∼2 nm in size.17,18 Another well-cited example of the properties of Au/Ag nanoparticles is their catalytic activity. It has been shown that they are good catalysts for quite a few chemical and electrochemical reactions.21 | ||
| Fig. 1 Colloidal solutions of Ag, Au and Ag–Au bimetallic nanoparticles. (Reproduced with permission from ref. 5. Copyright 2009 Springer Science + Business Media, LLC 2008.) | ||
The properties of Au/Ag nanoparticles can be varied over a wide range by changing the particle composition, size, crystal structure, shape, and surface chemistry.1,3 The particle properties can also be further modified by combining the two metals into a common entity as a bimetallic nanoparticle.22 In order to fully utilize these tunable properties in practical situations, a first and necessary step is the ability to synthesize nanoparticles with high monodispersity of the controlled particle attributes. Since particle properties are dependent on particle attributes, a high monodispersity of nanoparticles is required for the development of reliable property–attribute relationships and consistent application performance.23 In addition, high monodispersity is also a requirement for the self-assembly of the nanoparticles into superlattices, which are the framework for integrating nanoparticles into mesoscale or macroscale structures with new functionalities.24
There are several approaches developed to date to synthesize Au/Ag nanoparticles. Among them colloidal synthesis is one of the most commonly used methods because of some salient advantages:25 it offers a great variety of options for composition, size, crystal structure, shape, and surface chemistry control due to the flexibility in selecting the reaction system and synthesis conditions. In addition, this approach produces nanoparticles as a liquid dispersion, which is the desirable form for many applications. Furthermore, colloidal synthesis does not require expensive equipment and can be scaled up for quantity production relatively easily and inexpensively. In this article, we will review the most common colloidal synthesis methods for monodisperse Au, Ag, and bimetallic Au–Ag nanoparticles; in particular the efficacy of each method in tailoring the nanoparticle size and crystal structure. The size of the nanoparticles discussed in this article ranges from sub-nanometre to several tens of nanometres. The size of nanoparticles containing fewer than 150 atoms (∼1.7 nm of particle size) is usually described by the number of atoms per particle. In most papers, they are called nanoclusters (NCs). In comparison, the size of nanoparticles containing more than 150 atoms is usually described by their diameter. In this article, we use nanoclusters to refer to particles with fewer than 150 atoms and nanoparticles to refer to particles with more than 150 atoms, although such a distinction may not be used in some of the cited papers.
We will limit our discussion to the quasi-spherical nanoparticles. Readers who are interested in non-spherical nanoparticles may refer to several recent reviews on the shape controlled synthesis of metal nanoparticles.26–29 Actually the so-called spherical metal nanoparticles are not really spherical. It is known that Au and Ag, which adopt a face-centered cubic (fcc) lattice, possess different surface energies for different crystallographic planes. This anisotropy usually results in polyhedrons enclosed by low-index crystallographic planes being the most thermodynamically favorable morphology for the fcc metal nanoparticles.30 These polyhedral nanoparticles are frequently called spherical nanoparticles probably because they look like spheres in low resolution electron microscopic images. The real morphology of these quasi-spherical nanoparticles is determined by their crystallinity (or crystal structure).31 A fcc metal nanoparticle can be single-crystalline, single-twinned, multiple-twinned or polycrystalline. The crystal structure not only influences the morphology and morphology evolution of nanoparticles, but also the physical and chemical properties of the nanoparticles.32,33 The most common and technologically important examples are single-crystalline nanoparticles with a cuboctahedral or truncated octahedral morphology, decahedral multiple-twinned particles (MTPs) and icosahedral MTPs (Fig. 2).30 They are the focus of the review in this article.
 | ||
| Fig. 2 Schematic illustrations of the crystal structure of fcc metal nanoparticles. From left to right: single-crystalline nanoparticles, decahedral multiply twinned particles (MTPs), and icosahedral MTPs. | ||
Currently, “monodisperse nanoparticles” is an ambiguous term in the field of nanoparticle synthesis. A rigorous definition of monodisperse nanoparticles requires all the particles in an ensemble to be identical and indistinguishable, i. e. all the nanoparticles have the same size and crystal structure. However, this cannot be fulfilled in many of the current synthesis methods. Therefore, a relaxed definition of monodisperse nanoparticles is often adopted. For the nanoclusters, samples with a high yield of one type of “magic number” clusters are often referred to as “monodisperse”. For nanoparticles larger than 150 atoms, samples with a small standard deviation (σ) are often called “monodisperse nanoparticles”. 5% and 15% are two values that are typically used to define monodisperse nanoparticles although sometimes a standard deviation greater than 15% is also used.24,34 While size variability may be adequately quantified by statistics such as standard deviation, the quantification usually does not consider the uniformity of the nanoparticle crystal structure, or the lack of it. In this article, while we do not attempt to give a precise definition of “monodisperse nanoparticles”, we will take both size distribution and crystal structure uniformity into consideration when discussing the monodispersity of the nanoparticles.
Due to the large body of literature on this subject, this article is not intended to be comprehensive. We only include the most common methods of synthesis, with emphasis on those methods developed in the last decade. For each method, we only provide some exemplary papers to illustrate the method rather than to list all the references where the method was applied. Readers may like to read up on complementary review articles (and the review articles cited therein) to obtain a more comprehensive coverage in this field of research.2,3,35–38 Since the terminologies used to describe nanoclusters and nanoparticles, and their electronic properties and potential applications, are significantly different, we will discuss nanoclusters and nanoparticles in two separate sections. The article is organized as follows: we will begin the discussion with the synthesis of monometallic “magic number” Au/Ag nanoclusters. This is followed by the discussion of monometallic nanoparticles larger than 150 atoms. Since there are more studies on Au, Au will be used as an example to illustrate the synthesis of monometallic nanoclusters or nanoparticle for most methods. In the last two sections, we will discuss the synthesis of Au–Ag bimetallic nanoparticles, including homogeneous alloy nanoparticles and heterogeneous core/shell nanoparticles.
2. Stoichiometrically exact monometallic nanoclusters
The discovery of Au nanocluster compounds protected by phosphine molecules goes back several decades.36 Phosphine-protected Au NCs have limited stability and gradually decompose within a few days.36 The usability of these Au NCs is significantly affected by their poor stability and the difficulty of surface modification. More effective protecting ligands, such as thiols, have since substituted phosphines in the synthesis of Au NCs. The chemical synthesis of thiol-protected Au NCs was first reported by Brust et al.;39 recent advances in this field have greatly improved the technique, leading to extraordinarily stable Au NCs protected by thiols.3,40The thiol-protected Au NCs are often synthesized by reacting a thiolate–Au(I) polymer with a strong reducing agent (e.g., sodium borohydride (NaBH4)).3,39–41 The as-synthesized product is polydisperse in terms of the number of Au atoms per cluster. However, the product is abundant with clusters with discrete core masses of 5, 8, 14, 22, and 29 kDa,41–43 which can be isolated as stable compounds by various high-resolution separation techniques such as fractional precipitation, size-exclusion chromatography, and polyacrylamide gel electrophoresis.41–47 These ubiquitous size-selected clusters have a well-defined formula of [AuM(SR)N]Z, where M, N, and Z are the number of Au atoms in the core, the number of thiols on the surface, and the overall charge of the cluster, respectively.
The thiol-protected “magic number” Au NCs display extraordinary stability, which has been attributed to the closure of their electron shells.41–43,48,49 Au NCs can be treated as superatom complexes, and their stability and chemistry can be predicted by the “superatom electronic theory”.50,51 On the basis of the shell-closure of the superatom orbitals 1S, 1P, 1D…, the exceptional stability of the clusters is associated with a total electron count of n* = 2, 8, 18, 34, 58, 92… The protecting ligands, thiols, can electronically stabilize the Au NCs by withdrawing or delocalizing electrons from the Au core into their covalent bonds. Hence the requirement for an electronically closed shell superatom complex ([AuM(SR)N]Z) can be calculated by the equation: n* = M − N − Z, where the shell-closure electron count (n*) of the Au core has to satisfy one of the above-mentioned shell-closing numbers.51 The molecular formulas for the ubiquitous 5, 8, 14, 22, and 29 kDa clusters have recently been determined or suggested by various characterization techniques (e.g., mass spectroscopy and single-crystal X-ray diffraction) and/or theoretical calculations, as summarized in Table 1.
| Core mass/kDa | Formula | Shell-closure electron count (n*) | References |
|---|---|---|---|
| 5 | [Au25(SR)18]− | 8 | 52–55 |
| 8 | [Au38(SR)24]Z | 18 | 56–61 |
| 34 | [Au68(SR)34] | 34 | 62 |
| 58 | [Au102(SR)44] | 58 | 48, 49, 63 |
| 29 | [Au144(SR)59]Z | 92 | 64–66 |
While these NCs can be isolated by various separation techniques, it is still a great challenge to synthesize Au NCs with a specific size in high yield. In principle the synthesis of monodisperse thiol-protected Au NCs with a certain “magic number” can be achieved by manipulating the delicate balance between the thiol–Au interactions (thermodynamic factors) and the crystal growth process (kinetic factors). The following are highlights of some of the recent developments in this direction.
2.1 Controlling the reaction kinetics of thiolate–Au(I) polymers
As shown in Fig. 3, the first step of thiol-protected Au NC synthesis involves the conversion of Au(III) ions to polymeric thiolate–Au(I) intermediates. This is followed by the reductive decomposition of the thiolate–Au(I) polymers to Au NCs. A strong reducing agent (e.g., NaBH4) is generally used in the second step. The Au NCs formed from the fast reduction reaction are stabilized by kinetic factors to different degrees thereby leading to a broad size distribution. The monodispersity of the as-synthesized Au NCs can be improved through controlling the kinetics of the reactions. The reaction rate of the thiolate–Au(I) polymers is dependent on factors such as the nature of the thiol ligands, solvent conditions, ionic strength, reaction temperature, and reaction pH, and may be varied as such.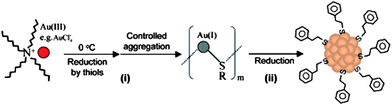 | ||
| Fig. 3 Schematic illustration of the synthesis of monodisperse Au25 clusters via thiolate–Au(I) intermediates. (Reproduced with permission from ref. 53. Copyright 2007 American Chemical Society.) | ||
Recently, Zhu et al.53 developed a two-phase method (Au ions in the organic phase and reducing agent (NaBH4) in the aqueous phase) to synthesize Au NCs with 25 atoms (Au25) at a very respectable yield (∼40% on the Au atom basis) by controlling the kinetics of thiolate–Au(I) polymer formation. Phenylethanethiol was selected as the protecting ligand because the bulky phenyl group could provide better steric protection for the clusters. The authors showed that a particular aggregation state of the thiolate–Au(I) polymers could be formed by carefully controlling the reaction temperature (0 °C) and the extent of stirring (slow). The well-controlled polymers finally led to the formation of Au25 clusters at a reasonably good yield. In comparison, thiolate–Au(I) polymers prepared at room temperature formed the Au25 clusters at a much lower yield (∼8%). The total structure of the as-synthesized phenylethanethiol-stabilized Au25 clusters was determined by single-crystal X-ray diffraction to be [Au25(SR)18]−,52 corresponding to a total electron count (n*) of 8. The Au NC has an icosahedral Au13 core which is completely shielded by six [–RS-Au-SR-Au-SR–] oligomers. The Au25 clusters exhibit interesting molecular-like properties, such as reversible magnetism13 and quantized charging.52–55
The two-phase method developed by Zhu et al. does not have general utility and cannot be easily extended to other thiols. Recently Wu et al.67–69 modified the kinetic-controlled approach to a one-phase system in tetrahydrofuran (THF). This one-phase method further improves the yield of the Au25 clusters and is applicable to other types of thiols (e.g., various hydrophobic and hydrophilic thiols with different functional groups). The authors considered the rate of formation of the thiolate–Au(I) polymers as the critical factor in the synthesis of monodisperse Au25 clusters. The choice of the solvent, THF, is another important consideration. THF can impact the size and the structure of the thiolate–Au(I) polymers, thereby affecting the subsequent growth of the Au25 clusters. However, the exact size and structure of the thiolate–Au(I) polymers are not clear at this time. The size-controlled synthesis of Au NCs with different “magic numbers” will be possible after the effects of various environmental factors such as the concentration ratio of thiols to Au, the reaction temperature, and the reaction pH, are completely understood through systematic experimentation.
2. 2 Thiol etching of polydisperse nanoclusters
The use of a strong reducing agent in the reductive decomposition of the thiolate–Au(I) polymers forms Au NCs which are kinetically stabilized. Some of these are metastable and can be digested by excess thiol at an elevated temperature (e.g., 55 and 80 °C), and stable Au NCs with certain “magic numbers” are formed finally. Au NCs with 25,42,70,71 38,59–61 and 14465 atoms have been prepared successfully by this method.Shichibu et al.70 first observed that glutathione-protected Au25 clusters in aqueous solution are resistant to thiol etching, while clusters of other sizes (e.g., Au10, Au15, Au18, Au22, Au29, and Au33) are vulnerable to the presence of excess thiol. AuM(SR)N clusters with M < 25 are completely oxidized to thiolate–Au(I) polymers while AuM(SR)N clusters with M > 25 are etched to Au25(SR)18 by the free glutathione molecules. A similar method was used by Toikkanen et al.61 recently for the synthesis of monodisperse hexanethiol-protected Au38 clusters in toluene. In the Toikkanen process, a polydisperse Au NC mixture was etched by excess hexanethiol. Only the Au38 clusters survived under these reaction conditions. The larger clusters were all etched by the excess thiol. Another variation was reported by Qian et al.65 in the synthesis of Au144 clusters. Instead of using hexanethiol, in which Au38 clusters are eventually formed, the authors used phenylethanethiol and the larger Au144 clusters were stabilized as the final product. It may be possible to synthesize monodisperse Au NCs with other “magic numbers” by using different thiols as the etching agents.
The thiol etching method can also be applied in a two-phase system, where initial products are synthesized in the aqueous phase, and the final products are stabilized in the organic phase, or vice versa. Qian et al.59,60 observed that monodisperse Au38 clusters could be formed by a two-phase ligand exchange process using 1-dodecanethiol to transfer the glutathione-protected polydisperse Au NCs from the aqueous phase to acetone. The phase transfer was carried out under an elevated temperature (80 °C). The exact formula of the as-prepared Au38 clusters was determined to be Au38(SR)24. In a subsequent work,71 polydisperse phenylethanethiol-protected Au NCs were first synthesized in toluene and then transferred to the aqueous phase by glutathione molecules. Monodisperse Au25 clusters were formed in this case. The two-phase method increases the possibility of obtaining monodisperse Au NCs with other “magic numbers” because of the greater latitude in the etching process afforded by the use of different solvents, different thiols or a combination of both.
3. Monometallic Au/Ag nanoparticles
3.1 One-step reduction of metal precursor
The simplest and most often used method to produce monometallic Au or Ag nanoparticles is to reduce the corresponding metal precursors in a solution in the presence of protecting agents or in a confined reaction environment. The crystal structure, size and size distribution can be varied by adjusting the kinetics of several competing processes. Generally, the size of the nanoparticles is determined by the number of atoms produced and the number of the nanoparticles formed, which are dependent on the kinetics of nucleation and growth of the nanoparticles.1 The crystal structure of the nanoparticles is determined by the competition between the kinetics of structural transformation of the nanoparticles and the kinetics of layer-by-layer growth.31,72,73 Improved size distribution of the nanoparticles can be delivered by the separation of nucleation and growth (e.g., a burst of nucleation) and diffusion-controlled growth.23,25 The kinetics of these competing processes can be altered by changing the nature and the concentration of the metal precursor, the reducing agent, and the capping agent, as well as environmental conditions such as temperature, pressure, and the solvent type, thus producing nanoparticles with different particle attributes. | ||
| Fig. 4 Gold nanoparticles formed under different conditions by the single-phase synthesis method where the gold precursor was reduced by amine–borane complexes in the presence of a thiol capping agent. (A) 2.1 ± 0.3 nm; (B) 3.5 ± 0.3 nm; (C) 5.3 ± 0.4 nm; (D) 6.2 ± 0.3 nm; (E) 7.1 ± 0.5 nm; (F) 8.3 ± 0.5 nm. All scale bars are 20 nm. (Reproduced with permission from ref. 82. Copyright 2007 American Chemical Society.) | ||
Other than using strong reducing agents based on boron, milder reducing agents could also be used to produce Au/Ag monometallic nanoparticles.32,93–107 The most common mild reducing agents are 1,2-hexadecanediol,96,97 oleylaminie,32,99–102,107 octadecylamine,93 and ethylene glycol.94,95 The reduction of Au/Ag ions with these mild reducing agents is usually carried out at an elevated temperature since their reducing power is temperature-dependent. Slower production of Au or Ag atoms and the use of elevated temperatures make it possible to control the kinetics of structural transformation and layer-by-layer growth, thus enabling the control of the crystal structure of the nanoparticles. Both single-crystalline nanoparticles and MTPs have been synthesized with these mild reducing agents by several groups. Particularly, we demonstrated that Au nanoparticles with different crystal structures can be formed by carefully controlling the conditions of the same synthesis protocol.96 Specifically the reduction of HAuCl4 was carried out in the solvent of 4-tert-butyl toluene, using 1,2-hexadecanediol as the reducing agent and oleylamine as the capping agent. The crystal structure of the Au nanoparticles could be varied by changing the HAuCl4 concentration while keeping other environmental factors constant. Exclusively single-crystalline nanoparticles, decahedral MTPs, and icosahedral MTPs were formed at low, intermediate, and high concentrations of the Au precursor, respectively (Fig. 5). The variation of the precursor concentration changes the deposition rate of the atoms onto the particle surface, thus altering the competition between layer-by-layer growth and structural transformation, and nanoparticles with different crystal structures are formed. The three types of nanoparticles synthesized in this work have a modest size distribution (10% < σ < 20%). However, nanoparticles with a uniform crystal structure could only be formed within a limited size range. The synthesis conditions need to be carefully controlled as undue variations can lead to the formation of a mixture of nanoparticles with different crystal structures.
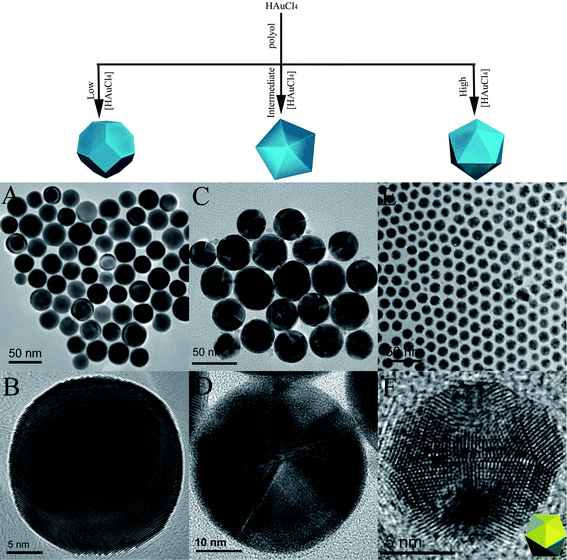 | ||
| Fig. 5 Tuning the crystal structures of Au nanoparticles in a modified polyol synthesis. A-B: TEM and HRTEM images of single-crystalline Au nanoparticles; C-D: TEM and HRTEM images of round decahedral Au MTPs; E: TEM image of icosahedral Au MTPs; F: HRTEM image of an icosahedral MTP along the two-fold symmetry axis; Inset in the bottom right corner is the schematic illustration of the icosahedron oriented as such. (Reproduced with permission from ref. 96. Copyright 2007 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
Selective etching was used by some groups to control the crystal structure of Ag nanoparticles. Xia and coworkers produced single-crystalline Ag nanoparticles by introducing chloride ions into the polyol reduction of AgNO3.108–111 Since the chemical reactivities of single crystal and twinned nuclei are different, the chloride ions can selectively remove multiple-twinned nuclei in an oxidative environment, thus leading to the formation of single-crystalline Ag nanoparticles in high yield. Tang et al.32 demonstrated the preparation of Ag nanoparticles with different structures by using different precursors in the oleylamine reduction of silver phosphine complexes. Single-crystalline nanoparticles and MTPs were obtained when (PPh3)3AgCl and (PPh3)3AgNO3 were used, respectively. However, the controlled formation of the MTPs in decahedral or icosahedral forms is not possible with this method.
In short, the size, crystal structure, and size distribution of Au/Ag monometallic nanoparticles can be varied by changing the synthesis conditions of a one-step reduction method. Even in successful examples, the one-step reduction controls mostly only one particular aspect of the particle attributes (e.g., size). This is understandable since the synthesis conditions for size, crystal structure, and size distribution control can be significantly different. This makes it difficult to produce nanoparticles with uniform crystal structure, narrow size distribution, and size and crystal structure control all in one step. More involved procedures have to be used to enable the independent control and customization of the nanoparticle attributes (e.g., size and crystal structure).
3.2 Seed-mediated growth method
Seed-mediated growth has been used by several groups to produce nanoparticles with tunable sizes.121–123 In this procedure, the reduction of the metal precursor and the formation of the final nanoparticles are fulfilled in two independent steps. In the first step, fine nanoparticles with a narrow size distribution are synthesized by one of the methods described in the previous section and these nanoparticles are used as seeds. In the second step, new atoms generated by the reduction of the metal precursor are deposited onto the seed nanoparticles to enlarge the particle size. The size of the final nanoparticles can be controlled by changing the ratio between the seed nanoparticles and the new atoms deposited. The environmental conditions and the reducing agents used in these two steps can be very different. Often, fast reduction of the metal precursor is used to create a burst of nucleation to form fine nanoparticles with a narrow size distribution. In the second step, a slow reduction of the metal ions is used to prevent the generation of new nuclei while increasing the size of the nanoparticles. By carrying out the reduction of the metal precursor in independent steps, the degree of freedom to vary the synthesis conditions can be vastly increased. Therefore, this method is very effective in producing nanoparticles with a narrower size distribution and customizable sizes (Fig. 6). | ||
| Fig. 6 Au nanoparticles with tunable sizes prepared from seed-mediated growth: (a) 5.5 ± 0.6, (b) 8.0 ± 0.8, (c) 17 ± 2.5, and (d) 37 ± 5 nm. (Reproduced with permission from ref. 121. Copyright 2007 American Chemical Society.) | ||
3.3 Post-treatment of synthesized nanoparticles
Treatment of the synthesized Au/Ag nanoparticles in selected chemical and/or thermal environments for a controlled period of time can also be used to adjust the size and size distribution of the nanoparticles. This provides another means to improve the size distribution and tune the size of the nanoparticles. Thus far, a number of post-treatment techniques have been reported. | ||
| Fig. 7 Narrowing the size distribution of Au nanoparticles by digestive ripening. (Modified with permission from ref. 134. Copyright 2007 American Chemical Society.) | ||
3.4 Step-wise synthesis
It is clear from the above discussion that an increase in the number of steps, i. e. using the seed-mediated growth and post-treatment of the synthesized nanoparticles can lead to an improved size distribution and more customizable sizes. If we further increase the number of steps prudently in the procedure, the degree of freedom can be further increased and this may lead to a better control of more nanoparticle attributes.135Fig. 8 illustrates a three-step procedure which we have developed to form icosahedral Ag MTPs. The synthesis procedure is designed in such a way that each step controls only one attribute of the nanoparticles. In the first step, icosahedral Ag nanoparticles with a modest size distribution are synthesized by a modified polyol synthesis. The synthesis conditions are optimized to form nanoparticles with only one crystal structure type. In the second step, the size distribution is narrowed by a digestive ripening procedure without changing the crystal structure of the nanoparticles. In the third and final step, the size of the nanoparticles is enlarged by seed-mediated growth. Since the prevailing thermodynamic and kinetic factors in each step are optimized to achieve the highest level of control for only one nanoparticle attribute, nanoparticles with uniform crystal structure, a narrow size distribution, and more tunable sizes can be realized with this strategy. Using this procedure, we have synthesized highly monodisperse icosahedral Ag MTPs from 9.6 to 20 nm.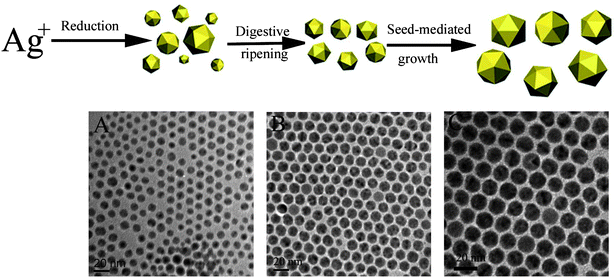 | ||
| Fig. 8 Step-wise procedure for the synthesis of monodisperse icosahedral Ag MTPs with tunable sizes showing the products in each step. A: TEM image of icosahedral Ag MTPs with a broad size distribution (9.1 ± 3.6 nm). B: TEM image of icosahedral Ag MTPs with a narrow size distribution (9.6 ± 0.5 nm). C: TEM image of monodisperse icosahedral Ag MTPs of 16.4 ± 0.5 nm. (Modified with permission from ref. 135. Copyright 2007 American Chemical Society.) | ||
4. Au–Ag alloy bimetallic nanoparticles
In addition to changing in the size and crystal structure of the monometallic nanoparticles, the combination of two metals into a bimetallic nanoparticle provides yet another means of property tuning since the properties of bimetallic nanoparticles may be varied through composition manipulation and the distribution of the constituent metals in the composite nanoparticles. The bimetallic nanoparticles can either be homogeneous alloy nanoparticles or heterogeneous core/shell nanoparticles. The different distributions of the two metals within a particle necessitate different methods of synthesis which are outlined below.4.1 Co-reduction
The co-reduction of Au and Ag precursors is the simplest method to prepare Au–Ag alloy nanoparticles.136–143 Since two metal precursors are involved in the reduction reactions, the influence of synthesis conditions on the rates of precursor reduction and the nucleation and growth of the alloy nanoparticles is more complex than in the case of monometallic nanoparticles. The size, size distribution, and crystal structure of the resulting alloy nanoparticles are difficult to control by this method. Consequently the size distribution of the Au–Ag alloy nanoparticles is rather broad (σ > 15%) and there is hardly any report on the control of the crystal structure of the alloy nanoparticles. Furthermore, changes in the nanoparticle composition are often coupled with changes in the particle size and size distribution. This makes it very difficult to map the properties of the alloy nanoparticles to their size and composition. Another issue with the co-reduction of Au and Ag precursors is the formation of silver halide precipitates since the majority of the Au precursors are halogen-containing. This not only introduces additional problems in composition control but also possible contamination of the alloy nanoparticle products. Consequently the preparation of Au–Ag alloy nanoparticles by the co-reduction method is carried out at very low concentrations of the precursors to avoid the precipitation of silver halide byproduct.1394.2 Annealing of Ag@Au core-shell nanoparticles
Homogeneous Au–Ag alloy nanoparticles have also been synthesized by annealing Ag@Au core–shell nanoparticles. Sun and coworkers synthesized homogeneous Au–Ag alloy nanoparticles by heating a dispersion of Ag@Au nanoparticles with a 13 nm core in oleylamine at 100 °C for an extended period of time under nitrogen atmosphere.144 This method takes advantages of the increased inter-diffusion rates of Au and Ag atoms in the core–shell nanoparticles at higher temperatures. The composition of the alloy nanoparticles can be varied by the thickness of the shell. Since this method does not form silver halide, it provides a better control of the nanoparticle composition because there is no potential silver loss.4.3 Digestive ripening
Digestive ripening is also suitable for the preparation of Au–Ag alloy nanoparticles.145 According to this method, Au–Ag alloy nanoparticles are synthesized by refluxing a mixture of polydisperse Au and Ag monometallic nanoparticles in 4-tert-butyl toluene for an extended period of time in the presence of a large excess of dodecanethiol and an argon blanket (Fig. 9). The size of the Au–Ag alloy nanoparticles synthesized by the Klabunde group was 5.6 ± 0.5 nm. The method is appealing because it can produce Au–Ag alloy nanoparticles with a very narrow size distribution from polydisperse Au and Ag monometallic nanoparticles. In addition, this method can generate Au–Ag alloy nanoparticles in high concentrations without the concern of AgCl precipitation. One limitation related to this method is that the size of the alloy nanoparticles cannot be tailored easily. Although the authors theorized that the size of the alloy nanoparticles may be varied by using other digestive agents, it is difficult to obtain alloy nanoparticles with custom sizes in practice because of the limited availability of digestive agents. | ||
| Fig. 9 Synthesis of monodisperse Ag–Au alloy nanoparticles by digestive ripening of a mixture of polydisperse Ag and Au nanoparticles. (Modified with permission from ref. 129, 131 and 145. Copyright 2007 American Chemical Society.) | ||
4.4 Replacement reaction
When Ag nanoparticles and Au precursors are mixed in a solution, a replacement reaction occurs between them because the standard electrode potential of Au(III)|Au is higher than that of Ag(I)|Ag. Hence the Ag nanoparticles are oxidized and the Au precursors are reduced to form homogeneous Au–Ag alloy nanoparticles under appropriate conditions.146 The formation of a homogeneous alloy nanoparticle relies on the rapid inter-diffusion of Au and Ag atoms which is facilitated by the reduced dimension of a nanoparticle, elevated temperatures, and the large number of vacancy defects generated by the replacement reaction. In this method, the size of the alloy nanoparticles can be controlled by the size of the starting Ag nanoparticles. The alloy composition can be varied by the ratio between Ag nanoparticles and the Au precursor. By decoupling size and composition control, this method makes it possible to independently change the size and the composition of the alloy nanoparticles. However, this method can only be used to produce Au–Ag alloy nanoparticles smaller than 10 nm. Larger starting Ag nanoparticles tend to form hollow or core/shell bimetallic nanoparticles instead.4.5 Step-wise method
We recently combined the effectiveness of the replacement reaction to form homogenous alloy nanoparticles with the step-wise method described in section 3.4 for a more versatile control of the composition, size, and crystal structure of the alloy nanoparticles.147 The procedure involves four discrete steps (Fig. 10). In the first step, Ag nanoparticles with a controlled crystal structure are synthesized from the chemical reduction of Ag ions. The crystal structure of the Ag nanoparticles can be controlled by the reduction chemistry and the synthesis conditions. The size distribution of the Ag nanoparticles is then narrowed by the digestive ripening method while keeping the crystal structure of the nanoparticles intact. Seed-mediated growth is applied next to vary the size of the monodisperse Ag nanoparticles to the desired range of values. In the last step, Au–Ag alloy nanoparticles are obtained by the replacement reaction between the monodisperse and size-adjusted Ag nanoparticles and a gold precursor. The size and crystal structure of the Au–Ag alloy nanoparticles are determined by the size and crystal structure of the starting Ag nanoparticles. The composition of the alloy nanoparticles, on the other hand, depends on the ratio of Ag nanoparticles to the Au precursor. Each step of the procedure is designed to control only one particle attribute. Therefore, the stepwise method not only enables the production of Au–Ag alloy nanoparticles with a uniform crystal structure and narrow size distribution, but also allows the size, crystal structure, and composition of the alloy nanoparticles to be independently tuned.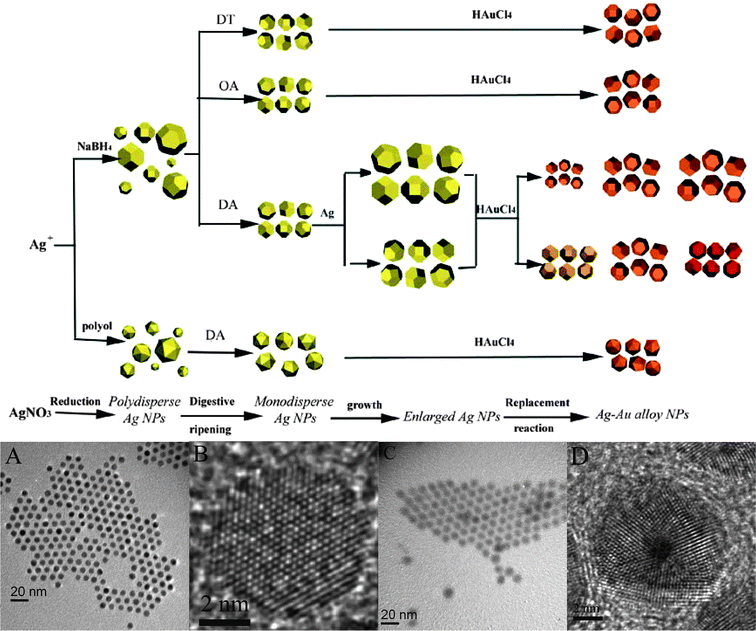 | ||
| Fig. 10 A step-wise procedure to synthesize Ag–Au alloy nanoparticles showing the products in different steps of the procedure. A–B: TEM and HRTEM images of single-crystalline Ag–Au alloy nanoparticles. C–D TEM and HRTEM images of icosahedral Ag–Au alloy multiply twinned nanoparticles. (Reproduced with permission from ref. 147. Copyright 2007 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
5 Au–Ag core/shell nanoparticles
5.1 Seed-mediated growth
Seed-mediated growth is the most commonly used method to prepare core/shell bimetallic nanoparticles.148–151 To do so nanoparticles of the first metal are synthesized using one of the methods in Section 3. Then the atoms of the second metal generated by precursor reduction are deposited onto the surface of the nanoparticles of the first metal to form core/shell nanoparticles. This procedure is similar to the seed-mediated growth method used to produce monometallic nanoparticles in Section 3.2. The only difference is that the depositing atoms in the second step are different from that of the seed. This method can be used to form either Ag@Au nanoparticles or Au@Ag nanoparticles. A mild reducing agent is also used to prevent the formation of monometallic nanoparticles of the shell metal as a separate phase. In the method, the size of the nanoparticle core is that of the monometallic seed nanoparticles. The thickness of the shell is dependent on the ratio between the seed nanoparticles and the depositing second metal atoms. The size distribution is usually determined by that of the core.5.2 Replacement reaction
The replacement reaction between Ag nanoparticles and a gold precursor can be used to produce core/shell nanoparticles with a Ag core (Fig. 11).152 The shell of the nanoparticles can either be all-Au or a Ag–Au alloy, depending on the ratio between Ag seeds to the Au precursor. There is no possibility of monometallic Au nanoparticle formation by this method because the Ag nanoparticles are the reducing agent and the Au atoms are deposited atop of the surface of the Ag nanoparticles. One point worthy of mention is that the replacement reaction between Ag nanoparticles and Au precursor could form Ag–Au alloy, core/shell, and hollow bimetallic nanoparticles. The structure of the final product depends strongly on the attributes of the starting Ag nanoparticles and the synthesis conditions. The multiplicity of the final products is caused by several concurrent events in the replacement reaction. They include the oxidative dissolution of the Ag nanoparticle template, the reduction of HAuCl4, the deposition of the reduced Au atoms, and the interdiffusion and rearrangement of atoms and vacancies. The rates of these processes on different sites of the particles may vary depending on the synthesis conditions and the structure of the starting Ag nanoparticle templates, thus leading to different end products. | ||
| Fig. 11 Synthesis of Ag@AgAu metal core/alloy shell nanoparticles by the replacement reaction between Ag nanoparticles and HAuCl4. (Reproduced with permission from ref. 152. Copyright 2007 Wiley-VCH Verlag GmbH & Co. KGaA.) | ||
One of the concerns in the synthesis of monodisperse nanoparticles with size and crystal structure control is the poor reproducibility of many methods of preparation. Synthesis of monodisperse nanoparticles is often referred to as an art rather than science. It is not unusual to find disparate experimental outcomes when the same synthesis protocol is used in different laboratories and even in the same laboratory sometimes. One of the main reasons for the lack of consistency and reproducibility is the poor control of transport processes in the synthesis. Many synthesis methods take advantage of burst nucleation to improve the uniformity of size and/or crystal structure. In these methods, a large number of atoms are produced instantaneously and the nucleation process occurs only within an extremely short period of time. In many synthesis procedures, burst nucleation is often achieved by injecting one reactant solution into the solution of another reactant. In some cases the solutions are also at different temperatures. The uniformity of temperature and reactant concentration is presumably overcome by the vigorous mixing of the two solutions. The reality can be significantly different. The coupling of transport processes to the kinetics of metal precursor reduction and to the rates of nucleation and growth of the nanoparticles is complex, especially when the transport processes are ill-characterized. For example, the rates of transport processes are influenced by factors such as the volume of the reactor, the concentration gradients in the reaction mixture, the agitation speed, or even the researchers' operating habits, etc. These factors are usually not sufficiently quantified in the reports, or have not been taken into consideration during the development of the synthesis protocol, thereby leading to variations in their extents in different situations. In addition, impurities in the reactants may also lead to the poor reproducibility in nanoparticle synthesis.108–111,153 It has been found that sometimes even a very low level of impurity can lead to significant variations in the nanoparticle product attributes. This could be the case when the impurities are catalytic, and are not a stoichiometric reactant in the nanoparticle synthesis.
6. Conclusions and outlook
After close to two decades of research efforts, a great many colloidal chemistry methods have been developed to synthesize Au/Ag monometallic nanoparticles. The simplest and the most common method to produce Au/Ag nanoparticles is the reduction of their precursors in the presence of capping agents or in a confined environment. The size, crystal structure, and size distribution of the nanoparticles can be varied by changing the synthesis conditions. The reduction of the precursor in two independent steps may lead to better size distribution and more customizable particle sizes. Post-treatment of the synthesized metal nanoparticles provides yet another means to improve the size distribution and to vary the particle size. A well-designed step-wise procedure can further enable the formation of nanoparticles with a uniform crystal structure, a narrow size distribution and tunable sizes over a wider range of values.The colloidal chemistry methods are also a rich resource for the preparation of Au–Ag homogeneous alloys and heterogeneous core/shell bimetallic nanoparticles. Homogeneous alloy nanoparticles are formed by the co-reduction of a mixture of the two metal precursors, annealing of core–shell nanoparticles, the digestive ripening of a mixture of the constituent monometallic nanoparticles, and the replacement reaction between Ag nanoparticles and a gold precursor. These methods are effective to control only one attribute of the alloy nanoparticles. A step-wise procedure involving a number of sequential steps is comparatively more versatile for the preparation of Au–Ag alloy nanoparticles with a uniform crystal structure, a narrow size distribution, and independently tunable crystal structure, size and composition. Bimetallic core/shell bimetallic nanoparticles, on the other hand, are best produced by seed-mediated growth or the replacement reaction between Ag nanoparticles and a gold precursor.
While much progress has been made in the synthesis of nanoparticles of Au and Ag, there are still substantial challenges. For the metal nanoclusters, only clusters with certain “magic numbers” have been produced with good yield. Nanoclusters with other “magic numbers” are either not obtainable or are difficult to produce at high yield. Although the size-controlled synthesis of nanoclusters with different “magic numbers” can in principle be possible by fine tuning the rate of formation and the size of the thiolate–Au(I) polymers; the objective is not realizable yet because of an insufficient understanding of the structure of the polymer intermediates and the effects of the synthesis conditions. For the nanoparticles, we still do not have the capability to produce nanoparticles with uniform crystal structure and narrow size distribution over a large size range and a wide variety of crystal structures, for instance the decahedral and icosahedral MTPs. Another challenge is to have controllable surface chemistry154–157 in addition to the crystal structure, size and composition control of the metal nanoparticles. Presently, a capping agent is used to control the size and the crystal structure of the nanoparticles but its persistence on the nanoparticle surface may compromise the intended application (e.g., catalysis). Hence we need to develop the ability to vary the crystal structure, size and size distribution with removable or modifiable capping agents. Another challenge is the scalability of the synthesis methods. The current synthesis methods usually produce the nanoparticles on a milligram scale, gram scale or even hundreds of grams scale production of nanoparticles is still difficult by these methods.
In order to achieve full control in the synthesis of monodisperse Au, Ag, and their bimetallic nanoparticles and the development of new methods of synthesis, a full understanding of all the events in the course of the reactions, such as the transport of heat and reactant species, the reduction of precursors, the diffusion of metal atoms and capping agents, the collision and nucleation of atoms, the interaction between the nuclei and the capping agents is essential. We need new high resolution characterization tools that will allow these events to be detected and followed in real time. Theoretical studies and computer simulations should be used to supplement the inadequacy of current experimental methods and to reduce the cost of experimentations. However, many simulation studies only examine certain phenomena and selected factors in a nanoparticle synthesis.72,73,158–160 Ideally simulations should consider the thermodynamics and kinetics of all possible events in the nanoparticle synthesis in order to establish a direct link between synthesis conditions and product attributes. In this regard first principle calculations may be the most satisfying for understanding nanoscale physics and chemistry from the atomic level up. They are, however, difficult to implement for large scale systems such as the hundreds and thousands of atoms in a nanoparticle. Hence some degree of simplification is needed to make the computation manageable by existing facilities, and some of these assumptions need experimental validation. At the end of the day the design of new synthesis protocols and the variation of synthesis conditions should be based on a deeper understanding of the underlying processes, rather than discoveries by empiricism and heuristics.
Acknowledgements
We acknowledge the financial support from Ministry of Education Academic Research Grant R279-000-260-112.References
- C. Burda, X. B. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- E. Hutter and J. H. Fendler, Adv. Mater., 2004, 16, 1685–1706 CrossRef CAS.
- Q. B. Zhang, Y. N. Tan, J. P. Xie and J. Y. Lee, Plasmonics, 2009, 4, 9–22 CrossRef CAS.
- X. H. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS.
- J. P. Xie, Q. B. Zhang, J. Y. Lee and D. I. C. Wang, ACS Nano, 2008, 2, 2473–2480 CrossRef CAS.
- J. P. Xie, Y. G. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS.
- S. Link, A. Beeby, S. FitzGerald, M. A. El-Sayed, T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2002, 106, 3410–3415 CrossRef CAS.
- H. X. Xu and K. S. Suslick, Adv. Mater., 2010, 22, 1078–1082 CrossRef CAS.
- J. P. Xie, Y. G. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- H. Hori, Y. Yamamoto, T. Iwamoto, T. Miura, T. Teranishi and M. Miyake, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 174411 CrossRef.
- M. Z. Zhu, C. M. Aikens, M. P. Hendrich, R. Gupta, H. F. Qian, G. C. Schatz and R. C. Jin, J. Am. Chem. Soc., 2009, 131, 2490–2492 CrossRef CAS.
- Y. Negishi, H. Tsunoyama, M. Suzuki, N. Kawamura, M. M. Matsushita, K. Maruyama, T. Sugawara, T. Yokoyama and T. Tsukuda, J. Am. Chem. Soc., 2006, 128, 12034–12035 CrossRef CAS.
- S. W. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS.
- J. F. Hicks, D. T. Miles and R. W. Murray, J. Am. Chem. Soc., 2002, 124, 13322–13328 CrossRef CAS.
- T. Laaksonen, V. Ruiz, P. Liljeroth and B. M. Quinn, Chem. Soc. Rev., 2008, 37, 1836–1846 RSC.
- R. W. Murray, Chem. Rev., 2008, 108, 2688–2720 CrossRef CAS.
- B. M. Quinn, P. Liljeroth, V. Ruiz, T. Laaksonen and K. Kontturi, J. Am. Chem. Soc., 2003, 125, 6644–6645 CrossRef CAS.
- O. M. Bakr, V. Amendola, C. M. Aikens, W. Wenseleers, R. Li, L. Dal Negro, G. C. Schatz and F. Stellacci, Angew. Chem., Int. Ed., 2009, 48, 5921–5926 CrossRef CAS.
- A. S. K. Hashmi and M. Rudolph, Chem. Soc. Rev., 2008, 37, 1766–1775 RSC.
- P. Mulvaney, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS.
- C. B. Murray, C. R. Kagan and M. G. Bawendi, Annu. Rev. Mater. Sci., 2000, 30, 545–610 CrossRef CAS.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS.
- Y. N. Xia, Y. J. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS.
- J. E. Millstone, S. J. Hurst, G. S. Metraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS.
- A. R. Tao, S. Habas and P. D. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- M. Grzelczak, J. Perez-Juste, P. Mulvaney and L. M. Liz-Marzan, Chem. Soc. Rev., 2008, 37, 1783–1791 RSC.
- Z. L. Wang, J. Phys. Chem. B, 2000, 104, 1153–1175 CrossRef CAS.
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 77, 371–423 CrossRef CAS.
- Y. Tang and M. Ouyang, Nat. Mater., 2007, 6, 754–759 CrossRef CAS.
- G. V. Hartland, Nat. Mater., 2007, 6, 716–718 CrossRef CAS.
- H. Bonnemann and R. M. Richards, Eur. J. Inorg. Chem., 2001, 2455–2480 CrossRef CAS.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS.
- G. Schmid, Chem. Soc. Rev., 2008, 37, 1909–1930 RSC.
- J. P. Wilcoxon and B. L. Abrams, Chem. Soc. Rev., 2006, 35, 1162–1194 RSC.
- A. C. Templeton, M. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- E. Boisselier and D. Astruc, Chem. Soc. Rev., 2009, 38, 1759–1782 RSC.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS.
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS.
- E. G. Mednikov and L. E. Dahl, Small, 2008, 4, 534–537 CrossRef CAS.
- V. L. Jimenez, M. C. Leopold, C. Mazzitelli, J. W. Jorgenson and R. W. Murray, Anal. Chem., 2003, 75, 199–206 CrossRef CAS.
- K. Kimura, N. Sugimoto, S. Sato, H. Yao, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2009, 113, 14076–14082 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- H. Tsunoyama, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2006, 128, 6036–6037 CrossRef CAS.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- R. L. Whetten and R. C. Price, Science, 2007, 318, 407–408 CrossRef CAS.
- D. E. Jiang, R. L. Whetten, W. D. Luo and S. Dai, J. Phys. Chem. C, 2009, 113, 17291–17295 CrossRef CAS.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Gronbeck and H. Hakkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- M. Zhu, E. Lanni, N. Garg, M. E. Bier and R. Jin, J. Am. Chem. Soc., 2008, 130, 1138–1139 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS.
- J. Akola, M. Walter, R. L. Whetten, H. Hakkinen and H. Gronbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS.
- D. E. Jiang, W. Luo, M. L. Tiago and S. Dai, J. Phys. Chem. C, 2008, 112, 13905–13910 CrossRef CAS.
- D. E. Jiang, M. L. Tiago, W. D. Luo and S. Dai, J. Am. Chem. Soc., 2008, 130, 2777–2779 CrossRef CAS.
- Y. Pei, Y. Gao and X. C. Zeng, J. Am. Chem. Soc., 2008, 130, 7830–7832 CrossRef CAS.
- H. F. Qian, M. Z. Zhu, U. N. Andersen and R. C. Jin, J. Phys. Chem. A, 2009, 113, 4281–4284 CrossRef CAS.
- H. F. Qian, Y. Zhu and R. C. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS.
- O. Toikkanen, V. Ruiz, G. Ronholm, N. Kalkkinen, P. Liljeroth and B. M. Quinn, J. Am. Chem. Soc., 2008, 130, 11049–11055 CrossRef CAS.
- A. Dass, J. Am. Chem. Soc., 2009, 131, 11666–11667 CrossRef CAS.
- Y. Gao, N. Shao and X. C. Zeng, ACS Nano, 2008, 2, 1497–1503 CrossRef CAS.
- O. Lopez-Acevedo, J. Akola, R. L. Whetten, H. Gronbeck and H. Hakkinen, J. Phys. Chem. C, 2009, 113, 5035–5038 CrossRef CAS.
- H. F. Qian and R. C. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS.
- C. A. Fields-Zinna, R. Sardar, C. A. Beasley and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 16266–16271 CrossRef CAS.
- Z. Wu, J. Suhan and R. C. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC.
- Z. W. Wu, C. Gayathri, R. R. Gil and R. C. Jin, J. Am. Chem. Soc., 2009, 131, 6535–6542 CrossRef CAS.
- Z. K. Wu and R. C. Jin, ACS Nano, 2009, 3, 2036–2042 CrossRef CAS.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS.
- H. F. Qian, M. Z. Zhu, E. Lanni, Y. Zhu, M. E. Bier and R. C. Jin, J. Phys. Chem. C, 2009, 113, 17599–17603 CrossRef CAS.
- F. Baletto, C. Mottet and R. Ferrando, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 6315.
- F. Baletto, C. Mottet and R. Ferrando, Phys. Rev. Lett., 2000, 84, 5544–5547 CrossRef CAS.
- J. Yang, J. Y. Lee and H. P. Too, Anal. Chim. Acta, 2007, 588, 34–41 CrossRef CAS.
- M. Brust and C. J. Kiely, Colloids Surf., A, 2002, 202, 175–186 CrossRef CAS.
- B. A. Korgel, S. Fullam, S. Connolly and D. Fitzmaurice, J. Phys. Chem. B, 1998, 102, 8379–8388 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428 CAS.
- A. Badia, S. Singh, L. Demers, L. Cuccia, G. R. Brown and R. B. Lennox, Chem.–Eur. J., 1996, 2, 359–363 CrossRef CAS.
- R. H. Terrill, T. A. Postlethwaite, C. H. Chen, C. D. Poon, A. Terzis, A. D. Chen, J. E. Hutchison, M. R. Clark, G. Wignall, J. D. Londono, R. Superfine, M. Falvo, C. S. Johnson, E. T. Samulski and R. W. Murray, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- D. Zanchet, B. D. Hall and D. Ugarte, J. Phys. Chem. B, 2000, 104, 11013–11018 CrossRef CAS.
- N. Zheng, J. Fan and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 6550–6551 CrossRef CAS.
- M. P. Rowe, K. E. Plass, K. Kim, C. Kurdak, E. T. Zellers and A. J. Matzger, Chem. Mater., 2004, 16, 3513–3517 CrossRef CAS.
- N. R. Jana and X. G. Peng, J. Am. Chem. Soc., 2003, 125, 14280–14281 CrossRef.
- C. K. Yee, R. Jordan, A. Ulman, H. White, A. King, M. Rafailovich and J. Sokolov, Langmuir, 1999, 15, 3486–3491 CrossRef CAS.
- A. C. Templeton, S. W. Chen, S. M. Gross and R. W. Murray, Langmuir, 1999, 15, 66–76 CrossRef CAS.
- T. Teranishi, I. Kiyokawa and M. Miyake, Adv. Mater., 1998, 10, 596–599 CrossRef CAS.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- B. H. Sohn, J. M. Choi, S. I. Yoo, S. H. Yun, W. C. Zin, J. C. Jung, M. Kanehara, T. Hirata and T. Teranishi, J. Am. Chem. Soc., 2003, 125, 6368–6369 CrossRef CAS.
- I. Hussain, S. Graham, Z. X. Wang, B. Tan, D. C. Sherrington, S. P. Rannard, A. I. Cooper and M. Brust, J. Am. Chem. Soc., 2005, 127, 16398–16399 CrossRef CAS.
- R. C. Doty, T. R. Tshikhudo, M. Brust and D. G. Fernig, Chem. Mater., 2005, 17, 4630–4635 CrossRef CAS.
- S. H. Chen and K. Kimura, Langmuir, 1999, 15, 1075–1082 CrossRef CAS.
- D. S. Wang, T. Xie, Q. Peng and Y. D. Li, J. Am. Chem. Soc., 2008, 130, 4016–4022 CrossRef CAS.
- Y. G. Sun, B. T. Mayers and Y. N. Xia, Nano Lett., 2002, 2, 481–485 CrossRef CAS.
- P. Y. Silvert, R. Herrera-Urbina and K. Tekaia-Elhsissen, J. Mater. Chem., 1997, 7, 293–299 RSC.
- Q. B. Zhang, J. P. Xie, Y. Yu, J. H. Yang and J. Y. Lee, Small, 2010, 6, 523–527 CrossRef CAS.
- Y. D. Yin, C. Erdonmez, S. Aloni and A. P. Alivisatos, J. Am. Chem. Soc., 2006, 128, 12671–12673 CrossRef CAS.
- R. Sardar and J. S. Shumaker-Parry, Chem. Mater., 2009, 21, 1167–1169 CrossRef CAS.
- H. Hiramatsu and F. E. Osterloh, Chem. Mater., 2004, 16, 2509–2511 CrossRef CAS.
- M. Aslam, L. Fu, M. Su, K. Vijayamohanan and V. P. Dravid, J. Mater. Chem., 2004, 14, 1795–1797 RSC.
- C. M. Shen, C. Hui, T. Z. Yang, C. W. Xiao, J. F. Tian, L. H. Bao, S. T. Chen, H. Ding and H. J. Gao, Chem. Mater., 2008, 20, 6939–6944 CrossRef CAS.
- M. Chen, Y. G. Feng, X. Wang, T. C. Li, J. Y. Zhang and D. J. Qian, Langmuir, 2007, 23, 5296–5304 CrossRef CAS.
- X. Z. Lin, X. W. Teng and H. Yang, Langmuir, 2003, 19, 10081–10085 CrossRef CAS.
- Y. Wang and H. Yang, Chem. Commun., 2006, 2545–2547 RSC.
- A. Sanchez-Iglesias, I. Pastoriza-Santos, J. Perez-Juste, B. Rodriguez-Gonzalez, F. J. G. de Abajo and L. M. Liz-Marzan, Adv. Mater., 2006, 18, 2529–2534 CrossRef CAS.
- X. M. Lu, H. Y. Tuan, J. Y. Chen, Z. Y. Li, B. A. Korgel and Y. N. Xia, J. Am. Chem. Soc., 2007, 129, 1733–1742 CrossRef CAS.
- X. M. Lu, H. Y. Tnan, B. A. Korgel and Y. N. Xia, Chem.–Eur. J., 2008, 14, 1584–1591 CrossRef CAS.
- B. Wiley, T. Herricks, Y. G. Sun and Y. N. Xia, Nano Lett., 2004, 4, 2057–2057 CrossRef CAS.
- B. Wiley, Y. G. Sun and Y. N. Xia, Langmuir, 2005, 21, 8077–8080 CrossRef CAS.
- S. H. Im, Y. T. Lee, B. Wiley and Y. N. Xia, Angew. Chem., Int. Ed., 2005, 44, 2154–2157 CrossRef.
- M. H. Kim, X. M. Lu, B. Wiley, E. P. Lee and Y. N. Xia, J. Phys. Chem. C, 2008, 112, 7872–7876 CrossRef CAS.
- C. Petit, P. Lixon and M. P. Pileni, J. Phys. Chem., 1993, 97, 12974–12983 CrossRef CAS.
- J. P. Wilcoxon, J. E. Martin and P. Provencio, Langmuir, 2000, 16, 9912–9920 CrossRef CAS.
- A. Taleb, C. Petit and M. P. Pileni, Chem. Mater., 1997, 9, 950–959 CrossRef CAS.
- J. P. Wilcoxon, J. E. Martin, F. Parsapour, B. Wiedenman and D. F. Kelley, J. Chem. Phys., 1998, 108, 9137–9143 CrossRef CAS.
- Y. G. Kim, S. K. Oh and R. M. Crooks, Chem. Mater., 2004, 16, 167–172 CrossRef CAS.
- J. Zheng, M. S. Stevenson, R. S. Hikida and P. G. Van Patten, J. Phys. Chem. B, 2002, 106, 1252–1255 CrossRef CAS.
- M. Q. Zhao and R. M. Crooks, Chem. Mater., 1999, 11, 3379–3385 CrossRef CAS.
- R. M. Crooks, M. Q. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS.
- F. Grohn, B. J. Bauer, Y. A. Akpalu, C. L. Jackson and E. J. Amis, Macromolecules, 2000, 33, 6042–6050 CrossRef.
- N. R. Jana, L. Gearheart and C. J. Murphy, Langmuir, 2001, 17, 6782–6786 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Chem. Mater., 2001, 13, 2313–2322 CrossRef CAS.
- J. P. Wilcoxon and P. P. Provencio, J. Am. Chem. Soc., 2004, 126, 6402–6408 CrossRef CAS.
- M. M. Maye and C. J. Zhong, J. Mater. Chem., 2000, 10, 1895–1901 RSC.
- M. M. Maye, W. X. Zheng, F. L. Leibowitz, N. K. Ly and C. J. Zhong, Langmuir, 2000, 16, 490–497 CrossRef CAS.
- T. Teranishi, S. Hasegawa, T. Shimizu and M. Miyake, Adv. Mater., 2001, 13, 1699–1701 CrossRef CAS.
- T. Shimizu, T. Teranishi, S. Hasegawa and M. Miyake, J. Phys. Chem. B, 2003, 107, 2719–2724 CrossRef CAS.
- M. Kanehara, J. Sakurai, H. Sugimura and T. Teranishi, J. Am. Chem. Soc., 2009, 131, 1630–1631 CrossRef CAS.
- S. Stoeva, K. J. Klabunde, C. M. Sorensen and I. Dragieva, J. Am. Chem. Soc., 2002, 124, 2305–2311 CrossRef CAS.
- S. I. Stoeva, B. L. V. Prasad, S. Uma, P. K. Stoimenov, V. Zaikovski, C. M. Sorensen and K. J. Klabunde, J. Phys. Chem. B, 2003, 107, 7441–7448 CrossRef CAS.
- A. B. Smetana, K. J. Klabunde and C. M. Sorensen, J. Colloid Interface Sci., 2005, 284, 521–526 CrossRef CAS.
- S. I. Stoeva, A. B. Smetana, C. M. Sorensen and K. J. Klabunde, J. Colloid Interface Sci., 2007, 309, 94–98 CrossRef CAS.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen and K. J. Klabunde, Chem. Mater., 2003, 15, 935–942 CrossRef CAS.
- B. L. V. Prasad, S. I. Stoeva, C. M. Sorensen and K. J. Klabunde, Langmuir, 2002, 18, 7515–7520 CrossRef CAS.
- Q. B. Zhang, J. P. Xie, J. H. Yang and J. Y. Lee, ACS Nano, 2009, 3, 139–148 CrossRef CAS.
- C. Wang, H. G. Yin, R. Chan, S. Peng, S. Dai and S. H. Sun, Chem. Mater., 2009, 21, 433–435 CrossRef CAS.
- O. M. Wilson, R. W. J. Scott, J. C. Garcia-Martinez and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 1015–1024 CrossRef CAS.
- M. J. Kim, H. J. Na, K. C. Lee, E. A. Yoo and M. Y. Lee, J. Mater. Chem., 2003, 13, 1789–1792 RSC.
- M. P. Mallin and C. J. Murphy, Nano Lett., 2002, 2, 1235–1237 CrossRef CAS.
- D. H. Chen and C. J. Chen, J. Mater. Chem., 2002, 12, 1557–1562 RSC.
- S. Link, Z. L. Wang and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 3529–3533 CrossRef CAS.
- M. J. Hostetler, C. J. Zhong, B. K. H. Yen, J. Anderegg, S. M. Gross, N. D. Evans, M. Porter and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 9396–9397 CrossRef CAS.
- S. W. Han, Y. Kim and K. Kim, J. Colloid Interface Sci., 1998, 208, 272–278 CrossRef CAS.
- C. Wang, S. Peng, R. Chan and S. H. Sun, Small, 2009, 5, 567–570 CrossRef CAS.
- A. B. Smetana, K. J. Klabunde, C. M. Sorensen, A. A. Ponce and B. Mwale, J. Phys. Chem. B, 2006, 110, 2155–2158 CrossRef CAS.
- Q. B. Zhang, J. Y. Lee, J. Yang, C. Boothroyd and J. X. Zhang, Nanotechnology, 2007, 18, 245605 CrossRef.
- Q. B. Zhang, J. P. Xie, J. Liang and J. Y. Lee, Adv. Funct. Mater., 2009, 19, 1387–1398 CrossRef CAS.
- I. Srnova-Sloufova, F. Lednicky, A. Gemperle and J. Gemperlova, Langmuir, 2000, 16, 9928–9935 CrossRef CAS.
- Y. W. Cao, R. Jin and C. A. Mirkin, J. Am. Chem. Soc., 2001, 123, 7961–7962 CrossRef CAS.
- L. H. Lu, H. S. Wang, Y. H. Zhou, S. Q. Xi, H. J. Zhang, H. B. W. Jiawen and B. Zhao, Chem. Commun., 2002, 144–145 RSC.
- S. Mandal, P. R. Selvakannan, R. Pasricha and M. Sastry, J. Am. Chem. Soc., 2003, 125, 8440–8441 CrossRef CAS.
- Q. B. Zhang, J. P. Xie, J. Y. Lee, J. X. Zhang and C. Boothroyd, Small, 2008, 4, 1067–1071 CrossRef CAS.
- D. K. Smith and B. A. Korgel, Langmuir, 2008, 24, 644–649 CrossRef CAS.
- G. A. DeVries, M. Brunnbauer, Y. Hu, A. M. Jackson, B. Long, B. T. Neltner, O. Uzun, B. H. Wunsch and F. Stellacci, Science, 2007, 315, 358–361 CrossRef CAS.
- A. M. Jackson, J. W. Myerson and F. Stellacci, Nat. Mater., 2004, 3, 330–336 CrossRef CAS.
- J. J. Kuna, K. Voitchovsky, C. Singh, H. Jiang, S. Mwenifumbo, P. K. Ghorai, M. M. Stevens, S. C. Glotzer and F. Stellacci, Nat. Mater., 2009, 8, 837–842 CrossRef CAS.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS.
- D. V. Talapin, A. L. Rogach, M. Haase and H. Weller, J. Phys. Chem. B, 2001, 105, 12278–12285 CrossRef CAS.
- I. L. Garzon, K. Michaelian, M. R. Beltran, A. Posada-Amarillas, P. Ordejon, E. Artacho, D. Sanchez-Portal and J. M. Soler, Phys. Rev. Lett., 1998, 81, 1600–1603 CrossRef CAS.
- T. Sugimoto, Adv. Colloid Interface Sci., 1987, 28, 65–108 CAS.
Footnote |
| † These two authors contributed equally to this article |
| This journal is © The Royal Society of Chemistry 2010 |