Engineering biofunctional magnetic nanoparticles for biotechnological applications†
Maria
Moros
a,
Beatriz
Pelaz
a,
Pilar
López-Larrubia
b,
Maria L.
García-Martin
c,
Valeria
Grazú
a and
Jesus M.
de la Fuente
*a
aInstituto de Nanociencia de Aragón, University of Zaragoza, Biofunctional Nanoparticles and Surfaces group (bioNANOsurf), Edif. I + D. C/Mariano Esquillor, Zaragoza, 50018, Spain. E-mail: jmfuente@unizar.es
bInstituto de Investigaciones Biomédicas, Alberto Sols, CSIC/UAM, Arturo Duperier 4, 28029, Madrid, Spain
cRM Ntra. Sra. del Rosario, C/Príncipe de Vergara, 53, 28006, Madrid, Spain
First published on 30th July 2010
Abstract
Synthesis and characterization of magnetic nanoparticles with excellent size control are showed here. Their functionalization using an amphiphilic polymer is also described. This strategy allows the stabilization of magnetic nanoparticles in aqueous solvents and in addition, the polymer shell serves as a platform to incorporate relevant biomolecules, such as poly(ethylene glycol) and a number of carbohydrates. Nanoparticles functionalized with carbohydrates show the ability to avoid unspecific interactions between proteins present in the working medium and the nanoparticles, so can be used as an alternative to poly(ethylene glycol) molecules. Results confirm these nanoparticles as excellent contrast agents for magnetic resonance imaging. Changes in the spin–spin transversal relaxation times of the surrounding water protons due to nanoparticle aggregation demonstrates the bioactivity of these nanoparticles functionalized with carbohydrates. To finish with, nanoparticle toxicity is evaluated by means of MTT assay. The obtained results clearly indicate that these nanoparticles are excellent candidates for their further application in nanomedicine or nanobiotechnology.
1. Introduction
Nanotechnology is considered by many as the next “big revolution”. This technological leap in controlling materials at the nanoscale, such as nanoparticles (NPs), has driven developments enabling the use of nanodevices. Nanoparticles have found applications in different fields such as electronics, communications, optics, chemical energy and of course, biology.1 Despite the benefits that nanoparticles have rendered to medicine, some applications remain challenging; for instance specific targeting of the recognition site, efficient drug delivery into the target cell or in vivo real-time monitoring of cellular events.2 Among a great number of different commonly synthesized NPs, magnetic NPs have excited a great deal of interest because of their intrinsic magnetic properties. Magnetic NPs are usually spherical nanocrystals with a size of 10–20 nm and very often with an iron oxide core; however other magnetic nanomaterials can also be obtained.3 Their magnetic properties make them excellent agents to label biomolecules in bioassays, as well as efficient contrast agents.3In this context, the use of inorganic nanoparticles as drug release systems and as molecular markers is nowadays receiving increasing attention.4,5 In the search for biotechnological and biomedical applications of these nanoparticles, different strategies have been reported for their functionalization with relevant biomolecules such as antibodies, peptides and DNA.6–9
Another important family of biomolecules due to its biological relevance is carbohydrates. Our group has significant experience in the functionalization of different kind of nanoparticles with carbohydrates.10–18 These biomolecules are mainly located in the outer membrane of most types of cells conjugated to sphingolipids (glycosphingolipids) or to proteins (glycoproteins). These carbohydrates are responsible for communication with other cells and with the extracellular matrix, and are involved in viruses and bacteria infections as well as in many other non pathological biorecognition processes.19,20 Taking into account the amount of processes where carbohydrates are involved and their biocompatibility, the high potential of these biomolecules in the nanomedicine field is clear.21 During the last 20 years, there has been a common interest in understanding the role of the carbohydrates implied in cellular recognition processes. Among these, carbohydrate–protein interactions acquire great importance.22,23 However, the study of these interactions is still very challenging due to their low affinity and the need of a multivalent presentation of the carbohydrates. Therefore, in order to study these interactions, a number of carbohydrates can be attached to inorganic nanoparticles, which are suitable candidates as they have a similar length scale to biomolecules.24
The high stability that carbohydrates provide to the nanoparticles must also be highlighted.25 One of the most challenging goals to achieve in biotechnological applications is to obtain non-aggregated NPs with a controlled size in biological media. Nanoparticles added to biological systems should not agglomerate, in order to avoid macrophage uptake and thrombosis. In this context, we report a reproducible way for obtaining magnetic nanoparticles functionalized with relevant monosaccharides. Herein we demonstrate the ability that these monosaccharides have to inhibit unspecific interactions with proteins, competing with PEG molecules for this purpose, but with the main advantage of their smaller length. We also show the potential of these multivalent platforms of carbohydrates for studying carbohydrate–protein interactions by observing changes in the aggregation state of magnetic NPs triggered by a target molecule, in this particular case a lectin. This aggregation induces changes in the relaxation time of water protons allowing us to monitor carbohydrate–protein interactions.26
2. Materials and methods
2.1. Materials
Iron(III) acetylacetonate [Fe(acac)3], 1,2-hexadecanediol, oleic acid, oleylamine, poly (maleic anhydride-alt-1-octadecene) (PMAO, MW = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–50000 g mol−1), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 4-aminophenyl β-D-glucopyranoside, 4-aminophenyl β-D-galactopyranoside, sodium dodecyl sulfate (SDS), β-mercaptoethanol, lysozime, bovine serum albumin (BSA), fetal bovine serum (FBS) and cellulose ester dialysis membranes (MWCO= 10000) were purchased from Sigma Aldrich. α-Methoxy-ω-amino poly(ethylene glycol) (PEG-MW 750 Dalton) was purchased from IRIS Biotech GmbH. Centricon centrifugal filter units (50000 MWCO cut off), and the Milli-Q reagent grade water system used to further purified house distilled water were from Millipore. Polyacrylamide gels (15%), Dulbecco's modified Eagle's medium (DMEM) were purchased from Lonza. MTT (3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide), penicillin, streptomycin and glutamine solutions were from Invitrogen. Buffers were prepared according to standard laboratory procedure. Other chemicals were reagent grade and used as received.
000–50000 g mol−1), N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC), 4-aminophenyl β-D-glucopyranoside, 4-aminophenyl β-D-galactopyranoside, sodium dodecyl sulfate (SDS), β-mercaptoethanol, lysozime, bovine serum albumin (BSA), fetal bovine serum (FBS) and cellulose ester dialysis membranes (MWCO= 10000) were purchased from Sigma Aldrich. α-Methoxy-ω-amino poly(ethylene glycol) (PEG-MW 750 Dalton) was purchased from IRIS Biotech GmbH. Centricon centrifugal filter units (50000 MWCO cut off), and the Milli-Q reagent grade water system used to further purified house distilled water were from Millipore. Polyacrylamide gels (15%), Dulbecco's modified Eagle's medium (DMEM) were purchased from Lonza. MTT (3-[4,5-dimethylthiazolyl-2]-2,5-diphenyltetrazolium bromide), penicillin, streptomycin and glutamine solutions were from Invitrogen. Buffers were prepared according to standard laboratory procedure. Other chemicals were reagent grade and used as received.
2.2. Synthesis of 8 nm iron oxide nanoparticles
Monodisperse Fe3O4 nanoparticles of 8 nm mean diameter were synthesized following the seed-mediated growth method described by Sun.27 First, 6 nm Fe3O4 nanoparticles seeds were synthesized by mixing and stirring under a flow of argon, Fe(acac)3 (0.71 g), 1,2-hexadecanediol (2.58 g), oleic acid (2 mL), oleylamine (2 mL), solubilized in phenyl ether (20 mL). The mixture was heated to 200 °C for 2 h and afterwards heated to reflux (265 °C) under argon atmosphere for 1 h. The mixture was allowed to cool to room temperature by removing the heat source. In order to wash the nanoparticles, ethanol was added, and the nanoparticles were collected with a magnet and redispersed in hexane three times. The NPs were then redispersed in 10 mL hexane containing 50 μl of oleic acid and 50 μl of oleylamine. In order to obtain 8 nm nanoparticles, 80 mg of the 5–6 nm Fe3O4 seeds in hexane were added to a mixture containing Fe(acac)3 (0.71 g), 1,2-hexadecanediol (2.58 g), oleic acid (0.5 mL), oleylamine (0.5 mL), and 20 mL of phenyl ether. The mixture was heated to 100 °C for 30 min to remove the hexane and then to 200 °C for 1 h. Under argon atmosphere the mixture was further heated to reflux (265 °C) for another hour. The mixture was allowed to cool to room temperature by removing the heat source and as described for the 6 nm nanoparticles, they were precipitated with ethanol and resuspended in hexane. NPs of 8 nm were precipitated with ethanol and weighted once completely dry.2.3. Water solubilisation of hydrophobic magnetic nanoparticles
250 mg of poly(maleic anhydride-alt-1-octadecene) (PMAO) was added to a flask containing 200 mL of chloroform. After the polymer was dissolved under magnetic stirring, 20 mg of the nanoparticles were added and the mixture was gently stirred for one hour at 25 °C. Afterwards, the solvent was removed under vacuum and a few millilitres of chloroform were added. Nanoparticles were then resuspended in 20 mL of NaOH 0.05 M and the sample was shaken either at room temperature or 60 °C in order to speed up the complete evaporation of chloroform. At this point the solution became clear as NPs were completely transferred into water. If the solution remains turbid, the pH should be raised with NaOH to approximately 12. NPs were then filtered on syringe filters of 0.22 μm.Two different strategies were used in order to remove the excess of unbound polymer: i) the NPs solution (10 mL) was centrifuged at 25000 rpm for 2 h. The smallest nanoparticles and the excess of unbounded polymer molecules remained in the supernatant liquid. The nanoparticles at the bottom were recovered and redispersed in water. The process was repeated twice. Then, this nanoparticle dispersion was filtered on syringe filters of 0.22 μm to remove larger particles and if necessary the pH was adjusted using a centrifugal filter. The membrane molecular weight cut-off was 50000 MW, in order to retain the particles above. By centrifuging the NPs for 5 min at 4000 S, it was possible to wash them with water and also concentrate them. ii) First, the pH of the NPs solution was lowered to 8 by a washing step with water using a centrifugal filter. Then the NPs solution was layered onto a sucrose gradient containing 5 layers of 5 mL each, from 0.5 M to 1.5 M sucrose. The gradient was centrifuged for 1.5 h at 24000 rpm. The biggest nanoparticles appear in the bottom of the sucrose layers, while the excess of unbound polymer and the smallest nanoparticles rest in the water phase or the rest of the sucrose layers. As when centrifuged without a gradient, to remove the excess of sucrose and to concentrate the NPs, they were extensively washed with water using a centrifugal filter. To check if the unbound polymer removal was successful, we performed agarose gel electrophoresis as reported before.28 The unbounded polymer should be easily detected under ultraviolet light excitation. A 1% (w/v) agarose gel was prepared by dissolving agarose powders in 0,5X tris–borate–EDTA buffer (TBE). The samples were loaded by mixing 80 μL of NPs in water with 20 μL of glycerol:TBE (1:4). Gels were run for 30 min at 80 V. Visible light was used to detect NPs whereas UV light under a blue filter was used to detect the blue emitting excess of unbounded polymer.
2.4. Functionalization of the nanoparticles with glucose, galactose or PEG
To study the adsorption of proteins on the NPs, 1 mg of PMAO modified NPs (27% Fe3O4) were incubated with 1.5 mg of EDC and 11 or 22 μmoles of 4-aminophenyl β-D-glucopyranoside in 250 μl of SSB buffer pH 9 (50 mM of boric acid and 50 mM of sodium borate). After 2 h at room temperature, the NPs were purified of the ligand excess by washing the sample with the desired PBS buffer pH 7.4 in a centrifugal filter.PMAO modified NPs (1 mg) were incubated with 1.5 mg of EDC and 5 or 13 μmoles of α-methoxy-ω-amino poly(ethylene glycol) (PEG-MW 750 Dalton) in 250 μl of SSB buffer pH 9. As above, the NPs were washed using a centrifugal filter.
For aggregation studies, 1 mg of PMAO modified NPs were incubated with 1 mg of EDC and 4 μmoles of 4-aminophenyl β-D-glucopyranoside or 4-aminophenyl β-D-galactopyranoside in 250 μl of SSB buffer pH 9. After two hours of incubation NPs were extensively washed with PBS buffer 1X pH 7.4 in a centrifugal filter.
2.5. General procedures for nanoparticles characterization
For transmission emission microscopy (TEM) examinations, a single drop (10 μL) of the aqueous solution (0.1 mg mL−1) of the different nanoparticles was placed onto a copper grid coated with a carbon film. The grid was left to dry in air for several hours at room temperature. TEM analysis was carried out in a JEM-1200EX electron microscope working at 80 kV. Particle size distribution was evaluated from several micrographs using an automatic image analyzer. The number of particles selected for consideration was around 100, which resulted in a stable size-distribution statistic.Thermogravimetric analysis (TGA) was performed using a TA STD 2960 simultaneous DTA-DTGA instrument in air or in a nitrogen atmosphere and at a heating rate of 10 °C min−1.
Magnetization measurements were performed as a function of temperature from 5 to 300 K and applied fields up to 0.6 Tesla with a commercial SQUID magnetometer system MPMS-7T, Quantum Design. Zero-field-cooling (ZFC) and field-cooling (FC) protocols were used in all samples.
Light scattering and ζ-potential measurements were performed on a Brookhaven Zeta PALS instrument at 25 °C. Each sample was measured three times, combining 10 runs per measurement.
2.6. Relaxivity studies
The longitudinal (T1) and transversal (T2) relaxation times have been measured at five different iron concentrations of NPs (0.17–8 mM) in water, at 0.47 T in a Relaxometer (Bruker Minispec spectrometer). Three different measurements of T1 or T2 were performed in every sample for statistical correction. T1 values were determined by the inversion-recovery method and T2 values were determined by the Carr–Purcell–Meiboom–Gill sequence. The longitudinal (r1) and transversal (r2) relaxivity values were calculated from T1 and T2 (the slope of 1/T as a function of millimolar Fe concentration).2.7. MRI studies
The magnetic resonance imaging (MRI) experiments were performed on a Bruker Pharmascan system (Bruker Medical Gmbh, Ettlingen, Germany) using a 7.0 T horizontal-bore superconducting magnet, equipped with a 1H selective birdcage resonator of 38 mm and a Bruker gradient insert with 90 mm of diameter (maximum intensity 36 G cm−1). All data were acquired using a Hewlett–Packard console running Paravision v 4.0 software (Bruker Medical Gmbh, Ettlingen, Germany) operating on a Linux platform. Two different phantoms with capillaries were prepared and imaged. One, containing PMAO NPs at different concentrations (0.03–0.3 mM Fe). The other phantom was imaged after 30 min of incubation of PMAO NPs (0.05 μmol Fe) functionalized with glucose or galactose with different concentrations of Concanavalin A (20–30 μg mL−1).One coronal slice (FOV= 3.0 × 3.0 cm, slice thickness = 2 mm) was imaged to obtain parametric maps generated on a pixel by pixel basis. Data were analyzed in a Linux platform with software written in-house in IDL (iterative data language, Research Systems, Boulder, CO). A series of nine T1-weighted (T1-W) images were acquired in order to obtain T1 maps based in a saturation-recovery experiment and the following parameters: TR= 20, 50, 100, 200, 400, 800, 1600, 3200, 6000 ms, TE = 6.7 ms, Av = 1, acquisition matrix= 128 × 128 corresponding to an in-plane resolution of 235 × 235 μm2. Maps were obtained by a least squares non-linear exponential fitting of the signal intensity to the expression: Sτ = S∞ [1 − 2 exp(−τ/T1)]
T 2 maps were based in a Carr–Purcell–Maiboom–Gill (CPMG) experiment in which T2-W images were acquired using a MSME sequence that collect an image per echo time and the following parameters: TR = 5000 ms, TE = 6.7–430 ms, number of echoes = 128, Av = 1, and the same geometric conditions as in T1 maps acquisition. Maps were obtained by a least squares non-linear exponential fitting of the signal intensity to the expression: Sτ = S0 exp(−τ/T2).
T 2-weighted (T2-W) spin-echo images were acquired with a multi slice multi echo (MSME) sequence in the same position as previous maps and the following parameters: Av = 3, FOV = 3.0 × 3.0 cm, acquisition matrix = 256 × 256 corresponding to an in-plane resolution of 117 × 117 μm2, Av = 1, TR/TE = 3000/15 ms for the phantom containing lectin, and TR/TE = 6000/50 ms for the phantom containing PMAO NPs.
2.8. Protein adsorption studies
Adsorption of bovine serum albumin (BSA) and lysozyme was carried out by mixing 0.5 mg of the different NPs with 25 μg of protein solution (125 μg mL−1 in PBS) in a final volume of 200 μL. The effect of pH on adsorption was evaluated using BSA solutions prepared in 10 mM sodium acetate/acetic acid buffer at pH 4.7 or in PBS buffer at pH 7.4.The mixtures were gently agitated at 37 °C in an orbital shaker for 90 min and afterwards NPs were centrifuged in a centrifugal filter with 100000 Da molecular weight cut off; the supernatant was recovered.
Proteins from the supernatants were resolved using SDS-PAGE as described by Laemmli.29 The gels consisted of a 15% polyacrylamide (w/v) and were used as purchased from Lonza. Before loading the samples on the gel, sample buffer containing (20% of SDS and 10% of mercapthoethanol) was added (10% of the total volume of the sample). The samples were boiled for 10 min. Electrophoresis was performed using vertical electrophoresis slab gel apparatus at a constant voltage of 150 V for 1.5 h . Molecular weight markers used: 190, 127, 77, 49, 38, 25, 18, and 12 KDa. The gel was silver stained using standard laboratory procedures.
2.9. Aggregation studies
For T2 relaxation times, a 0.47 T relaxometer (Bruker Minispec) was used. Measurements were made in 0.35 mL of an appropriate buffer containing 10 mM of sodium chloride, 1 mM of magnesium chloride and 1 mM of calcium chloride. The concentration of iron of the functionalized nanoparticles was calculated from ICP data.Concanavalin A final protein concentration was estimated spectrophotometrically at 280 nm by using an extinction coefficient, ε% = 11.4.30 The concentration of NPs was adjusted to 0.2 μmol Fe. Increasing amounts of Concanavalin A were added to different tubes of the NPs solution. Samples were kept at 37 °C for 5 min and T2 values were recorded stepwise. Experiments were carried out in parallel with samples containing glucose NPs, and galactose NPs as a control. When studying the reversibility of the aggregation, free glucose was added as a solid to avoid sample dilution; samples were kept at 37 °C for the whole experiment and T2 relaxation times measured throughout the time. Glucose was added to the same sample twice (0.7 mg and 3 mg) until T2 reach the initial value.
2.10. Cell culture
HeLa (human cervical carcinoma) and Infinity telomerase-immortalized primary human fibroblasts (hTERT-BJ1, Clontech Laboratories, Inc., Palo Alto, CA) were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 5% glutamine and 5% penicillin/streptomycin. Cell cultures were incubated at 37 °C and equilibrated in 5% CO2 and air.2.11. Viability testing
Cell viability and proliferation were analyzed by MTT colorimetric assay. For the cytotoxicity assay 5000 cells were seeded in each well of 96-well plates and grown for 24 h. After 24 h, the medium was replaced with fresh medium containing the different types of nanoparticles in varying concentrations. After cultivation again for 24 h, the medium was replaced with fresh medium and 20 μL of MTT dye solution (5 mg mL−1 in PBS) was added to each well. After 3 h of incubation at 37 °C and 5% CO2, the medium was removed and formazan crystals were dissolved in 100 μL of DMSO. The absorbance of each well was read on a microplate reader (Biotek ELX800) at 570 nm. The spectrophotometer was calibrated to zero absorbance using culture medium without cells. The relative cell viability (%) related to control wells containing cells incubated without nanoparticles was calculated by [A]test/[A]control × 100. Each measurement was repeated at least five times to obtain the mean values and the standard deviation.3. Results and discussion
3.1. Synthesis of water stable magnetic nanoparticles
Monodisperse iron oxide nanoparticles (NPs) having an average size of 8 nm were synthesized following the seed-mediated growth procedure published by Sun and co-workers.27,31 This synthetic technique was selected among the published methodologies due to its ability to provide monodispersed nanoparticles with a controlled and uniform size distribution.32,33 However, the obtained iron oxide nanoparticles only provide stable suspensions in organic solvents due to the presence of oleic acid and oleylamine chains on the surface of the nanoparticles. Stabilization of magnetic NPs in aqueous solutions is a critical requirement for their use in biological applications. So far, a number of approaches have been reported (silanization,34 protein adsorption,35etc.) aiming at this purpose. Recently, Parak and others reported the transference of NPs to aqueous media using an amphiphilic polymer shell. This methodology has been proven to generate very stable NPs in aqueous conditions, and in addition it provides extra functional groups onto the surface of the NPs for further biofunctionalization.28,36 However, the main disadvantage of this procedure is the low yield of the process, mainly due to the complexity of the purification of the polymer coated nanoparticles.Our group has improved and optimized this aqueous media transference for 8 nm magnetic iron oxide NPs using an amphiphilic polymer shell, poly(maleic anhydride-alt-1-octadecene) (PMAO). The hydrophobic part of this polymer has 18-carbon alkaline side residues that intercalate and interact hydrophobically with the oleic acid chains that cover the nanoparticles. The hydrophilic part of the polymer, the maleic anhydride moieties, is exposed to the NP outermost part conferring stability to them. It also provides the nanoparticles with useful reactive groups for further functionalization. Along the coating step, it is critical to optimize the NP:polymer ratio to avoid wrapping of multiple nanoparticles within the same polymeric shell. An optimal ratio has been proven to be from 10 to 14 mg PMAO/mg NP. A large excess of polymer molecules per nanoparticle together with a correct dilution of the sample are needed to avoid interparticle aggregation (Figure S1†). The hydrolysis of the external maleic anhydride groups of the polymer shell is another key step for the stabilization of the magnetic nanocrystals in aqueous solution. Their hydrolysis in water renders two carboxylic groups per monomeric unit of maleic anhydride. We have optimized this hydrolysis procedure by removing the solvent used during the coating of the NPs with PMAO, and resuspending the NPs in a small volume of chloroform in order to recover the NPs from the flask. Afterwards, a treatment of the NPs in basic conditions with NaOH 0.05 M, followed by the complete evaporation of the chloroform, provides NPs fully soluble in water without the need of additional sonication steps (Scheme 1). The excess of unbound polymer was removed via centrifugation at 25000 rpm for 2 h, twice. The complete purification of the NPs was confirmed by agarose gel electrophoresis (Fig. 1). Excess of unbound polymer was not observed in the purified NPs. Moreover, the thin migrations bands of NPs indicate a rather narrow size and surface charge distribution of the NPs.
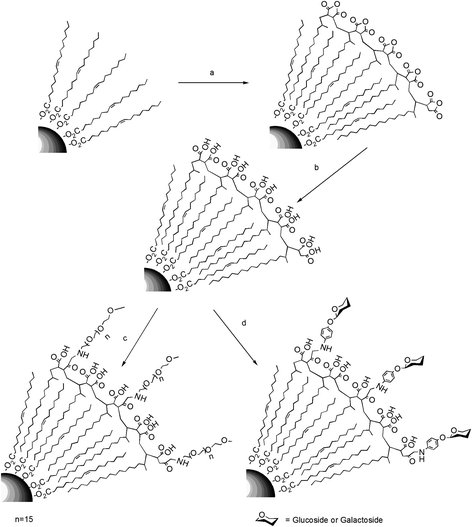 | ||
| Scheme 1 a) Functionalization of the magnetic NPs with PMAO. b) Hydrolysis of the polymer coating with NaOH 0.05 M. c) PEG and d) carbohydrate modification of the carboxylic acids from the surface of the polymer. | ||
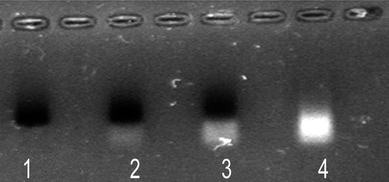 | ||
| Fig. 1 Gel electrophoresis of polymer-coated nanoparticles. Lane 1: NPs purified by centrifugation; lanes 2–3: NPs not purified; lane 4: supernatant of the centrifugation where the excess of unbound polymer appears. The bands of NPs were observed by their absorption colour and the presence of polymer by its fluorescence. | ||
Purified NPs have been characterized by different techniques. TEM images showed that, despite the polymer coating, nanoparticles are still monodisperse with a narrow size distribution (Fig. 2 and S2†). The percentage of organic coating has been evaluated by thermogravimetric analysis (TGA), showing an average coverage of 70% of organic material (Figure S3†). Magnetization measurements were carried out from dried purified NPs, providing a blocking temperature of 50 K (Figure S4†). Data confirmed these nanoparticles as superparamagnetic at room temperature with a saturation moment (Ms) reaching 50 emu gr−1 Fe3O4. It is well known that superparamagnetic iron oxide nanoparticles could be used as transversal relaxation (T2) MRI contrast agents.37,38 Therefore, the superparamagnetic behaviour of our PMAO-coated nanoparticles shown in Figure S4,† suggests that they can be an efficient T2 relaxation agent (Figure S5†). The NPs show high r2 relaxivity values when T2 times are measured as a function of NP iron concentration (r2 = 39 s−1mM−1 and 43 s−1mM−1 when measured with a relaxometer or MRI respectively). The obtained results clearly indicate that these NPs could be a good candidate for further application in molecular imaging (Figure S5†).
 | ||
| Fig. 2 Transmission electronic microscopic (TEM) image of the 8 nm magnetite hydrophobic nanoparticles transferred into water (PMAO-NPs). In the inset, a single NP with the polymer coat can be clearly observed. | ||
The main advantages of our coating approach lie in the preparation of nanoparticles containing only carboxylic groups on their surface and the improved efficiency of the NP synthesis. Previously reported methods achieved the opening of anhydride groups using nucleophiles such as alcohols, ammonia, primary or secondary amines, thiols, etc.39,40 In this line, hydroxyl or amino group terminated poly(ethylene glycol) methyl esters, amino-modified organic molecules or tris(hydroxymethyl) aminomethane (tris) had been used.28,41–43 The use of these kinds of molecules ensure the stability of the nanoparticles in water but at the cost of losing one of the carboxylic moieties obtained per anhydride. This nucleophilic addition methodology also requires the combination of either high temperatures or long sonication times to achieve solubilisation of the NPs.28,42 In contrast, the strategy reported in this manuscript ensures the stability of the nanoparticles by just generating carboxylic groups on the surface of the nanoparticles, facilitating their further functionalization. Our method also does not require sonication of the nanoparticles for their solubilization and hence, it reduces the synthesis time.
Besides, and as previously reported,36 we have observed that for our purposes there is not need to cross-link the polymer shell around each nanocrystal, which also allows shortening and simplifying the water transference protocol. Each monomeric unit of 1-octadecene of PMAO is reversibly bound via hydrophobic forces to oleic molecules attached to the surface of NPs. However, due to the high molecular weight of PMAO, which ranges from 30000 to 50000 Da, the multipoint attachment of each polymer molecule to the NPs surface is assured. When a large molecule is attached by multiple reversible bonds, the simultaneous cleavage of all the bonds is a very energy costly task; the energy cost increases with the number of multiple bonds.44,45 Therefore, the attachment of the PMAO molecules could be considered as “irreversible”, as detachment is only possible under very harsh conditions. In fact, the PMAO coating was not affected by sequentially washing steps of the water soluble NPs with solvents of increased apolarity (chloroform, hexane…), as it has been demonstrated by DLS (Table S1†).
The main advantage of the method reported in this manuscript concerns the purification of the stabilized NPs. Previously reported strategies proceed to remove the excess of unbound polymer using gel electrophoresis, size exclusion columns or sucrose gradients.28,36 It is important to note that although all processes are valid for removing the polymer, centrifugation at high speed is preferred as the synthesis becomes faster and easier, drastically improving the yields of the process.
3.2 Protein adsorption to the NPs
After the hydrolysis step, the surface of the polymer shell becomes negatively charged as each maleic anhydride unit is converted in two carboxylic acid groups. This allows stabilization of the NPs in water by electrostatic repulsion; this fact, combined with the steric stabilization provided by the polymer, results in NPs that are stable over a wide pH range (4–11), as it has been proven by DLS (Fig. S6 and S7†); moreover, they remain stable at high ionic concentration (up to 1 M NaCl) in water or physiological buffers (PBS, etc) for several months. However, the presence of free carboxyl groups could promote protein adsorption due to electrostatic attraction. This protein binding capacity consequently tends to the agglomeration of PMAO coated NPs when incubated with cell culture medium in the presence of 10% (v/v) of fetal bovine serum. This will strongly influence: i) the interaction of the NPs with cells, as the availability of the particles to be internalized seems to be strongly dependent on the size and morphology of the aggregates;46 and ii) their magnetic properties even in the absence of a magnetic field. Moreover, if these nanoparticles are going to be injected into the blood stream for drug delivery or diagnosis purposes, unspecific adsorption of plasma proteins will make them easily recognizable as “intruder objects”; thus, they will be cleared out by cells of the mononuclear phagocyte system (MPS) before they can perform their designed therapeutic function.Serum albumin (67 kDa) has been used as a model system for protein adsorption studies as it is the most abundant plasma protein in humans and other mammals. As it can be observed in Fig. 3a, as the pH value decreases from 7.4 to 4.7, the amount of adsorbed BSA on PMAO coated NPs increased significantly. The net charge of BSA varies with pH, being positive or negative at pH below or above its isoelectric point (pH 4.7) respectively.47 At pH 7.4 both BSA and PMAO NPs have a net negative charge, which results in the absence of BSA adsorption due to electrostatic repulsion (Fig. 3a lane 1). However, BSA has zero net surface charge whereas the PMAO NPs are still negative at pH 4.7. (Fig. S8†) At this pH value there is therefore a complete adsorption of BSA onto the NPs, as the electrostatic repulsion is not so important (Fig. 3a lane 5).
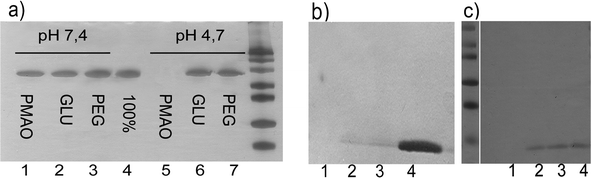 | ||
| Fig. 3 SDS PAGE analysis performed to study the protein adsorption on the NPs: a) supernatants of the BSA adsorption carried out on PMAO NPs (lane 1 and 5), glucose NPs (lanes 2 and 6) and PEG NPs (lanes 3 and 7) at pH 7.4 and 4.7 respectively. When compared with the input (100% of applied BSA, lane 4) it appears clear that at pH 7.4 none of the NPs adsorbed any protein, as all the protein remains in the supernatant. However, at pH 4.7, PMAO NPs adsorbed all the protein, but not PEG or glucose NPs, indicating that the PMAO coating needs further functionalization; b) supernatants of the lysozyme adsorption on the different NPs: PMAO NPs (lane 1), NPs functionalized with 11 μmoles of glucose per mg of NP (lane 2) and NPs functionalized with 5 μmoles of PEG per mg of NP (lane 3). Lane 4 corresponds with the total amount of lysozyme applied. As it can be observed, the adsorption of this protein can not be prevented in any of the cases. c) Supernatants of the lysozyme adsorption on the different NPs: PMAO NPs (lane 1), NPs functionalized with 22 μmoles of glucose per mg of NP (lane 2) and NPs functionalized with 13 μmoles of PEG per mg of NP (lane 3). Lane 4 corresponds to the total amount of lysozyme applied. In this case, lysozyme is only adsorbed on the PMAO NPs, where in the other ones it is completely prevented due to the correct functionalization of the surface. | ||
It is well known from several investigations that the plasma half-life of NPs increases when the NPs are small enough and have a neutral and hydrophilic surface, mainly due to a decrease of unspecific adsorption of blood proteins.48–50 It is also known that PEG molecules can provide a suitable surface to avoid unspecific interactions with proteins. However, their high price when they are adequately modified (amino, thiol ended, etc.), and their length that prone to mask specific biomolecules incorporated to the NPs surface, make necessary the search of alternatives to PEG molecules. For this reason and in order to avoid unspecific interaction of cell media or blood proteins, the carboxylic acids in the polymer shell were covalently functionalized with carbohydrates, which are neutral and very hydrophilic molecules, in particular glucose. PMAO-NPs passivated with PEG molecules (750 Da) were also prepared for comparison with the glyco-NPs. The density of the attached molecules could critically influence the unspecific protein adsorption due to steric hindrance. Therefore, the molar ratio of the glucose or PEG molecules attached to the NPs was optimized.
As it can be observed in Fig. 3a, when 11 μmoles of carbohydrate or 5 μmoles of PEG per mg of NP were used, the adsorption of BSA at pH 7.4 or pH 4.7 was completely prevented due to steric repulsion (lanes 2, 6 and lanes 3,7 respectively). However, the adsorption of a very small and positive protein, lysozyme from hen egg (12 kDa and pI = 11.1), could not be prevented (Fig. 3b). These results indicate a low density of PEG or glucose on the NPs surface. This fact implies that there are still negative charged areas on the NPs surface that in spite of being inaccessible for large proteins (BSA), are accessible for smaller ones. When the ratio of glucose or PEG was increased up to 22 μmoles of carbohydrate or 13 μmoles of PEG per mg of NP, adsorption of lysozyme was prevented in both cases (Fig. 3c). Adsorption of lysozyme on PMAO NPs was complete in all cases. Therefore, we can conclude that with the correct functionalization of the NP surface, either with PEG or a carbohydrate, adsorption of proteins from the culture media can be prevented.
3.3. Specific lectin-glyconanoparticle interaction studies
In addition to the passivation of the NPs surface, the functionalization of NPs with carbohydrates serves as a study of interactions in which carbohydrates are involved. In this sense, glyco-NPs are excellent platforms to be used in these studies, as they are of a similar size scale to biomolecules and allow a multivalent presentation of the ligands. Among these interactions, carbohydrate–lectin interactions are very important molecular recognition processes as they are involved in multiple relevant biological events, such as virus and bacterial infection, inflammation processes, metastasis, embriogenesis, etc.51 To probe the specificity of the prepared magnetic glyconanoparticles, we have studied their interactions with a relevant model lectin (Concanavalin A), monitoring the NPs lectin-triggered aggregation by using a tabletop NMR relaxometer and MR imaging.Concanavalin A (ConA), isolated from jack bean meal, exhibits a specific affinity for glucose-containing carbohydrates.52 When Con A is added to a solution containing glucose–NPs (GNPs), as ConA is a multimeric protein having more than one carbohydrate binding site, agglutination of NPs occurred. When high concentrations of ConA were added, the aggregation of glucose–NPs was very quick. The aggregates were visible by eye after 10 min from addition of ConA, and just by spinning the sample their presence became even clearer. The specificity of this glucose–NPs/ConA interaction was verified by addition of an excess of glucose. Glucose acts as a competitive inhibitor, binding to the Con A carbohydrate binding sites and displacing glucose conjugated to the magnetic nanoparticles. Specificity of the interaction has also been demonstrated by the fact that galactose–NPs do not interact with ConA as agglutination of these NPs has not been observed (Fig. 4).
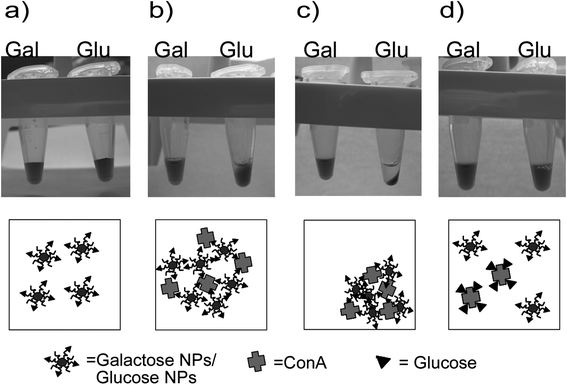 | ||
| Fig. 4 Glucose and galactose NPs (a) behaviour after addition of Concanavalin A, glucose NPs begin to precipitate (b) after ten minutes of incubation and just by spinning the sample the effect become clearer (c). When an excess of free glucose is added (d) the aggregation is reverted. | ||
PMAO coated NPs are superparamagnetic at room temperature. It is interesting to point out that the functionalization of the NPs surface with the different sugars did not alter the r2 relaxivity values. Moreover, the clustering of the NPs could be detected by a drastic shortening of the transverse relaxation time values (T2) of water protons.53T2 changes could be measured in a single tube T2 reader (a tabletop relaxometer) (Fig. 5) or in a high throughput fashion using MR imaging (Fig. 6). The decrease in T2 values will mainly depend on the factors that affect the degree of clustering of NPs. One of these parameters is the concentration of ConA added to the suspension of glucose–NPs (GNPs). Therefore, increasing concentrations of ConA were added to GNPs and T2 recorded after five minutes of incubation at 37 °C. As it can be observed in Fig. 5a and Fig. 6, T2 decreases as ConA concentration increases and consequently bigger clusters of NPs are formed (Fig. S9†). However, the addition of larger amounts of ConA resulted in the formation of NP aggregates that precipitate to the bottom of the sample tube. Once these aggregates separate from the sample, less NPs remained in solution producing an increase in T2 values (data not shown). To show the specificity of these interactions, an excess of free glucose was added and as it acts as competitive inhibitor, T2 values again reached the original values (Fig. 5b). Experiments performed with galactose–NPs and heat inactivated ConA did not lead to any significant change in T2 values, demonstrating again the specificity of the interactions.
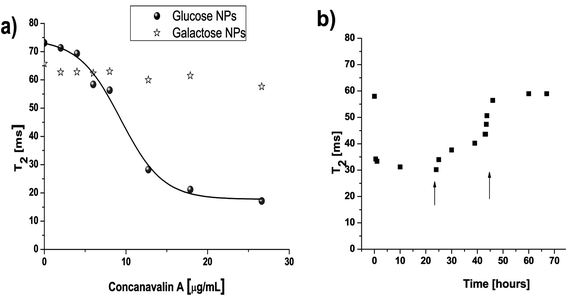 | ||
| Fig. 5 a) Addition of different concentrations of ConA to glucose–NPs resulted in a sigmoidal curve as a function of T2. b) If one of these samples containing NPs and ConA was measured throughout the time, the addition of two concentrations of free glucose (0.7 and 3 mg, indicated with an arrow) results in the return of the T2 to its original value. | ||
 | ||
| Fig. 6 T 2 weighted images of a phantom containing buffer, (a) glucose–NPs, and (b) galactose–NPs incubated with different concentrations of ConA. The signal intensity is inversely proportional to the relaxation rate. | ||
Weissleder and co-workers have already used this detection system to develop what they called “magnetic relaxation switches” (MRS) sensors.54,55 They had used them in order to detect several different targets such as nucleic acids (DNA, RNA), antibodies, proteases, viral particles, etc. From their work it is possible to conclude that two aspects are critical in order to achieve high sensitivity levels: i) the need to work with NPs with high r2 relaxativity values, with the aim of detecting small variations in this parameter when clustering occurs, and ii) the need to work with NPs with high colloidal stability. This last task is very difficult to control when working with commercial NPs, as they usually are stable only within a very narrow range of pH or ionic strength; moreover, reproducibility between different batches of NPs could not be assured. The glyco-nanoparticles which we have optimized fulfil both essential requirements for performing the assays. T2 values remained constant during the time at different pH values even when they were incubated with cell culture media. This allows the use of our NPs in working conditions that could not be possible with most commercial NPs. These characteristic features of GNPs can explain the excellent minimum amount of ConA that can be detected using these NPs (0.7 μg) compared to previously reported results using commercially available magnetic nanoparticles.56 We also have to take into account that about 50% of the protein subunits of commercial preparations of ConA are hydrolyzed, and that these subunits are less competent to self associate than native ones;57 therefore, better detection limits are expected when using not commercial preparations of lectins.
3.4. Viability studies
In vitro experiments in order to assess the influence of these nanoparticles in terms of cytotoxicity on HeLa cell line were done. Cells incubated with all the synthesized nanoparticles remained more than 85% viable after 24 h of incubation at concentration as high as 1 mg ml−1 according to MTT assay, which measures levels of metabolically active mitochondrial dehydrogenase enzymes (Fig. S10†). The use of few cell lines or techniques to evaluate NPs biocompatibility could result in misleading results and incorrect interpretation.48 Therefore, several cell lines and more techniques to evaluate the influence of these nanoparticles in terms of cytotoxity, morphology, adhesion capacity, endocytosis behaviour and cytoskeleton organization will be carried out. However, these preliminary in vitro cytotoxicity assays show that the obtained NPs are very promising for their use in several applications in the field of nanomedicine (contrast agents for tracking and diagnosis by MRI, multifunctional systems for controlled drug delivery, etc).4. Conclusions
Summarizing, herein we present the synthesis and characterization of superparamagnetic nanoparticles and their functionalization with different monosaccharides and PEG molecules. Magnetic characterization of these nanomaterials confirms their potential as contrast agents for MRI experimentation. Moreover, glucose and galactose as coating molecules have shown excellent properties as blocking molecules to inhibit unspecific protein interactions with nanoparticles. They represent an alternative to PEG molecules. We have also confirmed the bioactivity of glucose linked to the surface of the nanoparticles by their recognition by a specific lectin. Furthermore, it was possible to quantify the degree of agglutination of these NPs in a very sensitive way by measuring changes in T2 values caused by their specific clustering. This opens a gate to their use as platforms for the study of carbohydrate–protein interaction. The study of these interactions is of great importance in the nanomedicine field, as carbohydrates control many normal and pathological processes. Therefore, by coupling the presence of carbohydrate binding proteins to the induction of aggregates between the corresponding glyco-functionalized NPs, very sensitive aggregation-based sensors can be designed in order to detect glycosilated analytes (glycoproteins, recombinant hormones), search for new therapeutic agents that block the entry of virus and bacteria and analyze differences in the glycosilation pattern of cells (associated to hepatic diseases, cancer,…), etc.Acknowledgements
This work was supported by institutional funding from MITYC (CTQ2008-03739/PPQ) (Spain), Community of Madrid (Grant S-BIO2006-170) and NANOBIOMED-Consolider. JMF thanks ARAID for financial support. Authors also thank Iñigo Echaniz for technical support. The authors would also like to thank other BioNanoSurf members and Dr. F. Luvi for their contributions to the paper.Notes and references
- I. Brigger, C. Dubernet and P. Couvreur, Nanoparticles in cancer therapy and diagnosis, Adv. Drug Delivery Rev., 2002, 54, 631–651 CrossRef CAS.
- N. Sanvicens and M. P. Marco, Multifunctional nanoparticles-properties and prospects for their use in human medicine, Trends Biotechnol., 2008, 26, 425–433 CrossRef CAS.
- P. Sharrna, S. Brown, G. Walter, S. Santra and B. Moudgil, Nanoparticles for bioimaging, Adv. Colloid Interface Sci., 2006, 123–126, 471–485 CrossRef CAS.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Therapeutic nanoparticles for drug delivery in cancer, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS.
- K. K. Jain, Nanotechnology in clinical laboratory diagnostics, Clin. Chim. Acta, 2005, 358, 37–54 CrossRef CAS.
- G. Von Maltzahn, Y. Ren, J. Park, D. Min, V. R. Kotamraju, J. Jayakumar, V. Fogal, M. J. Sailor, E. Ruoslahti and S. N. Bhatia, In vivo tumor cell targeting with “Click” nanoparticles, Bioconjugate Chem., 2008, 19, 1570–1578 CrossRef CAS.
- U. Jeong, X. Teng, Y. Wang, H. Yang and Y. Xia, Superparamagnetic colloids: Controlled synthesis and niche applications, Adv. Mater., 2007, 19, 33–60 CrossRef CAS.
- M. K. Yu, Y. Y. Jeong, J. Park, S. Park, J. W. Kim, J. J. Min, K. Kim and S. Jon, Drug-loaded superparamagnetic iron oxide nanoparticles for combined cancer imaging and therapy in vivo, Angew. Chem., Int. Ed., 2008, 47, 5362–5365 CrossRef CAS.
- J. Park, G. von Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Micellar hybrid nanoparticles for simultaneous magnetofluorescent imaging and drug delivery, Angew. Chem., Int. Ed., 2008, 47, 7284–7288 CrossRef CAS.
- J. M. de la Fuente, A. G. Barrientos, T. C. Rojas, J. Rojo, J. Canada, A. Fernandez and S. Penades, Gold glyconanoparticles as water-soluble polyvalent models to study carbohydrate interactions, Angew. Chem., Int. Ed., 2001, 40, 2257 CrossRef.
- J. M. de la Fuente, D. Alcantara and S. Penades, Cell response to magnetic glyconanoparticles: Does the carbohydrate matter?, IEEE Trans. NanoBiosci., 2007, 6, 275–281 CrossRef.
- J. M. de la Fuente and S. Penades, Understanding carbohydrate-carbohydrate interactions by means of glyconanotechnology, Glycoconjugate J., 2004, 21, 149–163 CrossRef CAS.
- A. G. Barrientos, J. M. de la Fuente, T. C. Rojas, A. Fernandez and S. Penades, Gold glyconanoparticles: Synthetic polyvalent ligands mimicking glycocalyx-like surfaces as tools for glycobiological studies, Chem.–Eur. J., 2003, 9, 1909–1921 CrossRef CAS.
- J. D. M. de la Fuente and S. Penades, Glyco-quantum dots: a new luminescent system with multivalent carbohydrate display, Tetrahedron: Asymmetry, 2005, 16, 387–391 CrossRef CAS.
- K. El-Boubbou, C. Gruden and X. Huang, Magnetic glyco-nanoparticles: a unique tool for rapid pathogen detection, decontamination, and strain differentiation, J. Am. Chem. Soc., 2007, 129, 13392 CrossRef CAS.
- B. Srinivasan and X. Huang, Functionalization of magnetic nanoparticles with organic molecules: loading level determination and evaluation of linker length effect on immobilization, Chirality, 2008, 20, 265–77 CrossRef.
- S. I. Van Kasteren, S. J. Campbell, S. Serres, D. C. Anthony, N. R. Sibson and B. G. Davis, Glyconanoparticles allow pre-symptomatic in vivo imaging of brain disease, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18–23 CrossRef.
- V. Kekkonen, N. Lafreniere, M. Ebara, A. Saito, Y. Sawa and R. Narain, Synthesis and characterization of biocompatible magnetic glyconanoparticles, J. Magn. Magn. Mater., 2009, 321, 1393 CrossRef CAS.
- A. Varki, Biological Roles of Oligosaccharides-all of the Theories are Correct, Glycobiology, 1993, 3, 97–130 CrossRef CAS.
- R. A. Dwek, Glycobiology: Toward understanding the function of sugars, Chem. Rev., 1996, 96, 683–720 CrossRef CAS.
- J. M. De la Fuente and S. Penades, Glyconanoparticles: Types, synthesis and applications in glycoscience, biomedicine and material science, BBA-Gen. Subjects, 2006, 1760, 636–651 Search PubMed.
- H. Kogelberg, D. Solis and J. Jimenez-Barbero, New structural insights into carbohydrate-protein interactions from NMR spectroscopy, Curr. Opin. Struct. Biol., 2003, 13, 646–653 CrossRef CAS.
- P. Nangia-Makker, J. Conklin, V. Hogan and A. Raz, Carbohydrate-binding proteins in cancer, and their ligands as therapeutic agents, Trends Mol. Med., 2002, 8, 187–192 CrossRef CAS.
- G. M. Whitesides, The ‘right’ size in nanobiotechnology, Nat. Biotechnol., 2003, 21, 1161–1165 CrossRef CAS.
- S. C. McBain, H. H. P. Yiu and J. Dobson, Magnetic nanoparticles for gene and drug delivery, 2008, 3, 169–180 Search PubMed.
- L. Josephson, J. M. Perez and R. Weissleder, Magnetic nanosensors for the detection of oligonucleotide sequences, Angew. Chem., Int. Ed., 2001, 40, 3204 CrossRef CAS.
- S. H. Sun, H. Zeng, D. B. Robinson, S. Raoux, P. M. Rice, S. X. Wang and G. X. Li, Monodisperse MFe2O4 (M = Fe, Co, Mn) nanoparticles, J. Am. Chem. Soc., 2004, 126, 273–279 CrossRef CAS.
- R. Di Corato, A. Quarta, P. Piacenza, A. Ragusa, A. Figuerola, R. Buonsanti, R. Cingolani, L. Manna and T. Pellegrino, Water solubilization of hydrophobic nanocrystals by means of poly(maleic anhydride-alt-1-octadecene), J. Mater. Chem., 2008, 18, 1991–1996 RSC.
- U. K. Laemmli, Cleavage of Structural Proteins during Assembly of Head of Bacteriophage-T4, Nature, 1970, 227, 680–685 CAS.
- B. B. L. Agrawal and I. J. Goldstein, Protein-Carbohydrate Interaction.VII. Physical and Chemical Studies on Concanavalin A, the hemagglutinin of the Jack Bean, Arch. Biochem. Biophys., 1968, 124, 218–229 CrossRef CAS.
- S. H. Sun and H. Zeng, Size-controlled synthesis of magnetite nanoparticies, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS.
- A. G. Roca; M. P. Morales; C. J Serna. Synthesis of monodispersed magnetite particles from different organometallic precursors; 41st IEEE International Magnetics Conference; San Diego, CA, October 2006 Search PubMed.
- P. Tartaj, M. D. Morales, S. Veintemillas-Verdaguer, T. Gonzalez-Carreno and C. J. Serna, The preparation of magnetic nanoparticles for applications in biomedicine, J. Phys. D: Appl. Phys., 2003, 36, R182–R197 CrossRef CAS.
- S. Kalele, S. W. Gosavi, J. Urban and S. K. Kulkarni, Nanoshell particles: synthesis, properties and applications, Curr. Sci. India, 2006, 91, 1038–1052 Search PubMed.
- A. G. Tkachenko, H. Xie, D. Coleman, W. Glomm, J. Ryan, M. F. Anderson, S. Franzen and D. L. Feldheim, Multifunctional gold nanoparticle-peptide complexes for nuclear targeting, J. Am. Chem. Soc., 2003, 125, 4700–4701 CrossRef CAS.
- C. J. Lin, R. A. Sperling, J. K. Li, T. Yang, P. Li, M. Zanella, W. H. Chang and W. J. Parak, Design of an amphiphilic polymer for nanoparticle coating and functionalization, Small, 2008, 4, 334–341 CrossRef CAS.
- G. M. Lanza, P. M. Winter, S. D. Caruthers, A. M. Morawski, A. H. Schmieder, K. C. Crowder and S. A. Wickline, Magnetic resonance molecular imaging with nanoparticles, J. Nucl. Cardiol., 2004, 11, 733–743 CrossRef.
- C. Sun, J. S. H. Lee and M. Zhang, Magnetic nanoparticles in MR imaging and drug delivery, Adv. Drug Delivery Rev., 2008, 60, 1252–1265 CrossRef CAS.
- J. M. Rosenfel and C. B. Murphy, Hydrolysis Study of Organic Acid Anhydrides by Differential Thermal Analysis-II. Maleic Anhydride and Trimellitic Anhydride, Talanta, 1967, 14, 91–96 CrossRef.
- F. B. Zienty, A. A. Schleppnik and B. D. Vineyard, Base-Catalyzed Addition of Thiols to Alpha, beta-Unsaturated Anhydrides, J. Org. Chem., 1962, 27, 3140 CrossRef CAS.
- T. Pellegrino, L. Manna, S. Kudera, T. Liedl, D. Koktysh, A. L. Rogach, S. Keller, J. Radler, G. Natile and W. J. Parak, Hydrophobic nanocrystals coated with an amphiphilic polymer shell: A general route to water soluble nanocrystals, Nano Lett., 2004, 4, 703–707 CrossRef CAS.
- E. V. Shtykova, X. Huang, X. Gao, J. C. Dyke, A. L. Schmucker, B. Dragnea, N. Remmes, D. V. Baxter, B. Stein, P. V. Konarev and et al, Hydrophilic Monodisperse Magnetic Nanoparticles Protected by an Amphiphilic Alternating Copolymer, J. Phys. Chem. C, 2008, 112, 16809–16817 CrossRef CAS.
- W. W. Yu, E. Chang, C. M. Sayes, R. Drezek and V. L. Colvin, Aqueous dispersion of monodisperse magnetic iron oxide nanocrystals through phase transfer, Nanotechnology, 2006, 17, 4483–4487 CrossRef CAS.
- V. Grazu, L. Betancor, T. Montes, F. Lopez-Gallego, J. M. Guisan and R. Fernandez-Lafuente, Glyoxyl agarose as a new chromatographic matrix, Enzyme Microb. Technol., 2006, 38, 960–966 CrossRef CAS.
- C. Mateo, J. M. Palomo, M. Fuentes, L. Betancor, V. Grazu, F. Lopez-Gallego, B. C. C. Pessela, A. Hidalgo, G. Fernandez-Lorente, R. Fernandez-Lafuente and J. M. Guisan, Glyoxyl agarose: A fully inert and hydrophilic support for immobilization and high stabilization of proteins, Enzyme Microb. Technol., 2006, 39, 274–280 CrossRef CAS.
- B. Diaz, C. Sanchez-Espinel, M. Arruebo, J. Faro, E. de Miguel, S. Magadan, C. Yaguee, R. Fernandez-Pacheco, M. R. Ibarra, J. Santamaria and A. Gonzalez-Fernandez, Assessing Methods for Blood Cell Cytotoxic Responses to Inorganic Nanoparticles and Nanoparticle Aggregates, Small, 2008, 4, 2025–2034 CrossRef CAS.
- T. Peters, Serum-Albumin, Adv. Protein Chem., 1985, 37, 161–245 CAS.
- A. K. Gupta and M. Gupta, Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS.
- S. W. Choi, W. S. Kim and J. H. Kim, Surface modification of functional nanoparticles for controlled drug delivery, J. Dispersion Sci. Technol., 2003, 24, 475–487 CrossRef CAS.
- R. Michel, S. Pasche, M. Textor and D. G. Castner, Influence of PEG architecture on protein adsorption and conformation, Langmuir, 2005, 21, 12327–12332 CrossRef CAS.
- N. Yamazaki, S. Kojima, N. V. Bovin, S. Andre, S. Gabius and H. J. Gabius, Endogenous lectins as targets for drug delivery, Adv. Drug Delivery Rev., 2000, 43, 225–244 CrossRef CAS.
- I. J. Goldstein, C. E. Hollerman and E. E. Smith, Protein-Carbohydrate Interaction.II. Inhibition Studies on Interaction of Concanavalin a with Polysaccharides, Biochemistry, 1965, 4, 876–883 CrossRef CAS.
- J. M. Perez, L. Josephson and R. Weissleder, Use of magnetic nanoparticles as nanosensors to probe for molecular interactions, ChemBioChem, 2004, 5, 261–264 CrossRef CAS.
- J. M. Perez, L. Josephson, T. O'Loughlin, D. Hogemann and R. Weissleder, Magnetic relaxation switches capable of sensing molecular interactions, Nat. Biotechnol, 2002, 20, 816–820 CAS.
- T. Atanasijevic, M. Shusteff, P. Fam and A. Jasanoff, Calcium-sensitive MRI contrast agents based on superparamagnetic iron oxide nanoparticles and calmodulin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 14707–14712 CrossRef CAS.
- E. Y. Sun, R. Weissleder and L. Josephson, Continuous analyte sensing with magnetic nanoswitches, Small, 2006, 2, 1144–1147 CrossRef CAS.
- D. F. Senear and D. C. Teller, Effects of Saccharide and Salt Binding on Dimer-Tetramer Equilibrium of Concanavalin A, Biochemistry, 1981, 20, 3083–3091 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical, physical and magnetic characterization; R2 maps; stability of NPs at different conditions; size of glucose NPs in the presence of Concanavalin A; MTT assays of the samples are shown in figures S1–S10. Table S1 represents the hydrodynamic size of PMAO NPs after being washed with different solvents. See DOI: 10.1039/c0nr00104j |
| This journal is © The Royal Society of Chemistry 2010 |