Effect of synergy on the visible light activity of B, N and Fe co-doped TiO2 for the degradation of MO
Mingyang
Xing
,
Yongmei
Wu
,
Jinlong
Zhang
* and
Feng
Chen
Key Lab for Advanced Materials and Institute of Fine Chemicals, East China University of Science and Technology, 130 Meilong Road, Shanghai, 200237, P. R. China. E-mail: jlzhang@ecust.edu.cn; Fax: +86-21-64252062; Tel: +86-21-64252062
First published on 18th May 2010
Abstract
Single doped, co-doped and tri-doped TiO2 with B, N and Fe are successfully synthesized by using the hydrothermal method. The samples are characterized by X-ray diffraction (XRD), UV-vis diffuse reflectance spectroscopy (DRS), and X-ray photoelectron spectroscopy (XPS). The photocatalytic activities of the samples are evaluated for degradation of methyl-orange (MO, 20 mg L−1) in aqueous solutions under visible light (λ > 420 nm). The results of XRD suggest that all the catalysts present anatase crystal. All the doping catalysts show higher photoactivities than pure TiO2 under visible light irradiation. In the single nonmetal doped TiO2, the localized dopant levels near the valence band (VB) are responsible for the enhancement of photoactivies. Fe3+ impurity level formed under the conduction band (CB) induces the high photocatalytic activities of iron doped TiO2. In the co-doped and tri-doped catalysts, the B 2p and N 2p acceptor states contribute to the band gap narrowing by mixing with O 2p states combined with the overlapping of the conduction band by the iron “d” orbital, resulting in improvement of the photo-performance under visible light irradiation. Iron co-doped with boron catalyst shows low photoactivity under visible light due to the absence of Fe3+ impurity levels at the bottom of the conduction band. In addition, the XPS results indicate the presence of synergistic effects in co-doped and tri-doped catalysts, which contribute to the enhancement of photocatalytic activities.
1. Introduction
Titanium oxides, as one of the most promising photocatalysts, have been used for degradation of organic pollution, photocatalytic dissociation of water and solar energy conversion.1–4 Because the wide band gap of TiO2 hampers practical applications, many approaches have been undertaken to extend the activity of TiO2 into the visible range.5–11 There are a number of publications reporting the beneficial influence of the substitution of oxygen in TiO2 by nonmetals such as nitrogen, carbon, sulfur and boron on bringing activity of the photocatalysts into the visible range.12–19 Among all nonmetal-doped TiO2, nitrogen doped TiO2 nanomaterials have been found to exhibit superior photocatalytic activity under visible light due to its effective narrowing of the band gap of TiO2.16 Compared with single nitrogen doping, simultaneous doping of nitrogen and other atoms has attracted many researchers, due to its higher photocatalytic performance and peculiar characteristics. For example, Li et al.20,21 have found nitrogen co-doped with fluorine in TiO2 had a higher photo activity under visible light than TiO2 single doping with nitrogen or fluorine. Yang22 and co-workers found that TiO2 nanomaterials co-doped with carbon and nitrogen showed high photocatalytic activity due to the synergistic effect of carbon and nitrogen. Lu et al.23 have prepared the TiO2 co-doped with boron and nitrogen and ascribed its high photocatalytic activity under UV and visible light to the existence of synergistic effects between boron and nitrogen.Doping with transition metals is also used to improve the absorption properties of TiO2.24–29 Choi et al.30 conducted a systematic study on the photocatalytic activity of TiO2 nanoparticles doped with 21 transition metal elements and found that doping with Fe3+, Mo5+, Ru3+, Os3+, Re5+, V4+ and Rh3+ significantly increased photoactivity in the liquid-phase photodegradation of CHCl3, while Co3+ and Al3+ doping decreased the photoactivity. Among these transition metal elements, Fe3+ was considered as an interesting dopant of TiO2 and most of the investigations have been carried out on preparation of Fe-doped TiO2 using chemical and physical methods.31–40 Yamashita et al.31 have found significant improvement in the degradation of aqueous 2-propanol by Fe3+-ion-implanted TiO2 under visible light irradiation. However, iron doping can easily act as charge carriers which are recombination centers, harmful to the photo activity of TiO2. In order to minimize its role as recombination centers, TiO2 co-doped with iron and other nonmetal elements are prepared by many researchers. Kim41 and co-workers synthesized boron–iron co-doped anatase TiO2 using a modified sol–gel method and the presence of boron and iron caused a red shift in the absorption band of TiO2. Cong et al.42 have successfully synthesized the nano-TiO2 co-doped with nitrogen and iron(III) which showed high photocatalytic activity under visible light.
To date there are many published articles related to single doped and co-doped TiO2. However, the research on tri-doping, especially relating to iron, boron and nitrogen tri-doped TiO2, is rather rare. Herein we would like to prepare single doped, co-doped and tri-doped TiO2 with B, N and Fe using the hydrothermal method and investigate its photo-performance under visible light. The mechanisms for the photocatalytic activities of different doping catalysts are also investigated in this article.
2 Experimental
2.1 Preparation of photocatalysts
Tetrabutyl titanate (6.7 mL TBOT) was added to ethanol (30 mL), while stirring the solution for 0.5 h, and then Fe(NO3)3·9H2O (0.02, 0.04, 0.08 g corresponding to 0.2 at%, 0.5 at%, 1.0 at% of Fe/Ti mol%) was added to the above solution to obtain solution A. H3BO3 (0.12 g) and urea (0.6 ml) were dissolved in the ethanol (36 mL), and then deionized water (2.0 mL) and HNO3 (0.50 mL) was successively added to the solution under vigorous stirring to obtain solution B. Solution A was added drop wise to solution B, while stirring the mixture for 4 h. The resultant mixture was then transferred into a 100 mL Teflon-inner-liner stainless steel autoclave which was kept under 453 K for 12 h. After the hydrothermal treatment, the precipitate was washed, dried and ground to obtain the nano-particles of TiO2 tri-doped with nitrogen, boron and iron denoted as (mFe, N, B)–TiO2, where m describes the mol% of iron to titanium.(B,N)-TiO2 was prepared by a similar method without the addition of Fe(NO3)3·9H2O. By addition of 0.04 g Fe(NO3)3·9H2O in solution A, (Fe,N)-TiO2 and (Fe,B)-TiO2 were prepared by the similar procedure without the addition of H3BO3 and urea, respectively. 0.04 g Fe(NO3)3·9H2O was added in solution A to prepare Fe-TiO2 without the addition of H3BO3 and urea. N-TiO2 was synthesized without the addition of H3BO3 and Fe(NO3)3·9H2O and the catalyst B-TiO2 was prepared without the addition of urea and Fe(NO3)3·9H2O. Without the addition of Fe(NO3)3·9H2O, urea and H3BO3, pure TiO2 was prepared using the same method.
2.2 Characterization
X-Ray diffraction (XRD) patterns of all samples were collected in the range 20–80° (2θ) using a Rigaku D/MAX 2550 diffractometer (Cu-K radiation, λ = 1.5406 Å), operated at 40 kV and 100 mA. The crystallite size was estimated by applying the Scherrer equation to the full width at half-maximum (fwhm) of the (101) peak of anatase and the (110) peak of rutile, with α-silicon (99.9999%) as a standard for the instrumental line broadening. The instrument employed for XPS studies was a Perkin-Elmer PHI 5000C ESCA system with Al Kα radiation operated at 250 W. The shift of the binding energy due to relative surface charging was corrected using the C1s level at 285.0 eV as an internal standard. The UV-vis absorbance spectra were obtained for the dry-pressed disk samples using a Scan UV-vis spectrophotometer (Varian, Cary 500) equipped with an integrating sphere assembly, using BaSO4 as the reflectance sample. The spectra were recorded at room temperature in air within the range 200–800 nm.2.3 Measurements of photocatalytic activities
The photocatalytic activity of each sample was evaluated in terms of the degradation of methyl-orange (MO, 20 mg L−1). MO was chosen as a model pollutant to evaluate the photocatalytic activities. The photocatalyst (0.07 g) was added to a 100 mL quartz photoreactor containing 70 mL of a 20 mg L−1 MO solution. The mixture was stirred for 30 min in the dark in order to reach the adsorption–desorption equilibrium. A 1000-W tungsten halogen lamp equipped with a UV cut-off filters (λ > 420 nm) was used as a visible light source (the average light intensity was 60 mW cm−2.) and a 300-W high-pressure Hg lamp for which the strongest emission wavelength is 365 nm was used as a UV light source (the average light intensity was about 1230 μW cm−2). The lamp was cooled with flowing water in a quartz cylindrical jacket around the lamp and ambient temperature was maintained during the photocatalytic reaction. At the given time intervals, the analytical samples were taken from the mixture and immediately centrifuged, then filtered through a 0.22 μm Millipore filter to remove the photocatalysts. The filtrates were analyzed by recording variations in the absorption in UV-vis spectra of MO using a Cary 100 ultraviolet visible spectrometer.3 Results and discussion
3.1 X-Ray diffraction
XRD patterns of the samples are shown in Fig. 1. It can be seen that the diffraction peaks of all samples are ascribed to the peaks of TiO2 anatase phase. These observations indicate that there has been virtually no phase change in TiO2 during the doping, co-doping and tri-doping process. Furthermore, no significant characteristic peak of boron oxide and iron oxide can be found in boron and iron doped TiO2, due to lower amount of boron and iron. The crystal sizes of all the samples are estimated using the Scherrer equation:43Where B is the half-height width of the diffraction peak of anatase, K = 0.89 is a coefficient, θ is the diffraction angle, and λ is the X-ray wavelength corresponding to the Cu-Kα irradiation, and the detailed data are shown in Table 1. It is suggested that the particle size decreased obviously after the nitrogen doping which is consisted with our previous work.44 The crystallite sizes of catalysts doped with nitrogen are much smaller than that of the catalysts doped with iron or boron. Excluding the impact of nitrogen, the process of co-doping and tri-doping exhibit little effect on the particle size as shown in Table 1. In addition to this, there is no change in the “d” space values, which implies that the iron, boron and nitrogen modification in doping, co-doping and tri-doping samples do not change the average unit cell dimension.44
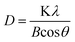
| Crystallite size/nm | d-Spacing/Å | |
|---|---|---|
| Fe-TiO2 | 8.01 | 3.5 |
| B-TiO2 | 8.86 | 3.5 |
| (Fe,B)-TiO2 | 8.22 | 3.5 |
| N-TiO2 | 6.58 | 3.5 |
| (Fe,N)-TiO2 | 6.14 | 3.5 |
| (Fe,B,N)-TiO2 | 6.32 | 3.5 |
 | ||
| Fig. 1 XRD patterns of Fe-TiO2, B-TiO2, N-TiO2, (Fe,B)-TiO2, (Fe,N)-TiO2, and (0.5Fe,B,N)-TiO2. | ||
3.2 UV-vis diffuse reflectance spectra
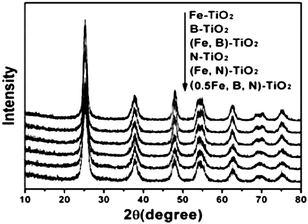
The diffuse reflectance spectra of doped, co-doped and tri-doped samples are shown in Fig. 2. The absorptions of Fe-TiO2, B-TiO2 and (Fe,B)-TiO2 are similar and stronger in the range of wavelengths from 400 to 600 nm. After nitrogen doping the absorptions of catalysts are drastic and stronger in the range of wavelengths from 400 to 800 nm. The absorptions of N-TiO2, (Fe,N)-TiO2 and (0.5Fe,B,N)-TiO2 are similar and present a stronger red shift compared to those of Fe-TiO2, B-TiO2 and (Fe,B)-TiO2 samples. It is expected that nitrogen doping contributed to the red shift because of the narrowing of the band gap by the N species.42,44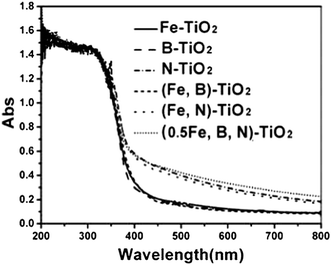 | ||
| Fig. 2 UV–vis diffuse reflectance spectra of Fe-TiO2, B-TiO2, N-TiO2, (Fe,B)-TiO2, (Fe,N)-TiO2, and (0.5Fe,B,N)-TiO2. | ||
3.3 XPS spectra
Fig. 3 shows the XPS spectra for the Fe 2p region and their fitting curves of doped, co-doped and tri-doped samples. It can be seen that there is a peak at approximately 709.5–710.5 eV, which is attributed to the presence of the Fe3+ state.45–47 The peak observed at a higher BE (713.3–714.3 eV) indicates the presence of Fe2+ ions.47,48 It can be speculated that Fe2+ is detected due to photocatalytic reduction from Fe3+ to Fe2+ by conduction-band electron of the catalysts during XPS measurement in vacuum.49 For the catalyst Fe-TiO2, the peak at 709.5 eV corresponds to the Fe3+ entered by substitution of part of the O sites (Ti–O–Fe).50 Another peak at 713.3 eV is attributed to the presence of Fe2+ (see Fig. 3(a)). When TiO2 is co-doped with iron and nitrogen, there is a new peak at 706.8 eV which is the result of the formation of Fe–N on the surface of the catalyst (see Fig. 3(c)). The peak at 706.4 eV ascribed to Fe–B also can be observed when iron and boron were doped simultaneously.51,52 To the catalyst of (Fe,B)-TiO2, the Fe2+ characteristic peak disappears compared with Fe-TiO2 (see Fig. 3(c)). The tri-doped catalysts show three peaks at 707.6, 710.5 and 714.3 eV which are attributed to the presence of Fe–B, Fe3+ and Fe2+ states, respectively (see Fig. 3(d)).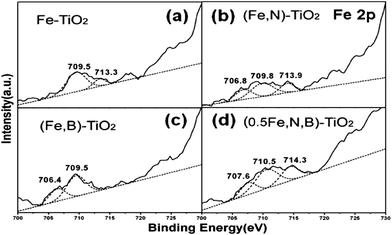 | ||
| Fig. 3 XPS spectra of Fe 2p for Fe-TiO2 (a), (Fe,N)-TiO2 (b), (Fe,B)-TiO2 (c), and (0.5Fe,B,N)-TiO2 (d) samples. | ||
The XPS spectra for the B1s region of B-TiO2 shows two peaks at the BE of 188.8 and 191.9 eV as shown in Fig. 4(a). The former is the characteristic peak of Ti–B and the later corresponds to boron weaved into the interstitial TiO2 structure, existing in the form of a Ti–O–B structure,53 which is consistent with the result reported by Jiang and co-workers.54 Some researchers found that BE peaks for B1s in FeB and Fe2B were 187.9 eV and 188.3 eV respectively.51,52 The peak at 190.5 eV for B1s of (Fe,B)-TiO2 in Fig. 4(b) reveals the existence of Ti–O–FeB. The oxygen bonded to FeB induces the reduction of B1s electron density, resulting in the increasing BE of B1s (a change from 187.9 eV to 190.5 eV). Fig. 4(c) reveals the XPS spectra of (B,N)-TiO2 and the BE peaks for B1s are 186.5 eV and BE at 189.7 eV which should be attributed to B1s in Ti–B and Ti–B–N, respectively.23,51 When TiO2 tri-doped with iron, nitrogen and boron, there appear four characteristic peaks at 185.9, 188.5, 190.8 and 192.6 eV seen from Fig. 4(d). The peak located at BE of 188.5 eV should be attributed to Ti–B–N. The peak at 190.8 eV indicates the existence of Fe–B in tri-doped catalysts. The peaks of BE above 192.6 eV are attributed to boron oxides such as B2O3.
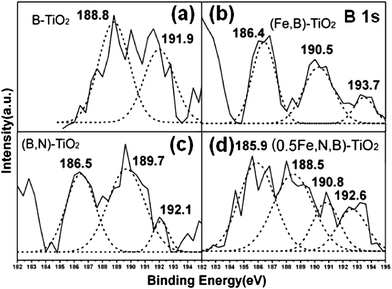 | ||
| Fig. 4 XPS spectra of B1s for B-TiO2 (a), (Fe,B)-TiO2 (b), (B,N)-TiO2 (c), and (0.5Fe,B,N)-TiO2 (d) samples. | ||
3.4 Evaluation of photocatalytic activity
The decomposition of MO (20 mg L−1) in visible light irradiation of TiO2 doped with different elements is carried out and compared with that of pure TiO2 (shown in Fig. 5). The results indicate that the photocatalytic activities are greatly improved by the doping of nitrogen, boron and iron. To the single doped samples, N-TiO2 shows the higher decomposition rate than other catalysts such as Fe-TiO2 and B-TiO2. For the co-doped samples, except for (Fe,B)-TiO2, the catalysts of (Fe,N)-TiO2 and (N,B)-TiO2 all show excellent photocatalytic activities which are higher than that of single doped samples. The photo degradation rate of catalyst is not improved by co-doped with iron and boron, whose degradation rate is even lower than iron single doped TiO2. The tri-doped TiO2 shows high photocatalytic activity under visible light and the photodegradation rate increases with increasing Fe/Ti ratios. The tri-doped catalyst with an Fe/Ti mole ratio of 0.5 at% presents the highest photocatalytic activity.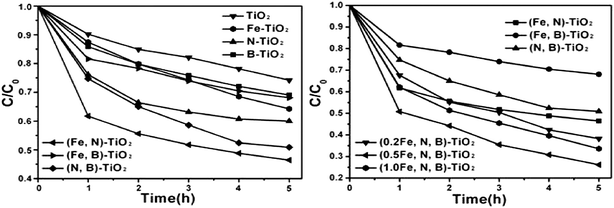 | ||
| Fig. 5 Photodegradation rate of MO under visible light illuminated for 5 h on pure TiO2, doping, co-doping and tri-doping samples. | ||
3.5 Mechanism for the photocatalytic activity under visible light irradiation
For the single doped catalysts, the photocatalytic activities of these are improved by the doping of metal or nonmetal elements compared with the pure TiO2 as shown in Fig. 6. The XPS spectra of Fe2p for Fe-TiO2 sample indicate the existence of Fe3+ and Fe2+ states as shown in Fig. 3(a). Fe3+ ions in the Ti4+ matrix form an impurity level under the CB and can act as a shallow trap to trap the photogenerated electron, which can restrain the recombination of photogenerated electrons and holes (the schemes as shown in Fig. 6(a)).54 Fe3+ ions trap photogenerated electrons to form Fe2+ which induces the detection of Fe2+ during XPS measurement in vacuum (without any hole scavenger as shown in Fig. 6(a)), that is, the detection of Fe2+ indirectly confirms the formation of Fe3+ impurity level under CB which can narrow the band gap of TiO2 and induce the improvement of photocatalytic activity of Fe-TiO2 under visible light. For the samples of B-TiO2 and N-TiO2, photocatalytic performance under visible light is also improved by the nonmetal doping due to the band gap narrowing of TiO2 as shown in Fig. 6(a). Oxygen atom in TiO2 is substituted by nitrogen or boron atom and a narrow substitute band (dopant level) is formed above VB. As a result, the binding energy of TiO2 is lowered by a significant shift.13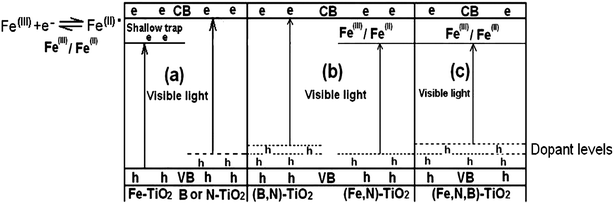 | ||
| Fig. 6 Various proposed schemes illustrating the possible changes that might occur to the band-gap electronic structure of anatase TiO2 on doping with various non-metals, metals: (a) B-TiO2 and N-TiO2 with localized dopant levels near the VB and Fe-TiO2 with an impurity level formed under the CB; (b) band-gap narrowing resulting from broadening of the VB by B co-doping with N-TiO2 and overlapping of the CB by Fe co-doping with N-TiO2; (c) (Fe,N,B)-TiO2 with localized dopant levels to broaden the VB by nonmetals doping and an impurity level formed to overlap the CB by iron doping. | ||
When TiO2 is co-doped by nonmetals such as nitrogen and boron, there exists a synergistic effect. In our experiment, the nitrogen and boron are simultaneously doped in TiO2 by a hydrothermal method. To the band-gap electronic structure of (B,N)-TiO2, the mixing of B 2p with O 2p states will effectively lead to the band gap narrowing of titania, and the mixing of the N 2p states of nitrogen atoms with the VB which results into the further decrease of band gap energy as shown in Fig. 6(b). Our XPS spectra of B1s for (B,N)-TiO2 results indicate the presence of synergetic effects between boron and nitrogen. The peak at the BE of 189.7 eV is attributed to Ti–B–N, which is the result of a synergistic effect. Lu et al.23 have found the role of Ti–B–N bonding in the formation of additional impurity states in the upper VB by the density functional theory (DFT) calculations method. Their calculation results suggested that B and N can be doped on the pristine surface of TiO2 to form Ti–B–N. The effect of B and N co-doping can change the surface electronic states of TiO2 dramatically. They found that the B and N co-doping introduces more surface electronic states and changes the gaps between these states, which may have an important influence on the lifetime of excitons. The formation of Ti–B–N leads to the construction of a favorable surface structure that facilitates the separation and transfer of charge carriers. Thereby in our case, it seems that the presence of Ti–B–N structure on the (B,N)-TiO2 surface acts as an efficient co-catalyst which facilitates the separation and transfer of charge carriers and that the N species contributes to the visible light absorption as shown in Fig. 2.42,44 The synergistic effects of creating visible light absorption and then promoting visible light induced carriers are responsible for the high visible-light photocatalytic activity of (B,N)-TiO2. The above results are contrary to the work of Lambert and co-workers, who did not find the synergistic effect of (B,N)-TiO2.6
The probable schematic diagram of (Fe,N)-TiO2 is proposed in Fig. 6(b) by considering the effects of nitrogen and iron ions co-doping. The co-doping of N and Fe induces the narrowing band gap of TiO2. The overlap of the CB due to Ti (d orbital) and Fe (d orbital) combined with the N impurity level formed near the VB results in the decrease of band gap as shown in Fig. 6(b).55 The Fe3+ ion can trap the photogenerated electrons while the nitrogen can trap the photogenerated holes, which enhances the utilization efficiency of photogenerated electrons and holes. For the catalyst of (Fe,N)-TiO2, the electrons can be excited from the N impurity level to the Fe3+ impurity level or from the VB to the Fe3+ impurity level or from the N impurity level to the CB under visible light irradiation. Therefore, the quantity of the photoincuced electrons and holes is improved compared with the pure TiO2 and single doped TiO2. The band gap narrowing and a large amount of photogenerated charges induce its enhancement of photo-performance.
It is worth noting that when the iron is co-doped with boron, the photocatalytic activity of the catalyst is not improved and its photodegradation rate of MO even lower than Fe-TiO2, as shown in Fig. 5. From the XPS spectra of Fe 2p for (Fe,B)-TiO2, the characteristic peak of Fe2+ has disappeared which suggests the absence of Fe3+/Fe2+ impurity levels under CB. That means the Fe doping can not contribute to the narrowing band gap of (Fe,B)-TiO2. For the catalyst of (Fe,B)-TiO2, the band gap can not be availably narrowed by the Fe and B co-doping resulting into its lower photocatalytic activity compared with (Fe,N)-TiO2. This is further supported by the results of Ti 2p XPS spectra as follows. As we all know, photogenerated electrons and holes could either recombine or move to the surface to react with species adsorbed on the surface. The formation of Fe3+ impurity levels can narrow the band gap and trap electrons to form Fe2+ defective sites. Some of the photogenerated electrons can also be reacted with Ti4+ to form a reduced state of the titanium ions due to the overlap of Ti CB (the CB consisting of Ti “3d” orbital and Fe “3d” orbital).56Fig. 7 shows the Ti 2p XPS of different samples. The spectrum of pure TiO2 exhibits a symmetrical profile, while the (Fe,N,B)-TiO2 exhibits a shoulder at the lower binding energy side of the main peak, as circled in Fig. 7. This shoulder peak has been previously observed for oxygen-treated TiO2 and it originates from the reduced state of the titanium ions.57,58 However, there is no shoulder peak observed for (Fe,B)-TiO2, as shown in Fig. 7, which indicates the absence of reduced titanium due to the disappearance of the Fe3+/Fe2+ impurity level which can trap the photogenerated electrons and transfer them to Ti4+ to form reduced titanium ions. Therefore, the photocatalytic activity of (Fe,B)-TiO2 is not enhanced by the co-doping of Fe and B due to the absence of Fe3+ impurity level which plays an important role in charge transfer.
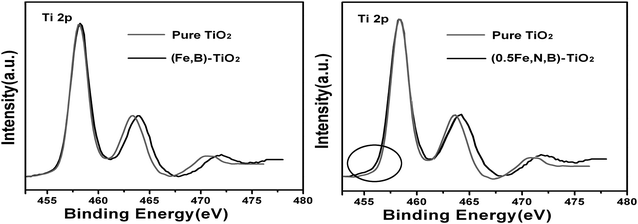 | ||
| Fig. 7 Ti 2p XPS spectra of pure TiO2, (Fe,B)-TiO2 and (Fe,N,B)-TiO2. | ||
Although the catalyst of (Fe,B)-TiO2 shows a low photoactivity and the N tri-doped with (Fe,B)-TiO2 presents excellent photo-performance in visible light irradiation, (Fe,N,B)-TiO2 presents high photoactivity due to the synergistic effects of metals and nonmetals. The results of the XPS measurements indicate the presence of synergistic effects between N and B due to the appearance of a Ti–B–N characteristic peak at 188.5 eV, as shown in Fig. 4(d). The B and N synergistic effect can induce a change of surface electronic states resulting in the promotion of visible light induced carriers and introduce new states around the upper VB close to the Fermi level. Meanwhile, the introduction of iron produces the impurity level at the bottom of the CB as shown in Fig. 6(c). The new states introduced by B and N combined with the Fe3+ impurity level induce the narrowing band gap of (Fe,N,B)-TiO2 which enhances the photoactivity of tri-doped TiO2. (Fe,N,B)-TiO2 shows the highest photodegradation rate when the Fe/Ti mole ratio is 0.5 at% due to the large excess amount of Fe3+ ions thought to occupy active sites on the surface of N-TiO2 and to function as a recombination center between electrons and holes.59
4 Conclusions
Single doped, co-doped and tri-doped TiO2 with iron, boron and nitrogen are successfully synthesized by hydrothermal method. All the doping catalysts have higher photocatalytic activities than pure TiO2 under visible light irradiation. Co-doped TiO2 presents higher photoactivities than single doped TiO2 while the tri-doping is better than co-doping. The (0.5Fe,N,B)-TiO2 catalyst shows the highest degradation rate in all the doped catalysts. The band gap of the doped catalysts is narrowed by the formation of nonmetal dopant levels near the VB and Fe3+ impurity level under the CB responsible for the enhancement of photocatalytic activities under visible light. In addition, the synergistic effect of Ti–B–N formed on the surface of the catalysts can act as an efficient co-catalyst to inhibit the recombination of photogenerated electrons and holes, which can improve the photo-performance of (B,N)-TiO2 and (Fe,N,B)-TiO2.Acknowledgements
This work has been supported by National Nature Science Foundation of China (20773039, 20977030); National Basic Research Program of China (973 Program, 2007CB613301, 2010CB732306), and the Ministry of Science and Technology of China (2007AA05Z303).References
- M. R. Hoffmann, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef CAS.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1 CrossRef CAS.
- C. Hu, J. C. Yu, Z. Hao and P. K. Wong, Appl. Catal., B, 2003, 42, 47 CrossRef CAS.
- J. C. Yu, W. Ho, J. Yu, H. Yip, P. K. Wong and J. Zhao, Environ. Sci. Technol., 2005, 39, 1175 CrossRef CAS.
- H. Kisch, S. Sakthivel, M. Janczarek and D. Mitoraj, J. Phys. Chem. C, 2007, 111, 11445 CrossRef CAS.
- S. In, A. Orlov, R. Berg, F. Garcia, S. Pedrosa-Jimenez, M. S. Tikhov, D. S. Wright and R. M. Lambert, J. Am. Chem. Soc., 2007, 129, 13790 CrossRef CAS.
- J. F. Zhu, Z. G. Deng, F. Chen, J. L. Zhang, H. J. Chen, M. Anpo and J. Z. Huang, Appl. Catal., B, 2006, 62, 329 CrossRef CAS.
- Y. M. Wu, J. L. Zhang, L. Xiao and F. Chen, Appl. Catal., B, 2009, 88, 525 CrossRef CAS.
- Y. M. Wu, J. L. Zhang, L. Xiao and F. Chen, J. Phys. Chem. C, 2009, 113, 14689 CrossRef CAS.
- M. Y. Xing, J. L. Zhang and F. Chen, Appl. Catal., B, 2009, 89, 563 CrossRef CAS.
- M. Y. Xing, J. L. Zhang and F. Chen, J. Phys. Chem. C, 2009, 113, 12848 CrossRef CAS.
- C. Burda, Y. B. Lou, X. B. Chen, A. C. S. Samia, J. Stout and J. L. Gole, Nano Lett., 2003, 3, 1049 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef CAS.
- B. Tryba, T. Tsumura, M. Janus, A. W. Morawski and M. Inagaki, Appl. Catal., B, 2004, 50, 177 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- T. Ohno, Z. Miyamoto, K. Nishijima, H. Kanemitsu and X. Y. Feng, Appl. Catal., A, 2006, 302, 62 CrossRef CAS.
- W. Zhao, W. Ma, C. Chen, J. Zhao and Z. Shuai, J. Am. Chem. Soc., 2004, 126, 4782 CrossRef CAS.
- K. Y. Jung, S. B. Park and S. K. Ihm, Appl. Catal., B, 2004, 51, 239 CrossRef CAS.
- D. Li, H. Haneda, S. Hishita and N. Ohashi, Chem. Mater., 2005, 17, 2588 CrossRef CAS.
- D. Li, H. Haneda, S. Hishita and N. Ohashi, Chem. Mater., 2005, 17, 2596 CrossRef CAS.
- D. M. Chen, Z. Y. Jiang, J. Q. Geng, Q. Wang and D. Yang, Ind. Eng. Chem. Res., 2007, 46, 2741 CrossRef CAS.
- G. Liu, Y. N. Zhao, C. H. Sun, F. Li, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 4516 CrossRef CAS.
- J. Kiwi and M. Graetzel, J. Phys. Chem., 1986, 90, 637 CrossRef CAS.
- K. E. Karakitsou and X. E. Verykios, J. Phys. Chem., 1993, 97, 1184 CrossRef CAS.
- J. F. Zhu, F. Chen, J. L. Zhang, H. J. Chen and M. Anpo, J. Mol. Catal. A., 2004, 216, 35 CAS.
- K. Nagaveni, M. S. Hegde and G. Madras, J. Phys. Chem. B, 2004, 108, 20204 CrossRef CAS.
- S. Klosek and D. Raftery, J. Phys. Chem. B, 2001, 105, 2815 CrossRef CAS.
- H. Kisch, L. Zang, C. Lange, W. F. Maier, C. Antonius and D. Meissner, Angew. Chem., Int. Ed., 1998, 37, 3034 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669 CrossRef.
- H. Yamashita, M. Harada, J. Misaka, M. Takeuchi, B. Neppolian and M. Anpo, Catal. Today, 2003, 84, 191 CrossRef CAS.
- J. A. Navio, G. Colon, M. I. Litter and G. N. Bianco, J. Mol. Catal. A: Chem., 1996, 106, 267 CrossRef CAS.
- Y. H. Zhang, S. G. Ebbinghaus, A. Weidenkaff, T. Kurz, P. J. Nidda, K. V. Klar, M. Guengerich and A. Reller, Chem. Mater., 2003, 15, 40283.
- J. Soria, J. C. Conesa, V. Augugliaro, L. Palmisano, M. Schiavello and A. Sclafani, J. Phys. Chem., 1991, 95, 274 CrossRef CAS.
- M. I. Litter and J. A. Navio, J. Photochem. Photobiol., A, 1996, 98, 171 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 108, 79 CrossRef CAS.
- E. Piera, M. I. Tejedor, M. E. Zorn and M. A. Anderson, Appl. Catal., B, 2003, 46, 671 CrossRef CAS.
- J. Navio, G. Colon, M. Macias, C. Real and M. Litter, Appl. Catal., A, 1999, 177, 111 CrossRef CAS.
- J. F. Zhu, F. Chen, J. L. Zhang, H. J. H. J. Chen and M. Anpo, J. Photochem. Photobiol., A, 2006, 180, 196 CrossRef CAS.
- C. Adan, A. Bahamonde, M. Fernandez-Garcia and A. Martinez-Arias, Appl. Catal., B, 2007, 72, 11 CrossRef CAS.
- R. Khana, S. W. Kima, T. J. Kima and C. M. Namb, Mater. Chem. Phys., 2008, 112, 167 CrossRef.
- Y. Cong, J. L. Zhang, F. Chen, M. Anpo and D. He, J. Phys. Chem. C, 2007, 111, 10618 CrossRef CAS.
- J. Lin, Y. Lin, P. Liu, M. J. Meziani, L. F. Allard and Y. P. Sun, J. Am. Chem. Soc., 2002, 124, 11514 CrossRef CAS.
- Y. Cong, J. L. Zhang, F. Chen and M. Anpo, J. Phys. Chem. C, 2007, 111, 6976 CrossRef CAS.
- L. Zhang, Y. F. Zhu, Y. He, W. Li and H. B. Sun, Appl. Catal., B, 2003, 40, 287 CrossRef CAS.
- J. G. Yu, H. Yu, C. H. Ao, S. C. Lee, J. C. Yu and W. Ho, Thin Solid Films, 2006, 496, 273 CrossRef CAS.
- F. Iacomi, D. Mardare, M. N. Grecu, D. Macovei and I. Vida-Simiti, Surf. Sci., 2007, 601, 2692 CrossRef CAS.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Appl. Surf. Sci., 2000, 165, 288 CrossRef CAS.
- T. Morikawa, T. Ohwaki, K. Suzuki, S. Moribe and S. Tero-Kubota, Appl. Catal., B, 2008, 83, 56 CrossRef CAS.
- Q. C. Ling, J. Z. Sun, Q. Y. Zhou, Q. Zhao and H. Ren, J. Photochem. Photobiol., A, 2008, 200, 141 CrossRef CAS.
- G. Mavel, J. Escard, P. Costa and J. Castaing, Surf. Sci., 1973, 35, 109 CrossRef CAS.
- D. J. Joyner, O. Johnson and D. M. Hercules, J. Am. Chem. Soc., 1980, 102, 1910 CrossRef CAS.
- Y. Huang, W. K. Ho, Z. H. Ai, X. Song, L. Z. Zhang and S. C. Lee, Appl. Catal., B, 2009, 89, 398 CrossRef CAS.
- D. Chen, D. Yang, Q. Wang and Z. Jiang, Ind. Eng. Chem. Res., 2006, 45, 4110 CrossRef CAS.
- J. Zhang, Y. Hu, M. Matsuoka, H. Yamashita, M. Minagawa, H. Hidaka and M. Anpo, J. Phys. Chem. B, 2001, 105, 8395 CrossRef CAS.
- H. B. Jiang and L. Gao, Mater. Chem. Phys., 2003, 77, 878 CrossRef CAS.
- L. Q. Wang, D. R. Baer and M. H. Engelhard, Surf. Sci., 1994, 320, 295 CrossRef CAS.
- Y. Adachi, S. Kohiki, K. Wagatsuma and M. Oku, J. Appl. Phys., 1998, 84, 2123 CrossRef CAS.
- T. Ohno, N. Murakami, T. Chiyoya and T. Tsubota, Appl. Catal., A, 2008, 348, 148 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |