Dendrimer-based organic/inorganic hybrid nanoparticles in biomedical applications
Mingwu
Shen
ab and
Xiangyang
Shi
*ab
aState Key Laboratory for Modification of Chemical Fibers and Polymer Materials, Donghua University, Shanghai, 201620, China
bCollege of Chemistry, Chemical Engineering and Biotechnology, Donghua University, Shanghai, 201620, China. E-mail: xshi@dhu.edu.cn
First published on 7th July 2010
Abstract
This review reports some recent advances on the synthesis, self-assembly, and biofunctionalization of various dendrimer-based organic/inorganic hybrid nanoparticles (NPs) for various biomedical applications, including but not limited to protein immobilization, gene delivery, and molecular diagnosis. In particular, targeted molecular imaging of cancer using dendrimer-based organic/inorganic hybrid NPs will be introduced in detail.
 Mingwu Shen | Mingwu Shen received her PhD degree in 2001 from Tsinghua University. Afterwards, she went to the University of Michigan, Ann Arbor as a visiting scholar and a research area specialist intermediate. Now she is an associate professor of biomedical engineering at Donghua University. Mingwu Shen's current research interests include dendrimer-based nanoparticles for medical imaging applications, and the development of nanofiber-based technology for biomedical and environmental applications. |
 Xiangyang Shi | Xiangyang Shi obtained his PhD degree in 1998 from the Chinese Academy of Sciences. From 2002–2008, he was appointed as a research fellow, research associate II, research investigator, and research assistant professor in Michigan Nanotechnology Institute for Medicine and Biological Sciences, University of Michigan, Ann Arbor. In September 2008, he joined Donghua University as a full professor. He has published more than 100 peer-reviewed papers. His current research interests are focused on dendrimer-based nanomedicine, and electrospun polymer nanofiber-based technology for applications in both regenerative medicine and environmental sciences. |
1. Introduction
In recent years, dendrimer-based organic/inorganic hybrid nanoparticles (NPs) have received immense scientific and technological interest because of their promising applications in a broad range of fields, such as catalysis, optics, electronics, and biomedical applications. Dendrimers are a class of highly branched, monodispersed, synthetic macromolecules with well-defined composition and structure.1–4 The unique properties of dendrimers as well as their excellent biocompatibility and non-immunogenicity5,6 lead to the synthesis of various dendrimer-based organic/inorganic hybrid composite NPs for a range of biomedical applications. The major advantage of using dendrimers to generate organic/inorganic hybrid NPs is their tunable surface chemistry, providing many opportunities for the functionalization of the NP surfaces.Dendrimers have been used as ideal templates or stabilizers for the synthesis and modification of various metal, metal oxide, and semiconductor NPs. Dendrimer-entrapped NPs (DENPs) are formed using dendrimers as templates, where each metal or other inorganic NP is entrapped within each dendrimer molecule, while dendrimer-stabilized NPs (DSNPs) are formed using dendrimers as stabilizers, where each metal or other inorganic NP is surrounded or protected by multiple dendrimer molecules on its surface. Detailed synthesis, characterization, and applications of DENPs and DSNPs in catalysis, optics, or other applications not related to biomedical sciences can be found in several reviews.7–11
The tunable surface chemistry of dendrimers allows their assembly onto various NPs. For example, through electrostatic interaction, dendrimers or biofunctionalized dendrimers can be assembled onto magnetic iron oxide NPs to stabilize or functionalize the NPs for biomedical applications.12–15 The dendrimer molecules assembled onto NPs can be further modified or functionalized, rendering the particles good colloidal stability, biocompatibility, and biofunctionality.14–16 In addition, amine-functionalized NPs can be used as cores for step-by-step divergent growth of dendrimers of different generations onto the particle surfaces.17–19 The formed nanoparticle core/dendrimer shell composite particles can be further used as a platform for biomedical applications.20
This review provides a glimpse of how various dendrimer-based organic/inorganic hybrid NPs have been designed and used in biomedical applications. Those contributions related to the dendrimer-based organic/inorganic hybrid NPs, but not being applied in biomedical sciences might be mentioned, but are not highlighted. In addition, we consider that a dendron is only a part of an entire dendrimeric nanostructure. In other words, the dendron is the reduced form of its original dendrimer. Therefore, those studies related to dendron-stabilized or dendron-modified organic/inorganic hybrid nanoparticles21–25 will not be included in this review. The review begins with a short introduction of various dendrimer-based organic/inorganic hybrid NPs, and ends with the discussion of the outlook and prospects. It should be stressed that this is not a comprehensive review, but rather discusses key developments of various dendrimer-based organic/inorganic hybrid NPs in biomedical applications, especially in the molecular imaging of cancer.
2. Dendrimer-entrapped nanoparticles (DENPs)
The unique interior feature of dendrimers allows them to be used as templates for synthesis of various NPs entrapped within the dendrimers. Likewise, the tunable surface chemistry of the dendrimers permits the surface functionalization with various biomolecules, providing a platform for biomedical applications. In this section, the synthesis of DENPs and the design of biofunctionalized DENPs for biomedical applications are discussed.2.1. Synthesis of DENPs
DENPs are often formed using fast reduction (e.g., sodium borohydride reduction) and nucleation chemistry. The formed DENPs are usually smaller than 5 nm. The formation of DENPs consists of two steps. In the first step, metal ions are preorganized by the dendrimer host through ligand–metal-ion interactions, salt formation, acid–base and donor–acceptor interactions, covalent bond formation, steric confinement, various types of weaker forces (van der Waals, hydrogen bonding, etc), and combinations thereof.7 Meanwhile, due to many possible overlapping equilibrium processes, the binding of metal ions within dendrimer host usually appears as a non-stoichiometric process. The preorganization results in dendrimer–metal complexes, which are in dynamic equilibrium with the template and metal ions. The dynamic equilibrium permits equal distribution of the metal ions between all the equivalent ligands in the dendrimer molecules and diffusion provides a homogenous distribution of metal ions among the dendrimer template. In the second step, a reduction of preorganized metal ions results in the formation of DENPs. A scheme for the synthesis of Au DENPs26–28 (Scheme 1) gives an example how the two steps lead to the formation of metal DENPs.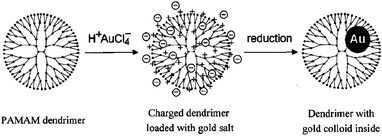 | ||
| Scheme 1 Dendrimer nanotemplating in aqueous solution. In the first step, the dendrimer is loaded with a precursor salt (H+AuCl4−), resulting in a charged dendrimer with the precursor as counterions. In the second step, the chemical reduction is performed which yields a colloid inside the dendrimer. Reprinted from Ref. 26. Not subject to US Copyright. Published 2000 American Chemical Society. | ||
A wide range of metal ions including Cu2+, Au3+, Ag+, Pd2+, Pt2+, Ni2+, Ru3+ can be preorganized with dendrimers, especially poly(amidoamine) (PAMAM) dendrimers to form metal DENPs.7,29–31 Dendrimer-entrapped semiconductor quantum dots (e.g., CdS and CdSe) can also be prepared by reacting Cd(II)–dendrimer complexes with S(II) and Se(II) ions.32–34 Likewise, bimetallic DENPs (e.g., NiAu with a size of 3 nm) can be prepared by co-reduction of the metal ions in the presence of dendrimer templates.35 In a recent report,36 Satoh et al. prepared titania NPs with a size of 1.8–2.4 nm using dendrimers as templates. The process involved complexation of a titania precursor with dendrimers, followed by a hydrolysis step.
Compared with regular/classic composite materials, DENPs possess nanometre-sized inorganic domains and organic hosts, which make them display unique physical and chemical properties that are characteristic of both the nanosized host and the nanodispersed guest.7,8 A range of physical properties come from the nanodispersed inorganic domains, instead of PAMAM dendrimers alone, which affords DENPs with many unique applications in a wide range of areas, including catalysis, optics, environmental, sensing, etc. While many unique properties and applications of DENPs are derived from the nanodispersed inorganic domain, the dendrimer in itself is also indispensable in the stabilization of the inorganic domains. In DENPs, most of the interaction between guest atoms and their microenvironment (metal–metal and metal–solvent interactions) are substituted with the metal–dendrimer and dendrimer–solvent interactions, which makes the DENP solutions stable for a relatively long time in appropriate solvent systems. In this context, prefunctionalized dendrimers have been used as templates for the synthesis of various metal DENPs in order to attain certain functionalities.37–42
2.2. Surface modification of DENPs for molecular imaging applications
For biological applications, the surface of DENPs has to be modified in order to avoid toxicity and non-specific cell membrane binding. Gold NPs have recently received immense scientific and technological interest because of their extensive applications in biology, catalysis, and nanotechnology.43,44 Although functionalized dendrimers have been used to prepare dendrimer-entrapped gold or other noble metal NPs with different functionalities,39,45 in most circumstances dendrimer-entrapped gold NPs (Au DENPs) are prepared using amine-terminated PAMAM dendrimers. This is due to the commercial availability of this material, but this yields particles with high cytotoxicity and non-specific membrane binding due to the amine surface on the dendrimers, limiting the biological application of these particles. Preparation of non-toxic, biocompatible Au DENPs is of great importance for applications in various biological systems.It is well documented that decreasing the surface charge of amine-terminated PAMAM dendrimers towards neutral reduces their toxicity.46–50 We have developed a new, facile approach to surface modification of Au DENPs by replacing the terminal amine groups of the dendrimers after the entrapment of Au NPs.51 Au DENPs formed using ethylenediamine (EDA) core amine-terminated generation 5 PAMAM dendrimers (G5·NH2) as templates were reacted with acetic anhydride or glycidol molecules to form acetamide or hydroxyl-functionalized Au DENPs (see Scheme 2). The formed Au DENPs after surface functionalization are stable, water-soluble, and display similar sizes (ranging from 2.0 ± 0.4 to 2.4 ± 0.5 nm, Fig. 1), size distributions, and optical properties to the original amine-terminated DENPs, while the surface charge changes to less positive or close to neutral (from {(Au0)51.2-G5·NH2} at +36.86 mV to {(Au0)51.2-G5·NGlyOH} at +23.47 mV and {(Au0)51.2-G5·NHAc} at +4.27 mV) and the biocompatibility is significantly improved. Using this approach, one can directly tailor the surface functionalities of preformed Au DENPs.
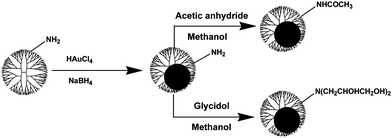 | ||
| Scheme 2 Reactions to modifying Au DENPs prepared using amine-terminated G5·NH2 dendrimers as templates. Reproduced from ref. 51 by permission of the Royal Society of Chemistry. | ||
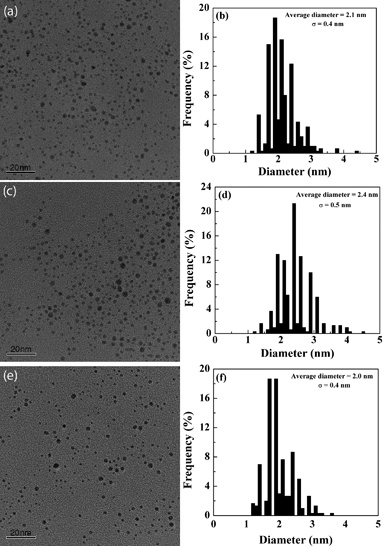 | ||
| Fig. 1 TEM micrographs of the {(Au0)51.2-G5·NH2} (a), {(Au0)51.2-G5·NHAc} (c), {(Au0)51.2-G5·NGlyOH} (e) DENPs; (b), (d) and (f) are size distribution histograms of the {(Au0)51.2-G5·NH2}, {(Au0)51.2-G5·NHAc}, and {(Au0)51.2-G5·NGlyOH} DENPs, respectively. Reproduced from ref. 51 by permission of the Royal Society of Chemistry. | ||
Using the same strategy, dendrimer-entrapped silver nanoparticles (Ag DENPs) can be acetylated.52 In sharp contrast to the acetylation of Au DENPs, Ag DENPs are more sensitive to the acetylation reaction. The size and size distribution significantly change when different degrees of acetylation reaction were applied. We show that the size of partially acetylated Ag DENPs displays a bimodal distribution (2.9 nm and 11.0 nm), whereas the pristine amine-terminated Ag DENPs and the completely acetylated Ag DENPs are relatively monodisperse with a size of 2.9 nm and 11.0 nm, respectively. It indicates that complete acetylation transfers Ag DENPs to dendrimer-stabilized Ag NPs (Ag DSNPs). This study suggests that changing the terminal amine groups of the host dendrimers to acetamide groups significantly affects the reorganization and nucleation of Ag NPs entrapped in the dendrimers. In contrast, in the case of Au DENPs with similar metal/dendrimer molar feed ratio, even after different acetylation, the size and size distribution do not change. This difference is presumably due to the higher reactivity of the silver (Ag0) than the gold (Au0). The higher reactivity of the silver clusters favors aggregation, resulting in the formation of larger NPs compared to those of the gold.27,53 The size and morphology changes of Au and Ag DENPs after acetylation were further confirmed by molecular dynamics (MD) simulations. Understanding the structural changes of Au and Ag DENPs upon acetylation is important for further efforts to develop multifunctional metal DENP-based nanodevices for various biological applications.
In the area of targeted cancer imaging and therapeutics, one great challenge is that the used NPs lack specific binding with cancerous cells. The ability to chemically functionalize preformed Au DENPs without significantly changing their sizes and size distributions led us to develop these Au DENPs as a multifunctional platform for cancer cell targeting, imaging, and treatment as we have done with dendrimers.47,54 We demonstrate that Au DENPs can be covalently linked with targeting ligands and imaging molecules for cancer cell targeting and imaging (Fig. 2a).55 One of the key steps in the preparation of folic acid (FA)- and fluorescein isothiocyanate (FI)-modified Au DENPs is to keep the surface charges on the particles neutral in order to avoid toxicity and non-specific binding. This can be accomplished by a final acetylation step to convert the remaining amine groups of G5·NH2 dendrimers to acetamides (Fig. 2a). Zeta potential measurements show that after the final acetylation step, the surface potentials of the formed {(Au0)51.2-G5-FI-Ac} (ξ = −1.11 mV) and {(Au0)51.2-G5-FI-FA-Ac} (ξ = −2.30 mV) DENPs (Ac denotes acetyl) are close to neutral, indicating the success of the acetylation reaction. The average numbers of FI and FA moieties conjugated onto each Au DENP were estimated to be 4.0 and 4.5, respectively. MD simulation studies show that most FA moieties (pink spheres in Fig. 2b) extend substantially outward, thus becoming available for interaction with the folic acid receptors (FAR) on the surface of the cells. Most FI moieties (green spheres in Fig. 2b) are distant from the Au sphere, minimizing the fluorescence quenching effect with the Au.
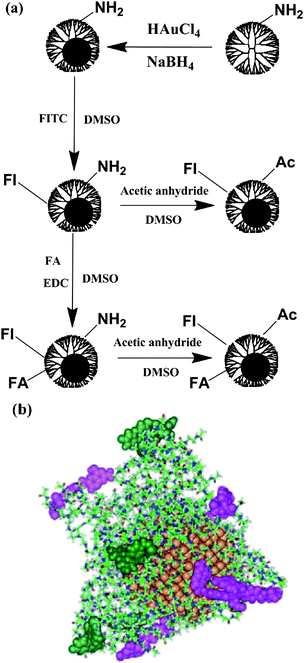 | ||
| Fig. 2 (a) Schematic representation of the reactions involved in modifying Au DENPs for cancer cell targeting and imaging. (b) Selected equilibrated configurations of {(Au0)51.2-G5-FI-FA-Ac} DENPs. Atoms in the PAMAM dendrimer platform are shown as sticks, gold atoms as gold spheres, atoms comprising FA as pink spheres, and atoms for FI as green spheres. Ref. 55; copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
Au DENPs linked with defined numbers of FA and FI molecules are water-soluble, stable, and biocompatible. FA has been extensively investigated for targeting various cancer cells, including ovary, kidney, uterus, testis, brain, colon, lung, and myelocytic blood that overexpress FAR.56–59 We show that the FA- and FI-modified Au DENPs can specifically bind to and be internalized by KB cells that overexpress high affinity FAR (Fig. 3) through flow cytometry and confocal microscopic imaging studies, similar to the dendrimers with the same surface functionalities but without Au NPs.60
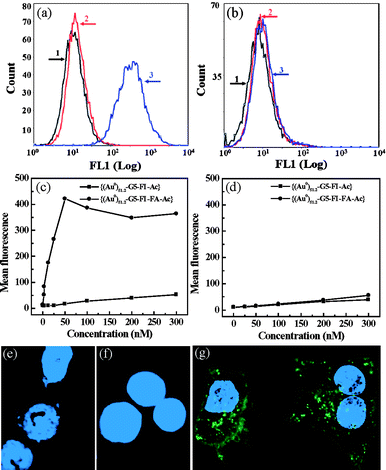 | ||
| Fig. 3 Flow cytometric and confocal microscopic studies of the binding of functionalized Au DENPs with KB cells. (a) and (b) binding of {(Au0)51.2-G5-FI-Ac} and {(Au0)51.2-G5-FI-FA-Ac} DENPs (25 nM) with KB cells with high- and low-levels of FAR, respectively. 1. PBS control; 2. {(Au0)51.2-G5-FI-Ac}; 3. {(Au0)51.2-G5-FI-FA-Ac}. (c) and (d) dose-dependent binding of {(Au0)51.2-G5-FI-Ac} and {(Au0)51.2-G5-FI-FA-Ac} DENPs with KB cells expressing high- and low-levels of FAR, respectively. (e)–(g) confocal microscopic images of KB cells with high-level FAR treated with PBS buffer (e), {(Au0)51.2-G5-FI-Ac} (25 nM) (f), and {(Au0)51.2-G5-FI-FA-Ac} (25 nM) (g) DENPs for 2 h, respectively. Ref. 55; copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
One major advantage of using functionalized Au DENPs as a platform to image cancer is their ability to differentiate cancer cells from surrounding cells or tissues due to the high electron density of Au element. Dendrimers without entrapped metal NPs cannot achieve this goal.47,54 By using the transmission electron microscopy (TEM) imaging technique, we can clarify the distribution of functionalized Au DENPs in different compartments inside targeted cells. This also aids in understanding the mechanism for targeted drug delivery and therapeutics, using dendrimer-based nanodevices. Upon 2 h incubation of functionalized Au DENPs, the FA-modified {(Au0)51.2-G5-FI-FA-Ac} DENPs were predominantly located in the lysosomes of KB cells with high-level FAR expression (Fig. 4a and 4b). We also observed that a small portion of {(Au0)51.2-G5-FI-FA-Ac} DENPs situated in vacuoles and the nucleus. However, we did not see any uptake of the {(Au0)51.2-G5-FI-Ac} DENPs without FA modification in the lysosomes of the same KB cells (Fig. 4c). A very small quantity of {(Au0)51.2-G5-FI-Ac} DENPs were observed in the vacuoles of some cells, and this was undetectable using confocal microscopy. The TEM studies highlighted the high specificity of FA-modified Au DENPs for targeting KB cells with high-level FAR expression, and corroborate the confocal imaging data. These findings document a facile approach to use Au DENPs as a platform for the targeting and imaging of cancer cells.
 | ||
| Fig. 4 TEM images of cellular uptake of Au DENPs. (a) and (c) TEM images of KB cells with high-level FAR treated with {(Au0)51.2-G5-FI-FA-Ac} (a and b) and {(Au0)51.2-G5-FI-Ac} DENPs (c) for 2 h, respectively. (b) A magnified area of the lysosome of the same cell shown in (a). The concentration for both Au DENPs is maintained at 50 nM. Ref. 55; copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
Besides the fact that preformed amine-terminated Au DENPs can be modified with targeting ligands and dye molecules for cancer targeting and imaging applications, dendrimers functionalized with targeting ligands and imaging molecules can also be used as templates to entrap metal NPs for targeted cancer imaging. In general, the synthesis of multifunctional Au DENPs from amine-terminated ones may have a significant challenge as synthetic manipulations often alter the surface characteristics of the particles or change the stability of the entrapped metal NPs, leading to aggregation or precipitation. Despite this, tumor targeted Au DENPs (<5 nm in diameter) remain very attractive for biomedical applications as they are likely to pass through large blood vessels or connective tissue matrices to target cancer cells and their vasculature, while at the same time remaining below the renal filtration threshold allowing excretion through the kidney.55 In a recent study, Baker and coworkers described the templated synthesis and characterization of monodisperse multifunctional Au DENPs from a partially acetylated FI-labeled G5 PAMAM conjugated with –cRGDyK peptides (G5-FI-RGD).40 Arg-Gly-Asp (RGD) peptide motifs have been identified by phage display studies as high affinity αVβ3 selective ligands,61 which is a specific marker of the neovasculature normally found on the luminal surface of endothelial cells during angiogenesis.62 Thus, these molecules appear to be an excellent ligand for targeting and treatment of cancer cells through interaction with αVβ3 integrin that is present on numerous cell types associated with cancer. In their study, 74 equivalent of HAuCl4 per dendrimer were mixed with an aqueous solution of G5-Fl-RGD to form a stable complex, followed by formation of {(Au0)74-G5-FI-RGD} DENPs via NaBH4 reduction. The specific targeting and internalization of the formed {(Au0)74-G5-FI-RGD} DENPs was demonstrated by flow cytometric analysis and confocal microscopy. In addition, preincubation with excess peptide almost completely blocked the binding and uptake of either the dendrimer conjugate or gold. This observation suggests that the Au DENPs are stable and specifically taken up by αVβ3 integrin over-expressing cell lines. In another study, Maly and coworkers42 utilized biotinylated, hydroxyl-terminated G7 PAMAM dendrimers as templates to synthesize Ag DENPs via NaBH4 reduction. They show that stable biotinylated Ag DENPs can be formed. Furthermore, the selective biorecognition function of the biotin is not affected by the AgNPs' synthesis step, which allows a potential application of Ag DENPs as biospecific labels in various bioanalytical assays, or potentially as fluorescent cell biomarkers.
Because of the strong X-ray attenuation characteristics of Au, AuNPs have recently been used as contrast agents for X-ray computed tomography (CT) imaging.63–68 In comparison with conventional iodine-based CT contrast agents, AuNPs have several major advantages. First, Au has a higher X-ray absorption coefficient than iodine and could theoretically induce greater contrast enhancement in CT imaging.68 Second, AuNPs are reported to be biocompatible.69,70 Third, properly treated AuNPs can have a longer circulation time than iodine-based molecular contrast agents and may provide enhanced signal contrast for a longer time after delivery to a living organism,66 which makes clinical observation and diagnosis of diseases more convenient and more patient-friendly. More importantly, it is relatively easy to modify the surface of AuNPs with various functional groups (e.g., targeting agents or specific biomarkers),55 which renders the resultant AuNPs a class of targeted CT imaging agents with enhanced imaging contrast and efficacy. In a recent study,71 we demonstrate that amine-terminated Au DENPs with dendrimer/Au atom molar ratios at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25, 1:50, 1:75, 1:100, 1:125, 1:150, 1:175, and 1:200 can be synthesized using G5·NH2 dendrimers as templates. By varying the molar ratio between gold salt to G5·NH2 at the above range, Au DENPs with a size range of 2–4 nm can be prepared. The synthesized Au DENPs are not only stable in water, PBS buffer, and cell culture media but also at different temperatures (from 4 to 50 °C) and different pH conditions (pH 5–8). X-Ray absorption coefficient measurements show that the attenuation of Au DENPs is much higher than that of the iodine-based contrast agent at the same molar concentration of the active element (Au versus iodine) (Fig. 5a). Furthermore, CT scanning showed significant enhancement in mice injected subcutaneously with Au DENPs (Fig. 5b), and intravenous injection of acetylated Au DENPs enabled the X-ray CT imaging of mice, rendering them a promising contrast agent in CT imaging applications. Taking into consideration the abundant amine groups on the surface of Au DENPs and their capability for further chemical coupling with various biological molecules,40,55 Au DENPs are expected to be a versatile platform for the construction of multifunctional theranostic nanoagents for targeted drug delivery and CT imaging of diseases.
:25, 1:50, 1:75, 1:100, 1:125, 1:150, 1:175, and 1:200 can be synthesized using G5·NH2 dendrimers as templates. By varying the molar ratio between gold salt to G5·NH2 at the above range, Au DENPs with a size range of 2–4 nm can be prepared. The synthesized Au DENPs are not only stable in water, PBS buffer, and cell culture media but also at different temperatures (from 4 to 50 °C) and different pH conditions (pH 5–8). X-Ray absorption coefficient measurements show that the attenuation of Au DENPs is much higher than that of the iodine-based contrast agent at the same molar concentration of the active element (Au versus iodine) (Fig. 5a). Furthermore, CT scanning showed significant enhancement in mice injected subcutaneously with Au DENPs (Fig. 5b), and intravenous injection of acetylated Au DENPs enabled the X-ray CT imaging of mice, rendering them a promising contrast agent in CT imaging applications. Taking into consideration the abundant amine groups on the surface of Au DENPs and their capability for further chemical coupling with various biological molecules,40,55 Au DENPs are expected to be a versatile platform for the construction of multifunctional theranostic nanoagents for targeted drug delivery and CT imaging of diseases.
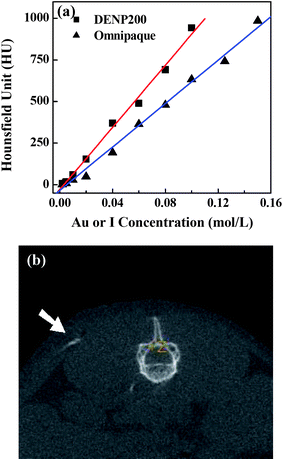 | ||
| Fig. 5 (a) X-Ray attenuation (HU) of DENP200 and Omnipaque as a function of the molar concentration of active element (Au or iodine). (b) A CT image of a mouse with 10 μL of DENP200 (0.02 mol L−1) subcutaneously injected into its back. The white arrow points to the Au DENPs injection region. Reprinted with permission from ref. 71. Copyright 2010 American Chemical Society. | ||
3. Dendrimer-stabilized nanoparticles (DSNPs)
DSNPs are referred to a nanostructure, where one metal or other inorganic NP is surrounded with multiple dendrimer molecules. In general, the DSNPs are formed in the presence of dendrimers and the formation process under appropriate conditions is in situ. Compared with DENPs that generally have a size smaller than 5 nm, the size of DSNPs are often larger than 5 nm. In additon, preformed NPs modified with dendrimers, also termed dendrimer-assembled NPs (DANPs) will be discussed in Section 4. Most of the research work related to DSNPs for biomedical applications is related to noble metal DSNPs (e.g., Au and Ag) and magnetic iron oxide DSNPs. Other inorganic metal sulfide DSNPs can also be formed using dendrimers or functionalized dendrimers as stabilizers.42,72–75 The formed metal sulfide DSNPs do not have associated biomedical applications, therefore details will not be discussed in this review.3.1. Synthesis and biomedical applications of metal DSNPs
Metal DSNPs are usually formed under mild reduction conditions to assist slow nucleation of the particles. The formation of metal DSNPs also depends on the structure of dendrimers used. In some cases, if low generation dendrimers (G1–G3) are used as templates, even fast reduction and nucleation can still afford the formation of metal DSNPs (instead of DENPs) because the limited terminal amines and open structures of dendrimers cannot entrap metal NPs inside the dendrimers.9,10,76Several groups pioneered the research on Au DSNPs.26,30,77,78 The preparation of Au DSNPs usually involves complexation of gold salts (e.g. HAuCl4) with PAMAM dendrimers, followed by physical or chemical reduction.9,10,77 It is well established that the size of the Au DSNPs is mainly dependent on the molar ratio between dendrimers and Au atoms.9 The mechanistic studies show that dendrimer terminal amines are extremely effective in the stabilization of Au NPs.27,78 Although there are a number of reports showing that DSNPs can be formed by simple thermo treatment,79–81 UV-irradiation,30,82 laser ablation83 or γ-ray irradiation84 of the dendrimer-metal complexes, a majority of the work related to the synthesis of DSNPs were performed by chemical reduction. There are also some reports related to a spontaneous formation of DSNPs by simply mixing the dendirmer solution with metal salts at room temperature.85–87
In our previous work,88 we utilized amine-terminated PAMAM dendrimers of generation 2 through 6 as stabilizers to synthesize Au DSNPs by hydrazine reduction chemistry. For all Au-DSNPs, the molar ratios between dendrimer terminal amines and Au atoms were kept consistent at 1:0.4. Fig. 6a shows the UV-Vis spectra of Au DSNPs prepared using G2·NH2 through G6·NH2 as stabilizers. The plasmon peak at around 525 nm, which is attributed to collective oscillation of free electrons in gold NPs,89,90 is clearly observed for all samples. Fig. 6b shows the fluorescence spectra of Au DSNPs and commercial Au colloid particles (5 nm and 100 nm). All Au DSNPs were found to be fluorescent and display strong blue photoluminescence. The maximum excitation and emission wavelengths were around 397 nm and 458 nm, respectively, in agreement with the literature data.91 In contrast, commercial gold colloids (5 nm and 100 nm) that are prepared using citric acid reduction and protection approach do not exhibit fluorescence emission, suggesting that the dendrimer stabilizers contribute to the fluorescence properties of the formed Au DSNPs.92 The fluorescent properties of the formed Au DSNPs make them potentially useful as fluorescent markers for cell labeling and biological sensing studies.
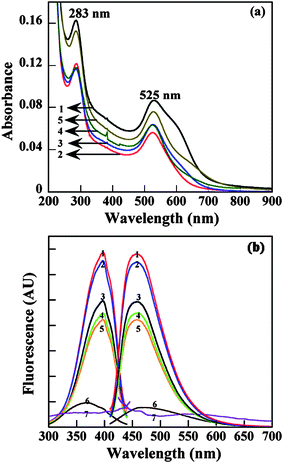 | ||
| Fig. 6 UV-Vis (a) and fluorescence (b) spectra of Au DSNPs. Curve 1, 2, 3, 4, and 5 correspond to {(Au0)6-G2·NH2}, {(Au0)12-G3.NH2}, {(Au0)24-G4.NH2}, {(Au0)57-G5·NH2}, and {(Au0)98-G6·NH2}, respectively. In (b), 6 and 7 indicate gold colloids with diameter of 5 and 100 nm, respectively. Reproduced from ref. 88 by permission of IOP Publishing Limited. | ||
The size distribution and morphology of the synthesized Au DSNPs were studied by TEM. Fig. 7 shows TEM images of Au DSNPs prepared using PAMAM dendrimers of different generations. The sizes of the formed Au DSNPs are 15.4 ± 5.8 nm, 12.0 ± 2.8 nm, 9.1 ± 3.2 nm, 8.6 ± 2.8 nm, and 7.1 ± 1.9 nm for {(Au0)6-G2·NH2}, {(Au0)12-G3.NH2}, {(Au0)24-G4.NH2}, {(Au0)57-G5·NH2}, and {(Au0)98-G6·NH2}, respectively. It is clear that all the Au DSNPs are relatively monodispersed except {(Au0)6-G2·NH2}. {(Au0)6-G2·NH2} displays larger size and higher polydispersity, which is attributed to limited number of amines of G2·NH2 dendrimers to stabilize Au NPs.93 The size of the Au DSNPs decreases with the increase of the number of dendrimer generations (Fig. 7f), suggesting the different nucleation and growth mechanisms for gold nanocrystals in the presence of PAMAM dendrimers. At basic pH conditions (pH ≈ 10.4 when dendrimers are dissolved in water), AuCl4− anions bind preferably to the protonated amines of PAMAM dendrimers through electrostatic interaction. Larger generation PAMAM dendrimers have denser structures that would significantly limit the nucleation, movement, and growth of gold nanocrystals. In contrast, smaller generation PAMAMs have relatively open structures, which hinder the growth of gold nanocrystals less significantly than larger generation PAMAMs. The synthesized Au DSNPs are significantly larger than Au DENPs reported in the literature.26–28,51,93 This is because the reduction potential of NH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH2 (−0.09 V) used in this work is significantly smaller than NaBH4 (−0.481 V) which was used by other groups.94,95 Therefore, the slower reaction rate favors the formation of larger Au DSNPs. The size dispersity of Au DSNPs synthesized using hydrazine reduction chemistry is comparable with those synthesized under UV or laser radiation.30,83 All the synthesized Au DSNPs are highly polycrystalline as demonstrated by both the high-resolution TEM images and selected area electron diffraction patterns.88 These larger Au NPs will be very useful in cellular labeling and imaging studies.
NH2 (−0.09 V) used in this work is significantly smaller than NaBH4 (−0.481 V) which was used by other groups.94,95 Therefore, the slower reaction rate favors the formation of larger Au DSNPs. The size dispersity of Au DSNPs synthesized using hydrazine reduction chemistry is comparable with those synthesized under UV or laser radiation.30,83 All the synthesized Au DSNPs are highly polycrystalline as demonstrated by both the high-resolution TEM images and selected area electron diffraction patterns.88 These larger Au NPs will be very useful in cellular labeling and imaging studies.
 | ||
| Fig. 7 Large scale TEM images of (a) {(Au0)6-G2·NH2}; (b) {(Au0)12-G3.NH2}; (c) {(Au0)24-G4.NH2}; (d) {(Au0)57-G5·NH2}; and (e) {(Au0)98-G6·NH2}. The plot of their sizes as a function of the number of dendrimer generations is shown in (f). Reproduced from ref. 88 by permission of IOP Publishing Limited. | ||
Zeta potential measurements confirmed that all the synthesized Au DSNPs are positively charged with zeta potentials ranging from 26.42 to 41.11 mV. This further indicates that after the formation of the hybrid nanostructures the terminal amines of dendrimers are still available to be protonated. The surface charge polarity of Au DSNPs is similar to the protonated corresponding dendrimers, which was confirmed by polyacrylamide gel electrophoresis measurements.88
The biological application of metal DSNPs is inherently related to the properties of metal NPs. For example, Ag DSNPs have been found to display antimicrobial activity without the loss of solubility and activity, even in the presence of sulfate or chloride ions.96 Radioactive Au198 DSNPs can be used to detect the organ/tissue biodistribution of the NPs.97 In most cases, metal DSNPs have been widely used as biomarkers for cellular imaging because of the high electron density contrast as compared with cellular or tissue structures.
In a previous work, Bielinska et al. utilized positively charged Au DSNPs as DNA carriers to transfect genes with similar efficiency to the corresponding dendrimers in the absence of Au NPs.98 The positively charged Au DSNPs easily penetrated into the polar zone of the cell membrane structure, thereby rendering them visible by TEM. The use of Au DSNPs makes the gene transfection process visible through TEM imaging, which is impossible for the use of dendrimers in the absence of metal NPs. The diffusion of various metal DSNPs is controlled rather by the surrounding dendrimers than by the stabilized metal NPs. Therefore, by varying the charge and lipophilicity of the dendrimers, the metal particle can be internalized and interact with various biologic entities.
In a recent study, we have shown that Ag DSNPs synthesized using amino-, hydroxyl-, and carboxyl-terminated EDA core generation 5 PAMAM dendrimers (G5·NH2, G5·NGlyOH, and G5·NSAH) as stabilizers are biocompatible, fluorescent and can be used as cell labeling markers.82 The Ag DSNPs were synthesized by UV irradiation of Ag(I)-dendrimer complexes and are water-soluble and stable. The fluorescent properties of Ag DSNPs are from the dendrimer templates, which has been demonstrated by our groups and others.88,92,99 The {(Ag0)25-PAMAM_G5·NH2}, {(Ag0)25-PAMAM_G5·NGlyOH}, and {(Ag0)25-PAMAM_G5·NSAH} DSNPs are fluorescent in the wavelength range of 400–500 nm. The fluorescence properties of Ag DSNPs allow us to image the intracellular uptake of Ag DSNPs using confocal microscopy, while the high electron density contrast of Ag metal allows one to image the cellular localization of the NPs using TEM. Representative TEM images collected for NIH3T3 and U937 cell lines incubated with {(Ag0)25-G5·NH2}DSNPs show that after 1 h incubation at 37 °C, Ag DSNPs can be observed in the form of randomly dispersed single particles or agglomerates on the surface of cellular membranes, in the cytoplasm, or trapped by the phagocytic or endocytic vesicles. It seems that internalization of the polycationic {(Ag0)25-G5·NH2} DSNPs may occur through two distinct mechanisms: phagocytosis and diffusion via cell walls. Similar results were obtained for the polyanionic {(Ag0)25-PAMAM_G5·NSAH} DSNPs, whereas cellular uptake of {(Ag0)25-G5·NGlyOH} with charge close to neutral was low. Combined with confocal microscopic imaging data, the results indicate that the uptake of the investigated silver DSNPs correlate with their surface charges and is not limited to specific cell lines.
For specific imaging of cancer cells, the surface charge of metal DSNPs should retain neutral or close to neutral in order to avoid non-specific cell membrane binding/repulsion or toxicity issues. Developing a facile approach to synthesizing neutralized metal DSNPs is important. In our recent work,86 we show that acetamide-functionalized Au DSNPs (diameter = 13 ± 4.5 nm) can be formed by acetylation of amine-terminated G5·NH2 PAMAM dendrimers complexed with Au(III) ions (AuCl4−). In addition, hydroxyl-functionalized Au DSNPs (diameter = 8.5 ± 0.9 nm) can be formed by simply mixing the glycidol hydroxyl-terminated G5 dendrimers (G5·NGlyOH) with HAuCl4. In both cases, no additional reducing agents were needed and the reactions were completed at room temperature. We also show that Alexa Fluor 594 dye-functionalized Au DSNPs (diameter = 16 ± 4.8 nm) can be formed by acetylation of Alexa Fluor 594-conjugated, amine-terminated G5 dendrimers complexed with HAuCl4. All of these functionalized Au DSNPs are water-soluble and stable. Fluorescence spectroscopy studies reveal that the Alexa Fluor 594-functionalized Au DSNPs retain similar fluorescence intensity to the Alexa Fluor 594-functionalized dendrimers that lack Au NPs. The formed acetylated Au DSNPs displayed a zeta potential value of +9.8 mV, which is close to neutral. These preparations of Au DSNPs provide a straightforward approach to synthesizing functionalized metal NPs for biomedical applications.
In comparison with Au DENP nanostructures composed of one or more metal NPs within one dendrimer molecule,26 it is possible to modify Au DSNPs with many more targeting ligands than those of Au DENPs, which significantly increases their targeting sensitivity and polyvalency effect.100 It is known that FA- and FI-modified G5 dendrimers with neutral surface charge can specifically target to cancer cells overexpressing FAR.50 It is anticipated that using FA- and FI-modified G5 dendrimers to synthesize Au DSNPs using the developed spontaneous approach86 may provide a means to create an Au NP-based multifunctional nanoplatform for cancer cell targeting and imaging. In our recent work,101 amine-terminated G5 PAMAM dendrimers pre-functionalized with FA and FI moieties were complexed with Au(III) ions, followed by acetylation of the amine groups on the dendrimer surfaces (Scheme 3). This one-step process led to the spontaneous formation of 6 nm-sized Au NPs stabilized by multifunctional dendrimers bearing both targeting and imaging functionalities. The multifunctional Au DSNPs were characterized by UV-Vis spectrometry, 1H NMR, and TEM. The formed Au DSNPs are water-soluble, stable, and biocompatible. Combined flow cytometry, confocal microscopy, silver staining, and inductively coupled plasma-mass spectrometry analyses show that the FA- and FI-functionalized Au DSNPs can specifically target to cancer cells expressing high-affinity FAR in vitro. The tunable dendrimer surface chemistry (e.g. surface functionalization and bioconjugation) in conjunction with the facile approach developed to form Au DSNPs open a new avenue to developing various biofunctionalized Au DSNPs for a range of interesting biomedical applications.
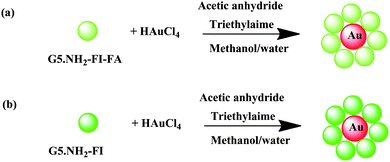 | ||
| Scheme 3 Schematic representation of the approaches to synthesizing {(Au0)7-G5·NHAc-FI-FA} (a) and {(Au0)7-G5·NHAc-FI} (b) DSNPs. Reproduced from ref. 101 by permission of The Royal Society of Chemistry. | ||
3.2. Synthesis and biomedical applications of iron oxide DSNPs
Iron oxide NPs has been widely used as a magnetic resonance (MR) imaging agent.14,102–105 In the presence of dendrimers, iron oxide NPs can be synthesized and simultaneously stabilized by dendrimers. Strable et al.106 synthesized stabilized ferromagnetic iron oxide NPs in the presence of carboxylated G4.5 PAMAM dendrimers (Scheme 4). In their work, the electrostatic interaction of negatively charged carboxylated PAMAM dendrimers with positively charged iron oxide NPs is considered to play an important role for the stabilization of the NPs. The carboxylated dendrimer provides both a good nucleation surface and strongly adsorbed passivating layer on the mineral surface. PAMAM dendrimers with other different functionalities (–NH2, –OH) cannot stabilize iron oxide NPs, indicating the role of electrostatic interaction for the NP stabilization. The diameter of the formed highly soluble iron oxide NPs is in the 20–30 nm regime.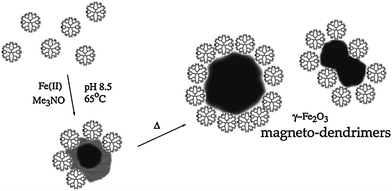 | ||
| Scheme 4 Schematic representation of the stabilization of maghemite nanoparticles by carboxyl-terminated PAMAM dendrimer (generation 4.5). Reprinted with permission from ref. 106. Copyright 2001 American Chemical Society. | ||
Using the synthesized magnetodendrimers, Bulte and coworkers102 labeled mammalian cells (e.g., human neural stem cells and mesenchymal stem cells) through a non-specific membrane adsorption process with subsequent intracellular localization in endosomes. The incubated magnetodendrimer doses as low as 1 μg iron/mL allow sufficient MR cell contrast without compromising the cell viability and differentiation. The labeled neural stem cell-derived oligodendroglial progenitors can be readily detected in vivo by MR imaging at least six weeks after transplantation. Using the same magnetodendrimers, Bulte and coworkers were able to track the olfactory ensheathing glia grafted into the rat spinal cord in vivo,107 to detect the murine and human skin stem/progenitor cells,108 and to monitor stem cell therapy in vivo by MR imaging.109 However, the utilized magnetodendrimers do not have any specific surface modifications, not enabling the specific interaction between the particles and the target cells.
4. Dendrimer-assembled nanoparticles (DANPs)
Preformed NPs that are modified with dendrimers or functionalized dendrimers could have improved biocompatibility, stability, and specific functionalities for biomedical application. Similarly, the dendrimer molecules assembled onto the preformed NPs could be further chemically modified to achieve desired biofunctionalities for biomedical applications. The major difference between DANPs and DSNPs that are generally formed via an in situ process is that the DANPs are formed on the basis of preformed NPs. The driving force to assemble dendrimers onto preformed NPs could be electrostatic interaction,13,110 covalent bonding,16,111,112 and the combination of different weak forces.In a recent study,112 fluorescein-doped magnetic mesoporous silica NPs were modified with 3-(triethoxysily)proplyl isocyamate-activated G2·NH2 PAMAM dendrimers, the amino terminal groups of the assembled dendrimers can be further neutralized by reacting with methyl acrylate to form esters. The formed composite NPs are biocompatible even at a NP concentration of 650 μg mL−1 and can be used as both drug delivery vehicles and fluorescent imaging agents.
For targeted imaging of cancer cells in vitro and in vivo, it would be ideal to assemble targeting ligand-modified dendrimer molecules onto the NPs (e.g., iron oxide NPs). In our previous study,13 we synthesized and characterized a group of FA-modified carboxyl-functionalized G3 PAMAM dendrimers that were used to assemble onto the superparamagnetic iron oxide (Fe3O4) NPs. The Fe3O4 NPs synthesized using controlled co-precipitation of Fe(II) and Fe(III) ions were assembled with the FA-modified dendrimers through electrostatic interaction in order to achieve specific targeting to KB cells that overexpress FAR. It appears that carboxyl-terminated PAMAM dendrimer-assembled Fe3O4 NPs can be uptaken by KB cells regardless of the repelling force between the negatively charged cells and the negatively charged particles. In the presence of a large amount of carboxyl terminal groups on the dendrimer surface, the receptor-mediated endocytosis of Fe3O4 NPs assembled by FA-modified dendrimers was not facilitated. This implies that the surface charge of dendrimer-stabilized magnetic iron oxide NPs in a biological medium is an important factor influencing their biological performance.
Earlier studies show that the targeted dendrimers with a neutral surface (e.g., with acetamide terminal groups) can specifically target cancer cells through ligand–receptor interaction.46,50,55 This leads to an idea to synthesize neutralized Fe3O4 NPs for specific MR imaging of tumors. In one of our recent studies,15 we demonstrated a unique approach that combines a layer-by-layer (LbL) self-assembly method with dendrimer chemistry to functionalize Fe3O4 NPs for specific targeting and imaging of cancer cells. In this approach, positively charged Fe3O4 NPs (8.4 nm in diameter) synthesized by controlled co-precipitation of Fe(II) and Fe(III) ions were modified with a bilayer composed of polystyrene sulfonate sodium salt and FA- and FI-functionalized G5 PAMAM dendrimers (G5·NH2-FI-FA) through electrostatic LbL assembly, followed by an acetylation reaction to neutralize the remaining surface amine groups of G5 dendrimers. Combined flow cytometry, confocal microscopy, TEM, and MR imaging studies show that Fe3O4/PSS/G5·NHAc-FI-FA NPs can specifically target cancer cells overexpressing FAR. Unfortunately, in vivo data show that most of these bilayer-modified Fe3O4 NPs accumulate in the liver of mice, which suggests that the particles lack in vivo stability (unpublished results). Development of a robust polymer shell coating onto Fe3O4 is necessary to achieve a successful in vivo MR imaging of a tumor. Approaches to accomplish this involve increasing the polymer layer thickness and/or chemically crosslinking the polymer shells.
In our very recent study,14 Fe3O4 NPs were assembled with multilayers of poly(glutamic acid) (PGA) and poly(L-lysine) (PLL), followed by assembly with G5·NH2-FI-FA dendrimers. The interlayers were then crosslinked through EDC(1-ethyl-3-[3-dimethylaminopropyl]carbodiimide hydrochloride) chemistry to covalently link the hydroxy groups of iron oxide, the carboxy groups of PGA, and the amino groups of PLL and the dendrimers. The remaining amino groups of the dendrimers are finally acetylated to neutralize the surface charge (Fig. 8a).The fabricated shell-crosslinked iron oxide (SCIO) NPs were characterized by TEM (Fig. 8b and 8c). The morphology of the FA-modified SCIO NPs does not show a significant change after the assembly and crosslinking of the polymers and dendrimers when compared with the pristine Fe3O4 NPs (Fig. 8b).13,15 A negatively stained TEM image using phosphotungstic acid (Fig. 8c) clearly shows that all Fe3O4 NPs are surrounded with the bright rings of the polymer multilayers, which confirms the successful self-assembly process. The formed SCIO NPs are water-dispersible, stable, and biocompatible. Following the injection of either targeted SCIO-FA or non-targeted SCIO-NonFA NPs, the in vivo MR imaging of tumor data (Fig. 9) show that the tumor MR signal intensity of mice injected with SCIO-FA NPs gradually decreases as a function of time. In sharp contrast, the tumor MR signal intensity of mice treated with SCIO-NonFA NPs does not decrease significantly with time post-injection. It is clear that 24 h after injection of the SCIO-FA NPs, the tumor MR signal intensity has decreased more significantly than the signal intensity in the tumors of the mouse treated with non-targeted SCIO-NonFA NPs and in the control mouse. After 48 h post-injection, the difference of the MR signal intensity of the tumors is smaller for both mice injected with SCIO-FA and SCIO-NonFA NPs. This approach to the functionalization of magnetic NPs may be applied to other small targeting molecules (e.g., peptides and growth factors), thereby providing a general cost-effective approach for in vivo MR detection of various biological systems.
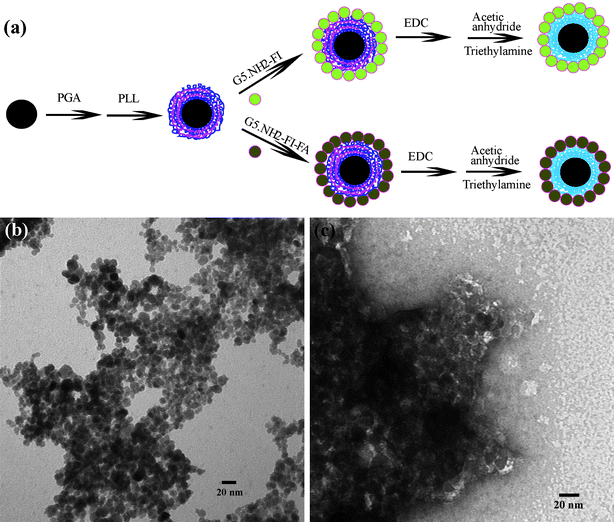 | ||
| Fig. 8 (a) Schematic representation of the procedure for fabricating multifunctional shell-crosslinked iron oxide NPs; (b) A TEM image of SCIO-FA NPs; and (c) a negatively phosphotungstic acid-stained TEM image of SCIO-FA NPs. Ref. 14; copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
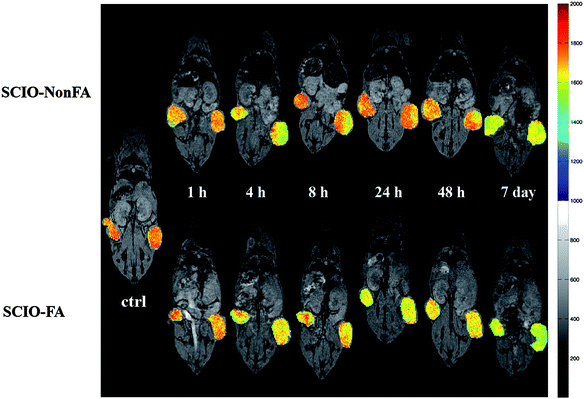 | ||
| Fig. 9 In vivo color maps of T2-weighted MR images of mice implanted with cancer cell line KB cells overexpressing FAR, at different time points after injection of SCIO-NonFA and SCIO-FA NPs, respectively. The color bar (from red to blue) indicates the MR signal intensity changes from high to low. Ref. 14; copyright Wiley-VCH Verlag GmbH & Co. KGaA. Reproduced with permission. | ||
The dendrimer assembly approach not only applies to 3-dimensional (3D) spherical particles, but also applies to 1D NPs (e.g., carbon nanotubes and Au nanorods),16,113 rendering it a unique strategy for fabrication of organic/inorganic hybrid NPs for biomedical applications. In one of our recent studies,113 FI- and FA-modified amine-terminated G5 PAMAM dendrimers (G5·NH2-FI-FA) were covalently linked to acid-treated multiwalled carbon nanotubes (MWCNTs) via a one-step EDC chemical coupling reaction, followed by acetylation of the remaining primary amine groups of the dendrimers. The resulting MWCNT/G5·NHAc-FI-FA composites are water-dispersible, stable, and biocompatible. In vitro flow cytometry and confocal microscopy data show that the formed MWCNT/G5·NHAc-FI-FA composites can specifically target to cancer cells overexpressing high-affinity FAR. The results of this study suggest that through modification with multifunctional dendrimers, complex carbon nanotube-based materials can be fabricated, thereby providing many possibilities for various applications in biomedical sensing, diagnosis, and therapeutics. In another study,16 Cui and coworkers utilized thiolated G4.0 PAMAM dendrimers to replace cetyltrimethylammonium bromides (CTAB) molecules on the surface of gold nanorods. Then, the amine groups on the surface of dendrimer assembled on Au nanorods were converted to carboxylic acids by reacting with excess glutaric anhydride, followed by conjugation with RGD peptides via EDC chemical coupling. The final RGD-modified Au nanorods are biocompatible and show highly selective targeting and destructive effects on the cancer cells and solid tumors overexpressing αvβ3 intergrin under near-infrared laser irradiation. In addition, in vivo studies show that the tumors implanted in four mice from the test group of ten disappear after treatment. This study suggests that the dendrimer assembly is a key step to fabricate multifunctional medical nanodevices for tumor targeting, imaging, and selective photothermal therapy.
5. Nanoparticle core/dendrimer shell composite particles formed via a divergent “grafting from” method
As generally known, dendrimers can be synthesized using both divergent and convergent synthetic approaches.4 In the divergent approach, starting from a core molecule (e.g., EDA), PAMAM dendrimers are synthesized via alternating reiterations of exhaustive Michael addition with methylacrylate (MA) followed by amidation with EDA.1–3 Using the same strategy, nanoparticle core/dendrimer shell composite particles can be synthesized via a divergent “grafting from” method starting from a NP core.17–20,114,115 In general, the NP cores are firstly modified to have a number of primary amine groups, rendering the reaction with MA possible. Compared with DENPs, DSNPs, and DANPs that are formed in the presence of preformed dendrimers, the dendrimer part in this type of hybrid nanoparticles is synthesized via a step-by-step manner, similar to the conventional divergent approach used to synthesize dendrimers.In an earlier study,18,19 Pan et al. developed an approach to growing PAMAM dendrimers onto the surface of Fe3O4 NPs to allow enhanced immobilization of bovine serum albumin (BSA). In their approach, Fe3O4 NPs were first modified by aminopropyltrimethoxysilane (NH2(CH2)3–Si–(OCH3)3, APTS) via silanization, rendering the NP surface with abundant primary amine groups. The APTS-modified NPs were further alternately reacted with MA and EDA, allowing divergent growth of PAMAM dendrimers onto the NP surface (Scheme 5). The dendrimer-modified Fe3O4 NPs are water dispersible and show increased binding with BSA with the generation number, mainly due to the increased number of surface amine groups with dendrimer generation. Using the dendrimer-modified Fe3O4 NPs, Pan et al.20 are able to transfect antisense surviving oligodeoxynucleotide (asODN) into human tumor cell lines such as human breast cancer MCF-7, MDA-MB-435, and liver cancer HepG2. Results showed that the asODN-dendrimer-Fe3O4 composites can enter into tumor cells within 15 min, causing marked down-regulation of the survivin gene and protein and cell growth inhibition in a dose-and time-dependent manner. G5 dendrimer-modified Fe3O4 NPs exhibits the highest efficiency for cellular transfection and inhibition. These results show that PAMAM dendrimer-modified magnetic NPs may be a good gene delivery system and have potential applications in cancer therapy and molecular diagnosis. Using the similar divergent “grafting from” approach, dendrimers can be modified onto MWCNTs. In a recent report,114 Pan et al. utilized oxidized MWCNTs with carboxyl groups to react with thionyl chloride, resulting in MWCNTs functionalized with chlorocarbonyl groups (MWCNT-COCl). Then, the MWNT-COCl was reacted with an excess of EDA, producing amine-functionalized MWCNT (MWCNT-NH2), which was used as the core to react sequentially with MA/EDA, generating PAMAM dendrimer-modified MWCNTs with different generations. The as-prepared nanocomposites exhibit excellent dispersibility in water. Using the dendrimer-modified MWCNTs, Pan et al.116 further demonstrated that FITC-labeled antisense c-myc oligonucleotides (asODN) can be effectively transfected into human breast cancer cell line MCF-7 cells and MDA-MB-435 cells, and liver cancer cell line HepG2 cells. The composites of dendrimer-modified MWCNTs with asODN are able to enter into tumor cells within 15 min (Fig. 10). Similar to dendrimer-modified Fe3O4 NPs, these composites can inhibit the cancer cell growth in time- and dose-dependent manners and down-regulate the expression of the c-myc gene and C-Myc protein. The intracellular gene transport and uptake via dendrimer-modified MWCNTs should be generic for the mammalian cell lines. As proved in literature,117,118 the MWCNTs can be used as drug carriers. Therefore, the dendrimer-modified MWCNTs should have potentials in applications in gene or drug delivery for cancer therapy and molecular imaging.
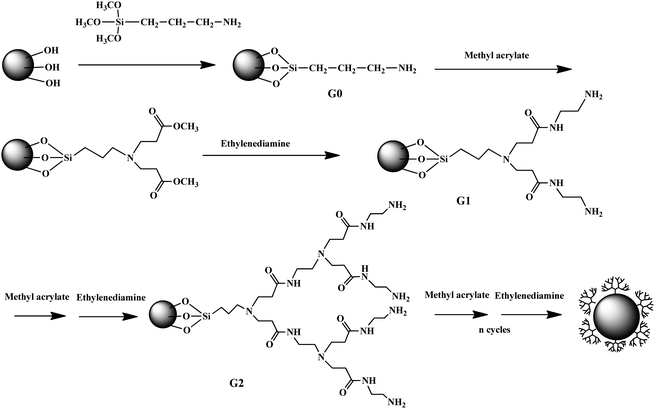 | ||
| Scheme 5 Magnetite nanoparticle modified with PAMAM dendrimers via a stepwise divergent approach. | ||
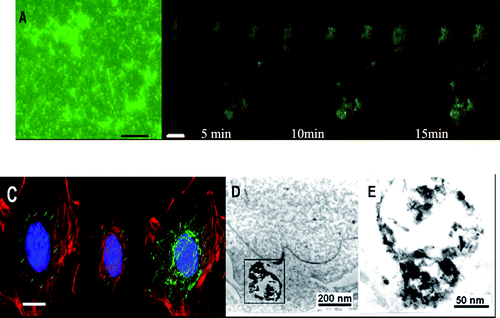 | ||
| Fig. 10 (A) Fluorescent microscopy image of FITC-asODN–dMNTs in medium, bar 500 nm; (B) laser confocal microscopy images of MCF-7 cells incubated with FITC-asODN–dMNTs for 5 min, 10 min and 15 min, respectively. Scale bar: 5.0 μm. (C) Tridimensional immunofluorescent images of FITC-asODN–dMNTs in MDA-MB-435 cells. Blue: DAPI stained cell nucleus. Red: cellular actin framework, scale bar: 5.0 μm. ((D) and (E)) HR-TEM images of as-ODN-dMNT composites in the cytoplasm of MCF-7 cells. (E) is the magnified image of local frame in (D). Reproduced from ref. 116 by permission of IOP Publishing Limited. | ||
6. Concluding remarks and outlooks
In summary, this review has described the fabrication of four kinds of different dendrimer-based organic/inorganic hybrid NPs for biomedical applications including DENPs, DSNPs, DANPs, and NP core/dendrimer shell composite particles formed via a divergent “grafting from” method. The unique aspects of dendrimer or dendrimer-like structure (the chemistry of the terminal groups, the generation-dependent size, the 3D structure, and the interior coordination chemistry) afford immense interest in the preparation of these nanocomposite materials. The approaches to preparing all types of hybrid NPs are variable and tunable depending on both the inorganic nanoparticle preparation chemistry and the dendrimer synthesis and modification chemistry. The inorganic part of DENPs and DSNPs that are formed in situ can be controlled by varying the reaction conditions, while preformed inorganic NPs can be assembled with dendrimers to form DANPs, or be modified with dendrimers formed via a divergent synthetic approach. The formed organic/inorganic hybrid NPs offers a wide range of opportunities for biomedical applications because of the functionalizable nature of the dendrimer materials and the controlled size, dimension, and crystalline structures of the inorganic NPs. These nanocomposites will find a variety of biomedical applications, including imaging, sensing, and treatment of various biological systems.Many efforts have been devoted to surface modification of dendrimer-based organic/inorganic hybrid NPs for biological applications. Since the discovery of metal dendrimer nanocomposite in 1998,29,31 the fabrication and biomedical applications of these NPs still remain an open area and offer great challenges. For example, dendrimer-stabilized magnetic NPs and dendrimer-modified magnetic NPs formed using a divergent “grafting from” method currently used in biological systems lack specificity to particular biological systems, which is largely due to the technical difficulty in modifying them with specific biological moieties. TEM imaging of in vivo tumor models is expected to be very difficult because of the 3-dimensional heterogeneity of tumor structures. Hyperthermia treatment of cancer using the current metal DENPs or DSNPs will be limited to skin tumors because the laser wavelength used for hyperthermia is short (e.g., 510–530 nm for Au DENPs and Au DSNPs in order to match their surface plasmon resonance absorption). Developing an Au DENP or DSNP-based Au nanoshell structure may be necessary to overcome the disadvantages, in the latter case, the Au nanoshell structure displays surface plasmon resonance absorption at the near infrared (NIR) wavelength range (700–1100 nm), which allows the laser used at this wavelength range to penetrate tissue and blood for deep tumor therapy. All these challenges will drive the effective collaboration of scientists working in chemistry, materials, engineering, and biomedical fields to develop effective dendrimer-based organic/inorganic hybrid nanostructures for various biological applications.
Acknowledgements
Financial support from the Shanghai Pujiang Program (09PJ1400600), the National Natural Science Foundation of China (20974019), the National Basic Research Program of China (973 Program, 2007CB936000), and the Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning were greatly acknowledged.References
- D. A. Tomalia, H. Baker, J. R. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J., 1985, 17, 117 CrossRef CAS.
- D. A. Tomalia, H. Baker, J. R. Dewald, M. Hall, G. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Macromolecules, 1986, 19, 2466–2468 CrossRef.
- D. A. Tomalia, A. M. Naylor and W. A. Goddard III, Angew. Chem., Int. Ed. Engl., 1990, 29, 138 CrossRef.
- D. A. Tomalia and J. M. J. Frechet, ed., Dendrimers and Other Dendritic Polymers, John Wiley & Sons Ltd, New York, 2001 Search PubMed.
- W. D. Jang, K. M. Kamruzzaman Selim, C. H. Lee and I. K. Kang, Prog. Polym. Sci., 2009, 34, 1–23 CrossRef CAS.
- S. H. Medina and M. E. H. El-Sayed, Chem. Rev., 2009, 109, 3141–3157 CrossRef CAS.
- R. M. Crooks, M. Zhao, L. Sun, V. Chechik and L. K. Yeung, Acc. Chem. Res., 2001, 34, 181–190 CrossRef CAS.
- R. W. J. Scott, O. M. Wilson and R. M. Crooks, J. Phys. Chem. B, 2005, 109, 692–704 CrossRef CAS.
- K. Esumi, Top. Curr. Chem., 2003, 227, 31–52 CAS.
- K. Esumi, in Encyclopedia of Nanoscience and Nanotechnology, ed. H. S. Nalwa, American Scientific Publishers, Valencia, 2004, pp. 317–326 Search PubMed.
- T. Goodson III, O. Varnavski and Y. Wang, Int. Rev. Phys. Chem., 2004, 23, 109–150 CrossRef CAS.
- K. J. Landmark, S. DiMaggio, J. Ward, C. Kelly, S. Vogt, S. Hong, A. Kotlyar, A. Myc, T. P. Thomas, J. E. Penner-Hahn, J. R. Baker, Jr., M. M. Banaszak Holl and B. G. Orr, ACS Nano, 2008, 2, 773–783 CrossRef CAS.
- X. Shi, T. P. Thomas, L. A. Myc, A. Kotlyar and J. R. Baker, Jr., Phys. Chem. Chem. Phys., 2007, 9, 5712–5720 RSC.
- X. Shi, S. H. Wang, S. D. Swanson, S. Ge, Z. Cao, M. E. Van Antwerp, K. J. Landmark and J. R. Baker, Jr., Adv. Mater., 2008, 20, 1671–1678 CrossRef CAS.
- S. Wang, X. Shi, M. Van Antwerp, Z. Cao, S. D. Swanson, X. Bi and J. R. Baker, Jr., Adv. Funct. Mater., 2007, 17, 3043–3050 CrossRef CAS.
- Z. Li, P. Huang, X. Zhang, J. Lin, S. Yang, B. Liu, F. Gao, P. Xi, Q. Ren and D. Cui, Mol. Pharmaceutics, 2010, 7, 94–104 CrossRef CAS.
- R. Abu-Reziq, H. Alper, D. Wang and M. L. Post, J. Am. Chem. Soc., 2006, 128, 5279–5282 CrossRef.
- B. Pan, F. Gao and L. Ao, J. Magn. Magn. Mater., 2005, 293, 252–258 CrossRef CAS.
- B. Pan, F. Gao and H. Gu, J. Colloid Interface Sci., 2005, 284, 1–6 CrossRef CAS.
- B. Pan, D. Cui, Y. Sheng, C. Ozkan, F. Gao, R. He, Q. Li, P. Xu and T. Huang, Cancer Res., 2007, 67, 8156–8163 CrossRef CAS.
- R. C. Advincula, Dalton Trans., 2006, 2778–2784 RSC.
- T. J. Daou, G. Pourroy, J. M. Greneche, A. Bertin, D. Felder-Flesch and S. Begin-Colin, Dalton Trans., 2009, 4442–4449 RSC.
- S.-H. Hwang, C. N. Moorefield, P. Wang, K.-U. Jeong, S. Z. D. Cheng, K. K. Kotta and G. R. Newkome, J. Am. Chem. Soc., 2006, 128, 7505–7509 CrossRef CAS.
- L. Tao, G. Chen, G. Mantovani, S. York and D. M. Haddleton, Chem. Commun., 2006, 4949–4951 RSC.
- R. Wang, J. Yang, Z. Zheng, M. D. Carducci, J. Jiao and S. Seraphin, Angew. Chem., Int. Ed., 2001, 40, 549–552 CrossRef CAS.
- F. Grohn, B. J. Bauer, Y. A. Akpalu, C. L. Jackson and E. J. Amis, Macromolecules, 2000, 33, 6042–6050 CrossRef.
- A. Manna, T. Imae, K. Aoi, M. Okada and T. Yogo, Chem. Mater., 2001, 13, 1674–1681 CrossRef CAS.
- Y.-G. Kim, S.-K. Oh and R. M. Crooks, Chem. Mater., 2004, 16, 167–172 CrossRef CAS.
- L. Balogh and D. A. Tomalia, J. Am. Chem. Soc., 1998, 120, 7355–7356 CrossRef CAS.
- K. Esumi, A. Suzuki, N. Aihara, K. Usui and K. Torigoe, Langmuir, 1998, 14, 3157–3159 CrossRef CAS.
- M. Zhao, L. Sun and R. M. Crooks, J. Am. Chem. Soc., 1998, 120, 4877–4878 CrossRef CAS.
- B. I. Lemon and R. M. Crooks, J. Am. Chem. Soc., 2000, 122, 12886–12887 CrossRef CAS.
- X. C. Wu, A. M. Bittner and K. Kern, J. Phys. Chem. B, 2005, 109, 230–239 CrossRef CAS.
- A. Fahmi, T. Pietsch, D. Appelhans, N. Gindy and B. Voit, New J. Chem., 2009, 33, 703–706 RSC.
- B. J. Auten, B. P. Hahn, G. Vijayaraghavan, K. J. Stevenson and B. D. Chandler, J. Phys. Chem. C, 2008, 112, 5365–5372 CrossRef CAS.
- N. Satoh, T. Nakashima, K. Kamikura and K. Yamamoto, Nat. Nanotechnol., 2008, 3, 106–111 CrossRef CAS.
- F. Divsar, A. Nomani, M. Chaloosi and I. Haririan, Microchim. Acta, 2009, 165, 421–426 CrossRef CAS.
- Y. Haba, C. Kojima, A. Harada, T. Ura, H. Horinaka and K. Kono, Langmuir, 2007, 23, 5243–5246 CrossRef CAS.
- M. R. Knecht, J. C. Garcia-Martinez and R. M. Crooks, Langmuir, 2005, 21, 11981–11986 CrossRef CAS.
- R. Shukla, E. Hill, X. Shi, J. Kim, M. C. Muniz, K. Sun and J. R. Baker, Jr., Soft Matter, 2008, 4, 2160–2163 RSC.
- E. Boisselier, A. K. Diallo, L. Salmon, J. Ruiz and D. Astruc, Chem. Commun., 2008, 4819–4821 RSC.
- J. Maly, H. Lampova, A. Semeradtova, M. Stofik and L. Kovacik, Nanotechnology, 2009, 20, 385101 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547–1562 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- S.-K. Oh, Y.-G. Kim, H. Ye and R. M. Crooks, Langmuir, 2003, 19, 10420–10425 CrossRef CAS.
- A. Quintana, E. Raczka, L. Piehler, I. Lee, A. Myc, I. Majoros, A. K. Patri, T. P. Thomas, J. Mule and J. R. Baker, Jr., Pharm. Res., 2002, 19, 1310–1316 CrossRef CAS.
- J. F. Kukowska-Latallo, K. A. Candido, Z. Cao, S. S. Nigavekar, I. J. Majoros, T. P. Thomas, L. P. Balogh, M. K. Khan and J. R. Baker, Jr., Cancer Res., 2005, 65, 5317–5324 CrossRef CAS.
- I. J. Majoros, A. Myc, T. P. Thomas, C. B. Mehta and J. R. Baker, Jr., Biomacromolecules, 2006, 7, 572–579 CrossRef CAS.
- I. J. Majoros, T. P. Thomas, C. B. Mehta and J. R. Baker, Jr., J. Med. Chem., 2005, 48, 5892–5899 CrossRef CAS.
- T. P. Thomas, I. J. Majoros, A. Kotlyar, J. F. Kukowska-Latallo, A. Bielinska, A. Myc and J. R. Baker, Jr., J. Med. Chem., 2005, 48, 3729–3735 CrossRef CAS.
- X. Shi, S. Wang, H. Sun and J. R. Baker, Jr., Soft Matter, 2007, 3, 71–74 RSC.
- X. Shi, I. Lee and J. R. Baker, Jr., J. Mater. Chem., 2008, 18, 586–593 RSC.
- P. E. Laibinis, G. M. Whitesides, D. L. Allara, Y. T. Tao, A. N. Parikh and R. G. Nuzzo, J. Am. Chem. Soc., 1991, 113, 7152–7167 CrossRef CAS.
- Y. Choi, T. Thomas, A. Kotlyar, M. T. Islam and J. R. Baker, Jr., Chem. Biol., 2005, 12, 35–43 CrossRef CAS.
- X. Shi, S. Wang, S. Meshinchi, M. E. Van Antwerp, X. Bi, I. Lee and J. R. Baker, Jr., Small, 2007, 3, 1245–1252 CrossRef CAS.
- I. G. Campbell, T. A. Jones, W. D. Foulkes and J. Trowsdale, Cancer Res., 1991, 51, 5329–5338 CAS.
- P. Garin-Chesa, I. Campbell, P. E. Saigo, J. L. Lewis, Jr., L. J. Old and W. J. Rettig, Am. J. Pathol., 1993, 142, 557–567 Search PubMed.
- D. Weitman, R. H. Lark, L. R. Coney, D. W. Fort, V. Frasca, V. R. Surawski and B. A. Kamen, Cancer Res., 1992, 52, 3396–3401 CAS.
- F. Ross, P. K. Chaudhauri and M. Ratnam, Cancer Res., 1994, 73, 2432–2443.
- X. Shi, S. H. Wang, I. Lee, M. Shen and J. R. Baker, Jr., Biopolymers, 2009, 91, 936–942 CrossRef CAS.
- R. Pasqualini, E. Koivunen and E. Ruoslahti, Nat. Biotechnol., 1997, 15, 542–546 CrossRef CAS.
- O. Cleaver and D. A. Melton, Nat. Med., 2003, 9, 661–668 CrossRef CAS.
- C. Alric, J. Taleb, G. Le Duc, C. Mandon, C. Billotey, A. Le Meur-Herland, T. Brochard, F. Vocanson, M. Janier, P. Perriat, S. Roux and O. Tillement, J. Am. Chem. Soc., 2008, 130, 5908–5915 CrossRef CAS.
- J. F. Hainfeld, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 Search PubMed.
- V. Kattumuri, K. Katti, S. Bhaskaran, E. J. Boote, S. W. Casteel, G. M. Fent, D. J. Robertson, M. Chandrasekhar, R. Kannan and K. V. Katti, Small, 2007, 3, 333–341 CrossRef CAS.
- D. Kim, S. Park, J. H. Lee, Y. Y. Jeong and S. Jon, J. Am. Chem. Soc., 2007, 129, 7661–7665 CrossRef CAS.
- R. Popovtzer, A. Agrawal, N. A. Kotov, A. Popovtzer, J. Balter, T. E. Carey and R. Kopelman, Nano Lett., 2008, 8, 4593–4596 CrossRef CAS.
- C. J. Xu, G. A. Tung and S. H. Sun, Chem. Mater., 2008, 20, 4167–4169 CrossRef CAS.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS.
- R. Guo, H. Wang, C. Peng, M. Shen, M. Pan, X. Cao, G. Zhang and X. Shi, J. Phys. Chem. C, 2010, 114, 50–56 CrossRef CAS.
- K. Sooklal, L. H. Hanus, H. J. Ploehn and C. J. Murphy, Adv. Mater., 1998, 10, 1083–1087 CrossRef CAS.
- J. Huang, K. Sooklal, C. J. Murphy and H. J. Ploehn, Chem. Mater., 1999, 11, 3595–3601 CrossRef CAS.
- X. Shi, K. Sun, L. Balogh and J. R. Baker, Jr., Nanotechnology, 2006, 17, 4554–4560 CrossRef CAS.
- B. Pan, F. Gao, L. Ao, H. Tian, R. He and D. Cui, Colloids Surf., A, 2005, 259, 89–94 CrossRef CAS.
- K. Esumi, R. Isono and T. Yoshimura, Langmuir, 2004, 20, 237–243 CrossRef CAS.
- L. Balogh, R. Valluzzi, K. S. Laverdure, S. P. Gido, G. L. Hagnauer and D. A. Tomalia, J. Nanopart. Res., 1999, 1, 353–368 CrossRef CAS.
- M. E. Garcia, L. A. Baker and R. M. Crooks, Anal. Chem., 1999, 71, 256–258 CrossRef CAS.
- X. Sun, S. Dong and E. Wang, Macromolecules, 2004, 37, 7105–7108 CrossRef CAS.
- X. Sun, X. Jiang, S. Dong and E. Wang, Macromol. Rapid Commun., 2003, 24, 1024–1028 CrossRef CAS.
- T. Vassilieff, A. Sutton and A. K. Kakkar, J. Mater. Chem., 2008, 18, 4031–4033 RSC.
- W. Lesniak, A. U. Bielinska, K. Sun, K. W. Janczak, X. Shi, J. R. Baker, Jr. and L. P. Balogh, Nano Lett., 2005, 5, 2123–2130 CrossRef CAS.
- K. Hayakawa, T. Yoshimura and K. Esumi, Langmuir, 2003, 19, 5517–5521 CrossRef CAS.
- K. Nie, J. Hu, W. Pang and Q. Zhu, Mater. Lett., 2007, 61, 3567–3570 CrossRef CAS.
- A. Sutton, G. Franc and A. Kakkar, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 4482–4493 CrossRef CAS.
- X. Shi, K. Sun and J. R. Baker, Jr., J. Phys. Chem. C, 2008, 112, 8251–8258 CrossRef CAS.
- M. Kavitha, M. R. Parida, E. Prasad, C. Vijayan and P. C. Deshmukh, Macromol. Chem. Phys., 2009, 210, 1310–1318 CrossRef CAS.
- X. Shi, T. R. Ganser, K. Sun, L. P. Balogh and J. R. Baker, Jr., Nanotechnology, 2006, 17, 1072–1078 CrossRef CAS.
- M. M. Alvarez, J. T. Khoury, T. G. Schaaff, M. N. Shafigullin, I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 1997, 101, 3706–3712 CrossRef CAS.
- C. F. Bohren and D. R. Huffman, Absorption and Scattering of Light by Small Particles, John Wiley, New York, 1983 Search PubMed.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS.
- D. Wang and T. Imae, J. Am. Chem. Soc., 2004, 126, 13204–13205 CrossRef CAS.
- K. Esumi, A. Suzuki, A. Yamahira and K. Torigoe, Langmuir, 2000, 16, 2604–2608 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, MacGraw-Hill, Inc, New York, 1992 Search PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press LLC, New York, 2002 Search PubMed.
- L. Balogh, D. R. Swanson, D. A. Tomalia, G. L. Hagnauer and A. T. McManus, Nano Lett., 2001, 1, 18–21 CrossRef CAS.
- M. K. Khan, S. S. Nigavekar, D. L. Minc, M. S. T. Kariapper, B. M. Nair, W. G. Lesniak and L. P. Balogh, Technol. Cancer Res. Treatment, 2005, 4, 603–613 Search PubMed.
- A. Bielinska, J. D. Eichman, I. Lee, J. R. Baker, Jr. and L. Balogh, J. Nanopart. Res., 2002, 4, 395–403 CrossRef CAS.
- W. I. Lee, Y. Bae and A. J. Bard, J. Am. Chem. Soc., 2004, 126, 8358–8359 CrossRef CAS.
- S. Hong, P. R. Leroueil, I. J. Majoros, B. G. Orr, J. R. Baker, Jr. and M. M. Banaszak Holl, Chem. Biol., 2007, 14, 107–115 CrossRef CAS.
- X. Shi, S. H. Wang, M. E. Van Antwerp, X. Chen and J. R. Baker, Jr., Analyst, 2009, 134, 1373–1379 RSC.
- J. W. M. Bulte, T. Douglas, B. Witwer, S.-C. Zhang, E. Strable, B. K. Lewis, H. Zywicke, B. Miller, P. van Gelderen, B. M. Moskowitz, L. D. Duncan and J. A. Frank, Nat. Biotechnol., 2001, 19, 1141–1147 CrossRef CAS.
- I. J. M. de Vries, W. J. Lesterhuis, J. O. Barentsz, P. Verdijk, J. H. van Krieken, O. C. Boerman, W. J. G. Oyen, J. J. Bonenkamp, J. B. Boezeman, G. J. Adema, J. W. M. Bulte, T. W. J. Scheenen, C. J. A. Punt, A. Heerschap and C. G. Figdor, Nat. Biotechnol., 2005, 23, 1407–1413 CrossRef CAS.
- F. Hu, L. Wei, Z. Zhou, Y. Ran, Z. Li and M. Gao, Adv. Mater., 2006, 18, 2553–2556 CrossRef CAS.
- J.-H. Lee, Y.-M. Huh, Y.-w. Jun, J.-w. Seo, J.-t. Jang, H.-T. Song, S. Kim, E.-J. Cho, H.-J. Yoon, J.-S. Suh and J. Cheon, Nat. Med., 2007, 13, 95–99 CrossRef CAS.
- E. Strable, J. W. M. Bulte, B. Moskowitz, K. Vivekanandan, M. Allen and T. Douglas, Chem. Mater., 2001, 13, 2201–2209 CAS.
- I. H. Lee, J. W. M. Bulte, P. Schweinhardt, T. Douglas, A. Trifunovski, C. Hofstetter, L. Olson and C. Spenger, Exp. Neurol., 2004, 187, 509–516 CrossRef.
- P. Tunici, J. W. M. Bulte, M. G. Bruzzone, P. L. Poliani, L. Cajola, M. Grisoli, T. Douglas and G. Finocchiaro, J. Gene Med., 2006, 8, 506–513 CrossRef CAS.
- J. W. M. Bulte, T. Douglas, B. Witwer, S.-C. Zhang, B. K. Lewis, P. van Gelderen, H. Zywicke, I. D. Duncan and J. A. Frank, Acad. Radiol., 2002, 9, S332–S335 CrossRef.
- B. L. Frankamp, A. K. Boal, M. T. Tuominen and V. M. Rotello, J. Am. Chem. Soc., 2005, 127, 9731–9735 CrossRef CAS.
- Z. Li, P. Huang, J. Lin, R. He, B. Liu, X. Zhang, S. Yang, P. Xi, X. Zhang, Q. Ren and D. Cui, J. Nanosci. Nanotechnol., 2010, 10, 1–9 CrossRef.
- J. Yu, H. Zhao, L. Ye, H. Yang, S. Ku, Y.N. and N. Xiao, J. Mater. Chem., 2009, 19, 1265–1270 RSC.
- X. Shi, S. H. Wang, M. Shen, M. E. Antwerp, X. Chen, C. Li, E. J. Petersen, Q. Huang, W. J. Weber, Jr. and J. R. Baker, Jr., Biomacromolecules, 2009, 10, 1744–1750 CrossRef CAS.
- B. Pan, D. Cui, F. Gao and R. He, Nanotechnology, 2006, 17, 2483–2489 CrossRef CAS.
- H. Liu, J. Guo, L. Jin, W. Yang and C. Wang, J. Phys. Chem. B, 2008, 112, 3315–3321 CrossRef CAS.
- B. Pan, D. Cui, P. Xu, C. Ozkan, G. Feng, M. Ozkan, T. Huang, B. Chu, Q. Li and R. He, Nanotechnology, 2009, 20, 125101 CrossRef.
- H. Ali-Boucetta, K. T. Al-Jamal, D. McCarthy, M. Prato, A. Bianco and K. Kostarelos, Chem. Commun., 2008, 459–461 RSC.
- Z. Liu, X. Sun, N. Nakayama-Ratchford and H. Dai, ACS Nano, 2007, 1, 50–56 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |