The role of ellipticity on the preferential binding site of Ce and La in C78-D3h—A density functional theory study
K.
Muthukumar
* and
J. A.
Larsson
Tyndall National Institute, University College Cork, Lee Maltings, Prospect Row, Cork, Ireland. E-mail: Kaliappan.Muthukumar@gmail.com; Fax: +00353 21 4270271; Tel: +00353 21 4904201
First published on 13th May 2010
Abstract
Endohedral metallofullerenes that encapsulate one or several atoms, or a cluster of atoms have molecular properties making them useful both in technology and in bio-medical applications. Some fullerenes are found to have two metal atoms incarcerated and it has been recently found that two Ce atoms are incorporated into the C78-D3h (78![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) cage. In this study, we report calculations on the structural and electronic properties of Ce2@C78 using density functional theory (DFT). While Ce2@C80-Ih (D3d) and La2@C80-Ih (D2h) have different ground state structures, we have found that Ce2@C78 has a D3h ground state structure just as La2@C78. The encapsulated Ce atoms bind strongly to the C78-D3h cage with a binding energy (BE) of 5.925 eV but not as strong as in Ce@C82-C2v nor in Ce2@C80-Ih. The elliptical nature of the cage plays a crucial role and accommodates the two Ce atoms at opposite ends of the C3 axis with a maximized inter atomic distance (4.078 Å). This means that the effect of the additional f-electron repulsion in M2@C78 with M = Ce compared to M = La, is less pronounced than in Ce2@C80 compared to La2@C80. We compare the results to the elliptical M2@C72 (#10611) (M = La, Ce), and with a range of additional Ce and La endohedral fullerenes and explain the role ellipticity has in the preferential binding site of Ce and shed light on the formation mechanism of these nanostructures.
:5) cage. In this study, we report calculations on the structural and electronic properties of Ce2@C78 using density functional theory (DFT). While Ce2@C80-Ih (D3d) and La2@C80-Ih (D2h) have different ground state structures, we have found that Ce2@C78 has a D3h ground state structure just as La2@C78. The encapsulated Ce atoms bind strongly to the C78-D3h cage with a binding energy (BE) of 5.925 eV but not as strong as in Ce@C82-C2v nor in Ce2@C80-Ih. The elliptical nature of the cage plays a crucial role and accommodates the two Ce atoms at opposite ends of the C3 axis with a maximized inter atomic distance (4.078 Å). This means that the effect of the additional f-electron repulsion in M2@C78 with M = Ce compared to M = La, is less pronounced than in Ce2@C80 compared to La2@C80. We compare the results to the elliptical M2@C72 (#10611) (M = La, Ce), and with a range of additional Ce and La endohedral fullerenes and explain the role ellipticity has in the preferential binding site of Ce and shed light on the formation mechanism of these nanostructures.
Introduction
Fullerenes, hollow carbon cages on the order of 1 nm in diameter, are future building blocks in many areas of applied and fundamental nanoscience.1–3 Fullerenes show potential for applications in nanotechnology due to the possibility of tuning their properties by doping and/or functionlization. In particular, the endohedral doping of the hollow carbon cage with metal atoms and clusters allows changing of the electronic and magnetic properties of the molecule without significant distortion of the geometry of the outer shell.4,5 Despite their outstanding applications in technology and in bio-medical sciences the limited availability of endohedral metallofullerens has hindered studies that describe their electronic and structural properties, the knowledge of which are essential prerequisites for many nanotechnological applications. The information about fullerenes that incarcerate a metal atom such as M@C60, M@C70, M@C82, (M = Sc, Y, La, Ce, etc.) are relatively abundant compared to the family of endohedral di-metallofullerenes that encapsulate two atoms inside the cage, (i.e., M2@C72, M2@C78 and M2@C80 (M = La or Ce)), which is attributed to the difficulty in separating them from co-formed species.6–16 Due to the many conceptual applications based on the electrically or magnetically controlled motion of the dopant within the cage there is considerable interest in these di-metallofullerenes. But for these applications to be viable their synthesis needs to have higher yields, which we will address using the findings from our simulations, and their separation from each other needs to be improved.The isolation of Ce2@C78 has been reported recently and it's visible–near infrared (NIR) spectrum show characteristic absorptions similar to La2@C78.17 Observation of eight peaks in the 13C nuclear magnetic resonance (NMR) spectrum of Ce2@C78 reveal that the cage is D3h symmetric. The single-crystal X-ray structure study of bis-silylated Ce2@C78 shows that the two Ce atoms are located on the C3 axis of the C78-D3h (78:5) cage17 (we will henceforth use ‘C78’ for the C78-D3h (78:5) cage unless otherwise stated). However, it should be noted that the NMR time scale is long compared to atomic movements and can thus in dynamic cases only give an average structure. Also, the position of the encapsulated metal atoms is sensitive to cage wall functionalization18 and the absence of proper theoretical computations has resulted in misleading conclusions for endohedral fullerenes in the past. For example the prediction of a Cs minimum,19 not C2v for La@C82,20,21 and similarly, D3d minimum,22 not D2h for La2@C80,23,24 as well as faulty predictions of the position of Gd12–14,25 and Eu26 in C82-C2v which have both been shown to be C2v symmetric.27 In fact, theoretical studies using DFT calculations are essential to understand the nature of La and Ce bonding in different fullerene cages,3,15,16,21–23,28–34 and have been used to explain the bonding sites of Ce in Ce@C8215 and Ce2@C80.16 Until now, theoretical studies explaining the structural and electronic properties of Ce2@C78 have not been reported and therefore we report in this paper the DFT characterization of the structural and electronic properties of Ce2@C78 and compare it with other dimetallofullerenes such as Ce2@C72, Ce2@C80 and La2@C78, and shed light on the crucial role the elliptical nature of C78 and C72 has for the preferential binding site of incarcerated metal atoms.
Computational method
We have used unrestricted DFT with the generalized gradient approximation (GGA) exchange–correlation functional PBE35 together with the resolution of identity (RI) approximation36 and a dense grid (m5) as implemented in Turbomole 5.10.37–40 We have found in our previous studies that the bonding is critically dependent on the theoretical description of the valence electrons and found for La and Ce that to get the correct chemistry of these lanthanoids a relativistic small core effective core potential (ECP) and associated triple zeta basis sets need to be used.15,16 Therefore for all our computations, a polarized valence triple zeta basis set (TZVP) was used, which includes d functions for carbon and g functions for cerium and lanthanum.41,42 These basis sets were used in conjunction with the ECP-28-MWB relativistic ECP for Ce and La.43 Mulliken charges have been computed using the double zeta basis set (DZVP) on carbon on the PBE/TZVP geometries.Results and discussion
C78 has five IPR (isolated pentagon rule) isomers D2 (78:1), C2v (78:2), C2v (78:3), D3h (78:4), D3h (78:5) in its pristine form and it has been established by 13C NMR measurements that La and Ce are encapsulated in the D3h (78:5) isomer.32,17 Our calculations confirms this for the case of the two D3h cages with Ce2@C78-D3h (78:5) being 2.747 eV more stable than Ce2@C78-D3h (78:4). Ce2@C78 is most stable as a triplet and the lowest singlet and quintuplet are 527 meV and 610 meV higher in energy, respectively. In this study, we have carried out geometry optimisations starting from several structurally inequivalent initial arrangements of the two Ce atoms inside the C78-D3h cage in an attempt to find all possible stationary points with regard to Ce positions. Regardless of the initial structure, the two Ce atoms end up at equivalent binding sites at the two opposite focal points of the elliptical C78-D3h cage during normal optimisation at 0 K (i.e. they repel each other as do the two Ce atoms in Ce2@C8016) resulting in a D3h symmetric ground state structure (see Fig. 1). More importantly, the Ce atoms favour only one particular binding site in C78-D3h similar to Ce in Ce@C82.15 In the ground state D3h symmetric structure of Ce2@C78, each Ce atom binds to the centre of a six-membered ring on the opposite side of a C3-axis with the Ce–C distances of 2.504 Å (6 bonds). The two carbon–carbon conjugated bonds of this six-membered ring in the empty C78-D3h are 1.393 and 1.443 Å, and binding Ce locally distorts the cage with elongations of 0.017 (6–5 bonds) and 0.053 (6–6 bonds) Å to these bonds, respectively. The two Ce atoms in Ce2@C78 are separated by 4.078 Å, which is longer than 3.905 Å found in Ce2@C80 due to the Ce atoms being bound at opposite focal points of this elliptical fullerene. The binding energy (BE) of each Ce atom in Ce2@C78 is 5.925 eV which is weaker than the BE of Ce in Ce@C82 and Ce2@C80 (see Table 1). Our calculations on La2@C78 result in a D3h symmetric ground state structure as reported in ref. 32, and our comparison of its La BE with La@C82 and La2@C80 indicates a similar trend as Ce illustrating that these two dopants bind with the cage in a similar fashion in C78-D3h, which has, remarkably, been found to be different in the case of M2@C80-Ih (M = La, Ce).16 Similarly, our calculations of the non-IPR Ce2@C72-D2 (#10611) show that Ce binds at the two focal points also in this elliptic cage as to attain maximum separation analogous to La in La2@C72.33,34 In C72-D2 the Ce (La) atoms bond to a partial double bond where two pentagons fuse with the BE of 6.668 eV (7.146 eV) and a Ce⋯Ce distance of 4.157 Å (similar to La⋯La in La2@C72). Comparison of the BEs of Ce and La in C72 shows that the bonding of these endohedral dopants is less favourable than in C82-C2v but more favourable than in C80-Ih.
 | ||
| Fig. 1 DFT optimised structure of the global D3h minimum Ce2@C78-D3h (side and top views). | ||
Both encapsulated La and Ce atoms bind with the cage through strong hybridisation between their s, p, and d orbitals and the cage π orbitals, while only Ce also binds through its f orbitals. Analysis of the electronic structure of Ce2@C78 shows that Ce retains most of its f character as Ce does in Ce@C82 and in Ce2@C80, which is evident from the presence of Ce f dominated frontier orbitals (HOMO-1 (54% of Ce f), HOMO (95% of Ce f) and LUMO (49% of Ce f)). HOMO-1 is Ce(f)⋯Ce(f) bonding as is LUMO, while the HOMO is Ce(f)⋯Ce(f) antibonding (see Fig. 2). The metal–cage interaction in endohedral metallofullerenes is maximized when Ce or La binds to a six-membered ring of C2v-symmetry, that has been found to be favourable for electrostatic and metal(d)–cage(π) interactions.15 In the case of di-metallofullerenes the dopant atoms repel each other in addition to the metal–cage interaction. This repulsion differs between La and Ce atoms in that Ce has an f-electron. Hence, the binding site of Ce in Ce2@C80 differs from La in La2@C80, as shown by Muthukumar and Larsson in ref. 16 where a change in binding site from the centre of a C2v symmetric six-membered ring for La2@C80 and Ce@C80 to on-top of an atom in Ce2@C80 accommodates an increased Ce⋯Ce distance due to the additional Ce(f)⋯Ce(f) repulsion (non-bonding interaction). In the case of Ce2@C78 both HOMO-1 and HOMO are singly occupied, and thus the Ce(f)⋯Ce(f) interaction is non-bonding, similar to the Ce atoms in Ce2@C80.16 Other occupied orbitals in the energy level spectrum down to 12 eV below HOMO are dominated by cage character (85–100%), but often have hybridisation from Ce s, p, d and f orbitals. Although, the electronic structure and the interaction between the two Ce atoms in Ce2@C78 remains similar to Ce2@C80, we observe a similar binding site for Ce and La in C78-D3h resulting in a D3h symmetric ground state structure for both species. The driving force behind the binding site change in Ce2@C80-Ih is the increased Ce⋯Ce distance that can be accommodated in the D3d configuration compared to D2h. In Ce2@C78, however, the Ce⋯Ce distance is sufficiently large due to the elliptic shape of the cage.
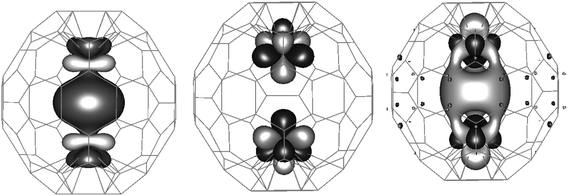 | ||
| Fig. 2 HOMO-1, HOMO and LUMO of Ce2@C78-D3h. HOMO-1 and HOMO are singly occupied in the triplet ground state. | ||
To elucidate the bonding nature of Ce and La in M2@C78 (M = La/Ce), calculations were also carried out for M@C78 (M = La/Ce) and the results show that the C3v symmetric configuration, for Ce@C78 and La@C78 where the metal atom binds to one of the six-membered ring on the C3 axis is found to be most stable (see Fig. 3). This is the same binding site the dopant atoms occupy in the D3h symmetric ground state of M2@C78, indicating that the presence of an additional Ce or La atom has no impact on the preferential binding site of the other Ce/La. It is surprising that the C2v configuration of M@C78 where the Ce (La) atom binds to the centre of a six-membered ring on a C2 axis of C78-D3h (located on the equator of the elliptical cage) is less stable than the C3v configuration by 554 meV (330 meV for La). This observation where Ce/La prefers a C3v six-membered ring rather than a C2v six-membered ring is peculiar since the C2v-symmetric six-membered rings in C78-D3h has a very similar charge pattern to what has been found for the C2v-symmetric Ce (La) binding site in C82 (see Fig. 3), which is the strongest binding of all studied metallofullerenes (see Table 1).15 This remarkable effect can be explained by comparing the degree of partial double bond character in the C–C bonds of the C2v six-membered rings in C78-D3h and C82-C2v: We have found that the 6–5 bonds of the C2v bonding site in C78 (1.470 Å) are much longer than in C82 (1.429 Å). Thus, the degree of double bond character of the C–C bonds of the six-membered ring is important for Ce binding, which is evident when the distortion in the 6–5 bond is compared between the C2v configuration of Ce@C78 and the ground state of Ce@C82: no change; compared to 0.043 Å elongations in the latter (similarly for La: i.e. no change; compared to 0.044 Å elongation). This analysis reveals a large difference between the C2v binding sites in the two cages, both with regard to geometry and electronic structure (lower electron density in the 6–5 bonds in C78) that strongly affects the bonds to Ce and La. Also, it should be noted that the BE of Ce in C3v-Ce@C78 is 0.798 eV lower than for Ce@C82. However, the Ce BE is 0.794 eV higher than for Ce@C60, although both are bound to C3v symmetric six-membered rings. A very similar trend has also been observed for La (see Table. 1). This means that the specific geometry, including the curvature and electronic structure around the binding site in different cages matter for the metal atom BE.
 | ||
| Fig. 3 Ground state structures of Ce endohedral fullerenes. The view is along the symmetry axes of the molecules, thus the second Ce atom is obstructed in the di-metallofullerenes. In the La analogues of these molecules, La occupy the same binding site as Ce, except in the case of M2@C80 (M = La/Ce). The 1st Schönflies symbol gives the symmetry of the molecule, while the 2nd one notes the symmetry of the cage isomer in its free undoped form. In C3v-Ce@C78-D3h the view is along the C3-axis. | ||
Comparison of Ce and La binding site in mono and dimetallofullerenes such as M@C60, M@C78, M@C80, M@C82, M2@C72, M2@C78, M2@C80, (M = La/Ce) indicates that in every cage studied there is one particular binding site, with a specific binding configuration, that is the most stable (of which there are several symmetry equivalent ones for M@C60, M@C80 and M2@C80, (see Fig. 3)), which these lanthanoids occupy constantly at room temperature (except for in C80-Ih where there are free dynamics of the encapsulated metal atoms at room temperature, due to the multitude of equivalent binding sites and the low barrier between them). Often the competitive binding sites within each cage are less favourable by 0.5 eV or more than the preferred binding site. This is in itself a remarkable feature of endohedral metallofullerenes, and is not intuitively obvious since the cage wall structure seems quite uniform at a casual glance. However, in the cages with only one principal symmetry axis, only one out of very similar units favourably binds the metal atom (e.g. in the case of C82-C2v). These selective metal–cage binding site interactions are governed by: the five- and six-membered ring structuring; the shape of the cage; the degree of double bond character in the C–C bonds that bind to the metal atom; and the presence of another element inside the cage. These effects are general for transition metal and lanthanoid endohedral doping and our investigation of M2@C72, M2@C78 and M2@C80 show that bonding of Ce/La in these cages is strongly affected by repulsion between the metal atoms. The f⋯f interaction between the two Ce atoms adds to the repulsion, which potentially changes the Ce binding site compared to La. This metal–metal repulsion leads to ground state configurations with a considerable separation distance, i.e. the metal atoms are bound at opposite ends of the cage. For Ce the metal atoms inter-distance needs to be slightly longer than for La to obtain an optimum structure, due to the above mentioned additional f⋯f repulsion. It has been found by Muthukumar and Larsson16 that this gives different ground state configurations for Ce2@C80-Ih, which is D3d symmetric with a Ce⋯Ce separation of 3.904 Å, and La2@C80-Ih that is D2h symmetric with a La⋯La separation of 3.868 Å. In the case of M2@C78-D3h, and M2@C72-D2, the metal atoms bind to the cage at their focal points and their separation is naturally longer because of the elliptical shape (see Fig. 4), which results in longer M⋯M distances: 4.078 Å in Ce2@C78-D3h and 4.157 Å in Ce2@C72-D2; and 4.077 Å in La2@C78-D3h and 4.153 Å in La2@C72-D2. These separations are more than enough to minimize the destabilization that comes with the repulsion, and result in a non-bonded interaction for the Ce(f)⋯Ce(f) orbitals.
 | ||
| Fig. 4 DFT optimised structures of cerium endohedral fullerenes (side views) of Ce2@C80-Ih, Ce2@C78-D3h and Ce2@C72-D2 (C72-D2 is more elliptical than C78-D3h). | ||
Three observations can be made to draw general conclusions about the growth mechanism of metallofullerenes in arc-discharge synthesis. (i) Most favourably during growth is the clustering of carbon atoms bound to a metal atom44 resulting in the formation of a C2v-symmetric six-membered ring binding site.15 This binding site is found in the two most abundant metallofullerenes M@C82-C2v and M2@C80-Ih. In these molecules the Ce/La BEs are found to be large, which is indicative of favourable binding. This has led us to conclude that the initial formation of a graphitic flake with a central C2v-symmetric six-membered ring governs the shape (and symmetry) of the entire cage when grown around a lanthanide atom,15 which almost exclusively results in the egg-shaped C82-C2v cage, and a C82–Cs isomer with the only difference in structure is at the far-end of the cage, furthest away from the metal atom's preferential binding site (in a 4:1 ratio). (ii) The interaction between lanthanide atoms has a huge impact on the cage formation. In the general case the presence of two metal atoms results in the formation of a spherical C80-Ih cage, which is evident by the abundance of M2@C80-Ih from the synthesis of endohedral metallofullerenes. Since the overall structure of the C82 cage for the half that metal atom binds to is very similar to that of C80-Ih (see Fig. 3), we suggest that M2@C80 is formed when the second metal atom becomes involved in the cage formation rather late in the process, i.e. a graphitic flake with a central C2v-symmetric six-membered ring is initially formed in the presence of one dopant atom, just like in the formation of C82, and the latter involvement of a second dopant atom dictates the formation of C80 of Ih symmetry. It is at least reasonable to assume that some interaction between the two metal atoms must control the formation of M2@C80-Ih since almost no M@C80-Ih is detected in the product (only M@C80-C2v (M = La) has been identified as a previously ‘missing metallofullerene’45). Thus, the percentage of M2@C80-Ih in the product could possibly be increased by optimizing the ratio of metal to carbon in the forming gas, which should be high but not too high (see below). In M2@C80-Ih the two metal atoms are always located at opposite positions on the cage inner wall, thus, maximizing the distance between each other. The repulsion between the metal atoms is most probably a driving force for the formation of elliptically shaped metallofullerenes such as M2@C78-D3h and M2@C72-D2, which leads us to the last observation. (iii) The interaction between lanthanide atoms can also create graphitic flakes, and ultimately fullerene cages, that does not have a central C2v-symmetric six-membered ring. The focal point binding sites in M2@C78-D3h (C3v-symmetric six-membered ring) and M2@C72-D2 (a partial double bond fusing two five-membered rings) attest to it. These binding sites also provide favourable interactions for the metal atoms, as can be seen from the large BEs (see Table 1), although not as favourable as in M@C82 and its C2v-symmetric six-membered ring. The stability of these focal point binding sites is further corroborated by the fact that our simulations also result in these binding sites when one endohedral dopant is removed creating the artificial M@C78-D3h (not found experimentally), and thus removing the M⋯M repulsion. The explanation for the increased binding energy for the C3v symmetric six-membered ring in C78-D3h and the two fused five-membered rings in C72-D2 compared to similar binding sites in more spherical fullerenes lies in the increased curvature of the graphitic carbon structure at the focal points. But it is important to note that these binding sites are only formed in the presence of two metal atoms since there are no metallofullerenes with only one metal dopant atom that has two fused five-membered rings, and Ce@C60 which has a C3v-symmetric six-membered ring binding site just like M2@C78-D3h (see Fig. 3) is produced in small quantities and has a small metal (Ce/La) BE. We speculate that the two elliptical di-metallofullerenes studied here could be targeted in synthesis by having a high percentage metal in the vapour forming gas; possibly metal concentrations so high that each clustering of carbon atoms around a metal atom happens in the presence of a second metal atom.
A reasonable measure of charge transfer in endohedral fullerenes is obtained from DFT computed electron densities through the proper usage of Mulliken atomic charges analysis. The analysis of Ce2@C78 charges shows that each Ce donates only 0.07 of its electrons to the cage. Although the total charge transfer is small we will show that Ce has oxidation state +3, as has been shown previously for Ce in Ce@C8215 and Ce2@C8016 (see discussion below). Further inspection of the charge transfer in Ce2@C78 reveals that Ce donates 1.49 of its s electrons, and a small amount of its f (0.13) electron to the C78 cage. This is compensated by considerable back-donation into 5d (-1.19) and also into 6p (-0.49) of Ce. The back donation from the cage to the Ce d orbitals is larger for Ce2@C78 than for Ce@C82 but similar to Ce2@C80. This analysis reveals that the electrons of Ce are not simply donated to the cage, but are localised in Ce–C bonds, which is the reason that Mulliken charges do not correspond directly to oxidation numbers in the case of polarized covalent bonding.15,16 A more detailed comparison with the charge transfer in Ce tri halides (CeF3 and CeCl3) has also confirmed this.16 Therefore it should be noted that models based on the simplified zwitterion, i.e. adding this number of electrons to the empty cage, without specific inclusion of the metal atom will not describe many of the endohedral fullerene properties properly as it explicitly ignores the localized hybridization between the metal atom(s) and the cage.
For M2@C78-D3h (M = Ce, La) the ground state is the only stationary point for the molecule, i.e. no local minima structures have been found in our geometry optimizations with the Ce atoms in a range of different initial configurations, and all simulations relax to the same D3h symmetric ground state structure. The vibrational spectrum has been calculated for the ground state of Ce2@C78, and has confirmed that it is a true minimum (see Fig. 5). We find absorption bands between 48 cm−1 and 1556 cm−1 for Ce2@C78-D3h and we find two e′′, two e′, an a1′ and an a2′′ mode below 200 cm−1 (amounting to six vibrational degrees of freedom) that correspond to bending modes of the Ce atoms. The frequencies observed between 213 and 1556 cm−1 correspond to cage vibrations, or mixed Ce and cage vibrations.
 | ||
| Fig. 5 Calculated vibrational spectrum of Ce2@C78-D3h. IR signal intensity plotted as a function of vibrational frequency. | ||
It is evident from our results that all experimentally observed metallofullerenes provide strong binding for the metal dopant atoms through a range of different binding sites with different graphitic structuring and local curvature. In all cases there is considerable dative covalent bonding with the cage through the metal atoms s and f electrons, with back donation to its unoccupied p and d orbitals. The C2v-symmetric six-membered ring binding site is the most favourable, but the metal atom can make do with other binding sites, as long as the complex charge transfer can be accommodated. We have found that not only the local structuring but also the C–C bond lengths (degree of double bond character) is important. This can give rise to strong variations in metal BEs between binding sites with the same local structure. The suitability of the binding site for metal binding is most likely purpose built in the presence of the metal atom(s) during the formation of the cages, i.e. the metal atom(s) catalyzes the growth of graphitic flakes of certain suitable shapes and forms, resulting in certain fullerene cages: We postulate that C82 is formed around one metal atom; C80-Ih is formed when a second metal atom is added to a ‘half-grown’ cage; and the C78-D3h and C72-D2 cages are formed when two metal atoms interact from the start of cage growth.
Conclusion
A comparative DFT study of M2@C78 (M = Ce, La) has been performed and our results reveal that Ce2@C78 retains the D3h symmetry of the C78-D3h (78:5) cage. The Ce atoms bind to a C3v symmetric six-membered ring of C78-D3h as in La2@C78. This situation is quite different from what has been observed for M2@C80 (M = Ce, La) where Ce binds on-top of a carbon atom on the C3 axis leading to a D3d ground state configuration, while La binds to the centre of six-membered rings leading to a D2h ground state structure. The difference between Ce2@C80 and La2@C80 stems from the additional repulsion between the two Ce atoms due to the Ce(f)⋯Ce(f) interaction. We have shown that the M⋯M interaction plays a vital role in determining the position of metal atom and its bonding with the cage. In M2@C78 and M2@C72 (M = Ce, La) the elliptical nature of the cage assist the dopant atoms to bond strongly with the cage at its focal points, thus avoiding the M⋯M interaction leading to their maximum separation. The two encapsulated dopant atoms are equivalent, and in the case of Ce retain much of their f character. Binding to a C3v symmetric six-membered ring (C78-D3h) results in lower binding energies than in C82-C2v and C80-Ih for both Ce and La. While bonding to a 5–5 bond (C72-D2) is considerably more favourable for both Ce and La, with bonding energies between those in C80-Ih and C82-C2v, which means that metal binding inside C82-C2v is still the strongest encountered in metallofullerene. The vibrational spectrum has been computed showing that the D3h configuration is the ground state isomer, and the low energy modes observed are due to the metal atom vibrations. We have found no additional local minimum configurations and, thus, the ground state is the only stationary state for M2@C78-D3h (M = Ce, La). The charge transfer analysis indicates that Ce is in a +3 oxidation state. Also, we have compared our results with the studies available for variety of La and Ce endohedral fullerenes and provided a most probable explanation for their formation mechanism.
Acknowledgements
This work was supported by Science Foundation Ireland and the FP6 Marie Curie Early Stage Training Network NANOCAGE (MEST-CT-2004-506854). The authors wish to acknowledge the SFI/HEA Irish Centre for High-End Computing (ICHEC) for the provision of computational facilities, and also SFI/HEA for the provision of local computing clusters.References
- H. Shinohara, Rep. Prog. Phys., 2000, 63, 843 CrossRef CAS.
- K. Muthukumar, J. A. Larsson in Advances in Nanotechnology and Applications, (ISBN: 1-4392-5489-3), CENTERA, Louisville, KY, USA ed. Y. V. Pathak, H. T. Tran, 2009, Vol. 1, Ch. 8, PP. 127 Search PubMed.
- A. A. Popov, J. Comput. Theor. Nanosci., 2009, 6, 292 CrossRef CAS.
- A. Stróżecka, K. Muthukumar, A. Dybek, T. J. Dennis, J. A. Larsson, J. Mysliveček and B. Voigtländer, Appl. Phys. Lett., 2009, 95, 133118 CrossRef.
- K. Muthukumar, Ph.D. Thesis, University College Cork, 2009.
- S. Nagase and K. Kobayashi, Chem. Phys. Lett., 1994, 228, 106 CrossRef CAS.
- J. Ding, L.-T. Weng and S. Yang, J. Phys. Chem., 1996, 100, 11120 CrossRef CAS.
- S. Lebedkin, B. Renker, R. Heid, H. Schober and H. Rietschel, Appl. Phys. A: Mater. Sci. Process., 1998, 66, 273 CrossRef CAS.
- L. Wang, K. Schulte, R. A. J. Woolley, M. Kanai, T. J. S. Dennis, J. Purton, S. Patel, S. Gorovikov, V. R. Dhanak, E. F. Smith, B. C. C. Cowie and P. Moriarty, Surf. Sci., 2004, 564, 156 CrossRef CAS.
- K. Schulte, L. Wang, P. J. Moriarty, J. Purton, S. Patel, H. Shinohara, M. Kanai and T. J. S. Dennis, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 115437 CrossRef.
- J. Ding and S. Yang, J. Am. Chem. Soc., 1996, 118, 11254 CrossRef CAS.
- L. Senapati, J. Schrier and K. B. Whaley, Nano Lett., 2004, 4, 2073 CrossRef CAS.
- L. Wang and D. Yang, Nano Lett., 2005, 5, 2340 CrossRef CAS.
- L. Senapati, J. Schrier and K. B. Whaley, Nano Lett., 2005, 5, 2341 CrossRef CAS.
- K. Muthukumar and J. A. Larsson, J. Phys. Chem. A, 2008, 112, 1071 CrossRef CAS.
- K. Muthukumar and J. A. Larsson, J. Mater. Chem., 2008, 18, 3347 RSC.
- M. Yamada, T. Wakahara, T. Tsuchiya, Y. Maeda, M. Kako, T. Akasaka, K. Yoza, E. Horn, N. Mizorogi and S. Nagase, Chem. Commun., 2008, 558 RSC.
- M. Yamada, C. Someya, T. Wakahara, T. Tsuchiya, Y. Maeda, T. Akasaka, K. Yoza, E. Horn, M. T. H. Liu, N. Mizorogi and S. Nagase, J. Am. Chem. Soc., 2008, 130, 1171 CrossRef CAS.
- W. Andreoni and A. Curioni, Appl. Phys. A: Mater. Sci. Process., 1998, 66, 299 CrossRef CAS.
- Y. Maeda, Y. Matsunaga, T. Wakahara, S. Takahasi, T. Tsuchiya, M. O. Ishitsuka, T. Hasegawa, T. Akasaka, M. T. H. Liu, K. Kokura, E. Horn, K. Yoza, T. Kato, S. Okubo, K. Kobayashi, S. Nagase and K. Yamamoto, J. Am. Chem. Soc., 2004, 126, 6858 CrossRef CAS.
- P. Jin, C. Hao, S. Li, W. Mi, Z. Sun, J. Zhang and Q. Hou, J. Phys. Chem. A, 2007, 111, 167 CrossRef CAS.
- H. Shimotani, T. Ito, Y. Iwasa, A. Taninaka, H. Shinohara, E. Nishibori, M. Takata and M. Sakata, J. Am. Chem. Soc., 2004, 126, 364 CrossRef CAS.
- K. Kobayashi, S. Nagase and T. Akasaka, Chem. Phys. Lett., 1995, 245, 230 CrossRef.
- J. Zhang, C. Hao, S. Li, W. Mi and P. Jin, J. Phys. Chem. C, 2007, 111, 7862 CrossRef CAS.
- E. Nishibori, K. Iwata, M. Sakata, M. Takata, H. Tanaka, H. Kato and H. Shinohara, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 113412 CrossRef.
- B.-Y. Sun, T. Sugai, E. Nishibori, K. Iwata, M. Sakata, M. Takata and H. Shinohara, Angew. Chem., Int. Ed., 2005, 44, 4568 CrossRef CAS.
- N. Mizorogi and S. Nagase, Chem. Phys. Lett., 2006, 431, 110 CrossRef CAS.
- W. Andreoni and A. Curioni, Appl. Phys. A, 1998, 66, 299 CrossRef CAS.
- M. Yamada, T. Wakahara, Y. Lian, T. Tsuchiya, T. Akasaka, M. Waelchli, N. Mizorogi, S. Nagase and K. M. Kadish, J. Am. Chem. Soc., 2006, 128, 1400 CrossRef CAS.
- W. Andreoni and A. Curioni, Phys. Rev. Lett., 1996, 77, 834 CrossRef CAS.
- Z. Slanina, S. L. Lee, F. Uhlik, L. Adamowicz and S. Nagase, Int. J. Quantum Chem., 2005, 104, 272 CrossRef CAS.
- B. Cao, T. Wakahara, T. Tsuchiya, M. Kondo, Y. Maeda, G. M. A. Rahman, T. Akasaka, K. Kobayashi, S. Nagase and K. Yamamoto, J. Am. Chem. Soc., 2004, 126, 9164 CrossRef CAS.
- H. Kato, A. Taninaka, T. Sugai and H. Shinohara, J. Am. Chem. Soc., 2003, 125, 7782 CrossRef CAS.
- Z. Slanina, Z. Chen, P. V. R. Schleyer, F. Uhlk, X. Lu and S. Nagase, J. Phys. Chem. A, 2006, 110, 2231 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119 CrossRef CAS.
- K. Eichkorn, O. Treutler, H. Öhm, M. Haser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652 CrossRef CAS.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346 CrossRef CAS.
- M. V. Arnim and R. Ahlrichs, J. Comput. Chem., 1998, 19, 1746 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2002, 4, 4285 RSC.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef CAS.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348 CrossRef CAS.
- S. Suzuki, H. Torisu, H. Kubota, T. Wakabayashi, H. Shiromaru and Y. Achiba, Int. J. Mass Spectrom. Ion Processes, 1994, 138, 297 CrossRef.
- H. Nikawa, T. Yamada, B. Cao, N. Mizorogi, Z. Slanina, T. Tsuchiya, T. Akasaka, K. Yoza and S. Nagase, J. Am. Chem. Soc., 2009, 131, 10950 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |