Flame spray pyrolysis: An enabling technology for nanoparticles design and fabrication
Wey Yang
Teoh
a,
Rose
Amal
a and
Lutz
Mädler
*b
aARC Centre of Excellence for Functional Nanomaterials, School of Chemical Engineering, The University of New South Wales, Sydney, NSW 2052, Australia
bFoundation Institute of Materials Science (IWT), Department of Production Engineering, University of Bremen, 28359, Bremen, Germany. E-mail: lmaedler@iwt.uni-bremen.de; Fax: +49-421-218-5378; Tel: +49-421-218-7737
First published on 17th May 2010
Abstract
Combustion of appropriate precursor sprays in a flame spray pyrolysis (FSP) process is a highly promising and versatile technique for the rapid and scalable synthesis of nanostuctural materials with engineered functionalities. The technique was initially derived from the fundamentals of the well-established vapour-fed flame aerosols reactors that was widely practised for the manufacturing of simple commodity powders such as pigmentary titania, fumed silica, alumina, and even optical fibers. In the last 10 years however, FSP knowledge and technology was developed substantially and a wide range of new and complex products have been synthesised, attracting major industries in a diverse field of applications. Key innovations in FSP reactor engineering and precursor chemistry have enabled flexible designs of nanostructured loosely-agglomerated powders and particulate films of pure or mixed oxides and even pure metals and alloys. Unique material morphologies such as core–shell structures and nanorods are possible using this essentially one step and continuous FSP process. Finally, research challenges are discussed and an outlook on the next generation of engineered combustion-made materials is given.
 Wey Yang Teoh | Wey Yang Teoh is an ARC Australia Postdoctoral Fellow (APD). He obtained his doctorate from the School of Chemical Engineering, University of New South Wales in 2007 and spent a brief attachment at the Swiss Federal Institute of Technology (ETH) Zürich in 2003. He continued his postdoctoral work at the laboratory of Prof. Rose Amal, where he is a Program Leader in flame spray pyrolysis with focusing on energy, environmental and bioapplications. He will assume the role of Assistant Professor at the School of Energy and Environment, City University of Hong Kong. |
 Rose Amal | Rose Amal is an ARC Australian Professorial Fellow (APF) and University of New South Wales (UNSW) Scientia Professor. She heads a group of over 30 research members at the Particles and Catalysis Research Group, School of Chemical Engineering, UNSW. She is the Director of the ARC Centre of Excellence for Functional Nanomaterials, a multi-institution research centre. She is also the inaugural Director of the Centre for Energy Research and Policy Analysis (CERPA), the new Energy Research Institute at UNSW. Her research interests are in particle technology and nanomaterials, spreading over a wide range of applications from energy, environmental to bioapplications. |
 Lutz Mädler | Lutz Mädler (Dr -Ing. Univ. Freiberg/Sa., Germany in 1999) was born in Zwickau, Germany. He received his Habilitation in 2003 at Swiss Federal Institute of Technology (ETH Zurich). He was Senior Lecturer at the Department of Chemical Engineering, University of California, before he joined the Department of Production Engineering at the University of Bremen leading the Particle and Process Technology division in 2008. He is also Director at the Foundation Institute of Materials Science. His research focuses on integrated aerosol processes for nanomaterials and surface films for sensors, catalysts, and optical devices, and on nano-bio-interactions. |
1. Introduction
The large scale production of nanoparticles by flame aerosol technology is a critical aspect contributing to the progress of a wider Nanotechnology. In fact, the technology existed long before the “Nano” buzz, but has since rapidly evolved. Industry leaders such as Cabot, Cristal (formerly SCM and Millenium Chemicals), DuPont, Evonik (formerly Degussa) and Ishihara, manufacture flame-made materials in millions of tons, valued at more than $15 billion/yr.1 This includes most notably, carbon blacks, fumed SiO2, TiO2, Al2O3 as well as other ceramic nanoparticles. While many of these nanoparticles are used in commodity applications such as reinforcing agents (carbon blacks, SiO2, carbon-coated SiO2), pigments (TiO2) and flowing aids (SiO2), emerging niche markets requiring more complex and functional materials are expected to spearhead the next phase of growth. This includes for example, polishing agents in microelectronics (doped CeO2), catalysts (Pt/Al2O3, V2O5/TiO2, Ru/Co3O4–ZrO2), dental fillers (Ta2O5–SiO2) and other specialty applications – see ref. 2 and references therein, many of which are multielements in nature. Given their complexities and the variety of available permutations, the design of sophisticated and functional flame materials is far from trivial and in fact system specific. Nevertheless, their importance and lucrativeness continue to drive the evolution of flame synthesis.Much is to be learned from the evolution of flame aerosol technology, developed through years of industrial significance: from reactor design to process sophistication and versatility. A classic example is the industrial manufacture of carbon blacks which evolved from the “smouldering” process to the current norm of jet-like flame aerosol reactors.3 Formed via the pyrolysis of hydrocarbon sprays, the carbon black syntheses take place under fuel rich conditions where about half the hydrocarbons are burnt to supply the energy that pyrolyses the other half to carbon black.4 Modern aerosol reactors for the production of ceramic oxides bear a strong resemblance to carbon black jet furnaces.5 The fundamental differences between the two, besides the choice of metal elements present in the precursors, lies in the combustion regime in which they operate: fuel rich to obtain carbon particles and lean oxidising conditions to obtain metal oxides. In some cases, combination of the two is also possible (e.g. C/TiO2, C/SiO2), but this is governed by the combustion engineering in tandem with the corresponding particle formation, materials chemistry and thermodynamics of the system.
Depending on the precursor state (aqueous-based, solvent-based, vapour) and combustion conditions, flame aerosol syntheses fall into three general categories:6,7 (a) vapour-fed aerosol flame synthesis (VAFS) where a metal precursor is supplied in the form of vapour like SiCl4 and TiCl4 to make most of today's ceramic commodities;8 (b) flame-assisted spray pyrolysis (FASP) where precursor is supplied in the state of low combustion enthalpy solution (<50% of total combustion energy) usually in aqueous solvent, and because of this, its combustion needs to be assisted by an external hydrogen or hydrocarbon flame;9 and (c) flame spray pyrolysis (FSP) where the precursor is also in liquid form, but with significantly higher combustion enthalpy (>50% of total energy of combustion), usually in an organic solvent.10 FSP (the focus of this review), which is the youngest of the three processes, has important technological elements such as self-sustaining flame, usage of liquid feeds and less volatile precursors, proven scalability, high temperature flames and large temperature gradients. It is also the most heavily explored in recent years for the production of complex and functional nanomaterials. Some of the most notable applications are in the areas of catalysis (as reviewed by Strobel et al.7 on general flame aerosols), optics and photonics,11–15 sensors,16–18 health care,19–21 magnetic materials,22–25 electroceramics for fuel cells26,27 and composite materials.28–33 A key feature in the use of FSP as a convenient tool in synthesising these nanomaterials is the ability to upscale its production, while closely preserving its tailored properties.
Despite being only at the dawn of FSP research, the applications and markets for these nanoparticles are so encouraging that pioneer start-ups based on the FSP process are already in operation. Quite recently, Johnson Matthey Co., a major manufacturer of catalysts, announced their systematic exploration of FSP-made catalysts at its Research Center in UK.34 Many opportunities await discovery, both in particles design as well as their applications. The key element lies in the continuous integration and bridging of knowledge between combustion engineering, aerosol technology and materials science. An inevitable complexity lies in the specificity of every new materials system.
Hence this review presents up-to-date knowhow in FSP, highlighting the many innovations in this area, from reactor design (Section 2) to particles design and synthesis (Section 4). Through this, it aims to provide a clearer, and more importantly, a holistic view on the relationship between synthesis properties and the resultant particle properties (see Fig. 1). Intrinsic properties specific to the precursor formulation (covered in Section 3) such as enthalpy of combustion, solvent–metal precursor specificity and combustion environment (excess or restrictive oxygen) are some of the governing factors, while externally tunable parameters such as the rate of enthalpy of combustion, quenching rate as mediated by natural heat loss or through external quenching unit, thermophoretic gradient, mixing stages of precursors etc. define the extrinsic properties. Understanding the intricate inter-relationship between synthesis properties and the limiting boundaries of flame profile, particle formation, materials chemistry and thermodynamics is critical towards the fabrication of functional nanomaterials with specific characteristics. These include the product particles size, homogeneity, morphologies, configurations and crystallite properties.
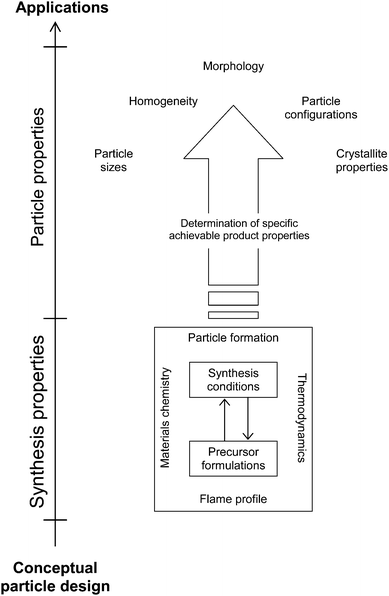 | ||
| Fig. 1 General paradigm and strategy for the synthesis of functional nanoparticles by FSP, from conceptual design to targeted end applications, taking into account the tunable synthesis parameters and their respective boundary conditions in achieving the product particles with desirable characteristics. | ||
It should be reiterated here that the current manuscript reviews exclusively FSP, with only brief accounts of VAFS and FASP that will be provided in Section 2, in view of their relevance to FSP. Readers interested in further information on other flame aerosol techniques are referred to reviews by Pratsinis5 which covered extensively the fundamentals and the then state-of-the-art in VAFS of ceramics nanoparticles, and Mädler6 on the spray aerosol techniques (with and without flame). Since the general applications of flame-made materials have been reviewed earlier by Strobel et al.,2,7 they shall not be repeated here. Rather, this review focuses on the general “art and science” of nanoparticle fabrication by FSP based on the most current knowledge in the area.
2. Flame aerosol reactors and flame spray pyrolysis
The origin of flame aerosols for useful materials can be traced as far back to ancient lampblacks production by which vegetable oils were combusted in oxygen-starved flames, yielding carbon soot fume.3 For centuries, this was the dominant form of carbon black, a rather coarse material probably due to prolonged and uneven residence times5 in these rather primitive pyrolysis reactors. Much later, the discovery of natural gas in US and gasification of tars in Europe enabled the production of carbon blacks with much finer texture to meet the demand as tire reinforcing agents.4 The so-called gas black or channel black was made by a VAFS process whereby gaseous hydrocarbons (natural gas) were combusted in gas jets and soot collected on water-cooled channel surfaces – justifying the name of “channel blacks” (Fig. 2, I). The high demand for carbon blacks driven by the growing tire market before and during World War II further motivated the development and scale-up of today's FSP “furnace process”. Here, liquid hydrocarbon fuel sprays, rather than natural gas, were combusted within a closed furnace, followed by control-quenching of the oxygen-starved flames by water sprays to yield “furnace blacks”4 (Fig. 2, III).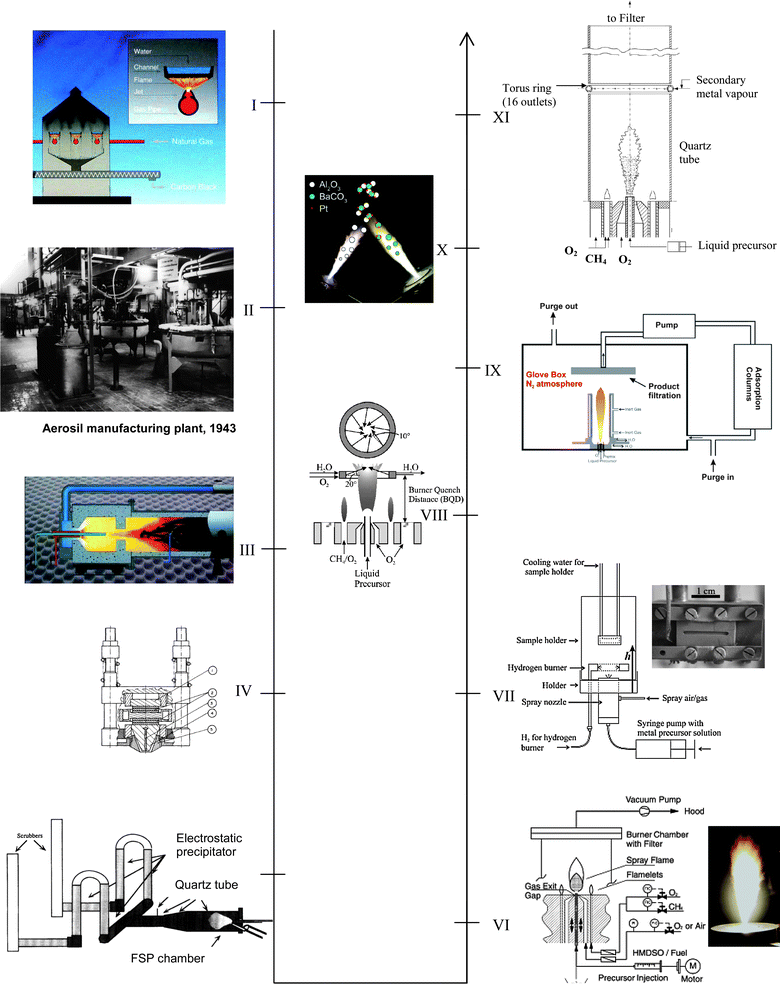 | ||
| Fig. 2 Evolution of aerosol flame reactors, including FSP reactor and its modifications. (I) 1940s: Early industrial-scale production of carbon blacks as commodity particles – “channel black process”. (II) 1940–50s: Production of fumed SiO2 (1943) from the flame hydrolysis of metal halide vapours (picture showing the first Aerosil production plant,36 source: courtesy of Evonik Degussa GmbH). The process further led to the manufacture of other commodity oxide ceramics such as Al2O3 (1953) and TiO2 (1954) using the same vapour-type flame technique. (III) 1970s: Furnace black process using a modern “jet-type” flame reactor. Figure adapted from ref. 36. (IV) 1977: First reported development of ultrasonic-assisted Flame Spray Pyrolysis by Sokolowski et al.,10 where metal acetylacetonate in organic solvent was used as the liquid precursor. (V) 1996/97: Gas-assisted atomiser FSP reported by Bickmore et al.28 and Karthikeyan et al.64 mark the pioneering of systematic studies in FSP. Figure adapted with permission from ref. 28. (VI) 2002: Pressure-assisted FSP reported by Mädler et al.65 that further led to systematic studies of various simple to complex metal oxides and metal/metal oxide systems using similar type reactor. (VII) 2004: A one-step film deposition technique by thermophoretic coating that further led to a series of supported catalysts and gas-sensor fabrications.77 (VIII) 2005: A flame height-selective rapid-quenching technique to control precisely the metal deposit size on metal oxide support.70 (IX) 2006: Reducing FSP by limiting the ambient O2 in flame reactor that allows production of metallic and carbon-coated nanoparticles.24 (X) 2006: A multi-nozzle FSP to synthesise multi-component particles with controllable phase segregation.89 (XI) 2008: In situ sequential coating of preformed FSP nanoparticles by introducing secondary metal vapour via a torus pipe ring with multiple hollow openings.94 Note: Figures are adapted with permissions from the respective references unless otherwise stated. | ||
At about the same time, the production of fumed SiO2 by VAFS was conceived where the vapour phase SiCl4 was hydrolysed in an oxy-hydrogen flame yielding pristine silica aerosols (Fig. 2, II).35 This patented process is known as the Aerosil process, after its commercial product name. Within 10 years after the first fumed SiO2 was produced, the manufacture of fumed Al2O3 (from AlCl3) and TiO2 (from TiCl4, chloride process) by VAFS followed, demonstrating the versatility of the flame process for product ceramic particles.36 A very important innovation of the Aerosil-like process is the commercial manufacture of lightguides for telecommunications, where SiO2–GeO2 particles (from SiCl4 and GeCl4) were flame deposited directly onto and into substrate rods and subsequently extruded into continuous thin optical fibers of extremely high purity.37 Synthesis of other ceramic oxide particulates by VAFS e.g. UO2, SnO2, Fe2O3, ZrO2, ZnO and its composites,3,38 carbon-coated oxides39,40 and noble metal/metal oxides41 have also been reported from their respective halide, carbonyl or acetylacetonate vapours.
Despite its early industrial significance, very little was understood regarding particle formation within VAFS. It was only in the late 80s that Dobbins and Megaridis42 introduced the concept of thermophoretic sampling and essentially opened up the “black box” of particle formation in vapour-fed flames. The rapid insertion and retraction of a TEM (transmission electron microscopy) copper grid into the particle-laden flame allows direct sampling of the evolution of particle morphology along the flame axis, from spherical particles to fractal-like aggregates. From there, quantitative description of the flame particles growth by coagulation and sintering, including population balance and coalescence models43 contributed decisively to the development of computational tools for the design of VAFS reactors – to the point that particle sizes could be predicted from first principles without any adjustable parameters in premixed flames.44 Further accounting for the detailed particle residence time distribution by interfacing computational fluid mechanics with particle dynamics in diffusion flames45 allowed for quantitative understanding of a number of experimental observations: The effect of process variables (flame temperature, reactant concentration, mixing or flowrate) on product particle size distributions (example see ref. 46).
While VAFS was undoubtedly a milestone achievement in terms of flame aerosol synthesis, its usage of metal vapours is rather demanding. The continuous demands for process versatility require the replacement of metal vapour precursor, which can often be costly, difficult to handle or in many cases, simply not available. This prompted the spraying of liquid precursors directly into the flame i.e. flame-assisted spray pyrolysis (FASP). The strategy uses less volatile and economical precursors, particularly those of aqueous metal salts (e.g. nitrates and acetates). Since the precursor exists as liquid, an additional dispersion nozzle feature by ultrasonication or pressure-assistance, is necessary prior to combustion. Many of these precursors, especially aqueous nitrates have low combustion enthalpy or endothermic in nature and as such do not form self-sustaining flames. Hence an external flame source, usually oxy-hydrogen or oxy-hydrocarbon, is required to support the combustion.5 One of the disadvantages of FASP is the yield of submicron particles with inhomogeneous morphology and size especially when operating at low rates of combustion enthalpy (defined as the ratio of combustion rate (kJ min−1) to total gas flow (ggas min−1), <4.7 kJ g−1gas),47 where there is insufficient energy for the formation of metal vapour (see Section 3). In such cases, dense particles are formed as a result of drying and solid-state densification48 while hollow particles are due to precipitation on surface droplets prior to evaporation of the carrier liquids.49–52 The many types of FASP-synthesised particles include simple metal oxides – TiO2,53 ZnO,54 perovskites – SrMnO3, NiMn2O4,55 La1−xCexCoO3,56 La(Co, Mn, Fe)O357,58 and doped-metal oxides Y2O3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Eu3+,14,50,52,59–61 BaMgAl10O17:Eu2+.62,63
:Eu3+,14,50,52,59–61 BaMgAl10O17:Eu2+.62,63
In an FSP process, the exclusive use of highly exothermic liquid precursors (hereby referred to the mixture of metal precursor and solvent), particularly those based on organic solvents gives rise to self-sustaining flames. First introduced by Sokolowski et al.10 (Fig. 2, IV) who reported the synthesis of Al2O3 nanoparticles by combusting an ultrasonically-dispersed spray of aluminium acetylacetonate in benzene–ethanol. The concept was only further developed two decades later by Bickmore et al.28 who used a single pilot-scale FSP to synthesise spinel MgAl2O4, while Karthikeyan et al.64 reported the synthesis of Al2O3, Mn2O3, ZrO2 and Y2O3–ZrO2 using a multiple diffusion-flamelet spray gun (Fig. 2, V). Pressure-assisted FSP ignited by six surrounding oxy-methane flamelets was introduced by Mädler et al.65 and was very quickly superseded by an oxy-methane ring.66 A variant of the more general FSP (or FASP, depending on its combustion enthalpy criteria), the emulsion combustion (ECM) utilises an organic solvent–water metal precursor stabilised in an emulsion phase.6,67 The presence of incombustible water phase in the ECM precursor not only lowers the combustion enthalpy density, but may also result in poorer gas-phase mixing and reaction as compared to a homogenous solvent-based only precursor, thereby affecting the particles homogeneity.67
An integral part of the FSP reactor design is the aerosols collection, which can be accomplished either by electrostatic precipitation or vacuum-assisted filtration. In the latter case, fume nanoparticles are collected on a range of high temperature resistant porous media, ranging from glass fiber65 or PTFE membrane68 sheet filters, to Gore-Tex67 or PTFE-coated fiber69 bag type filters.
While the design of FASP and FSP reactors bear many similarities (and in fact may be inter-transferable), the core difference between the two classifications does not exactly lie in the reactor design, but more essentially in the origin of majority combustion enthalpy content of the system. The bulk of flame enthalpy in FSP originates from the combustion of liquid precursor, whereby the external flame serves only as an ignition source. This provides direct control over primary particle size by controlling the combustion enthalpy rate and metal concentration of the precursor alone.65,66 More details on particle formation will be presented in Section 3, considering also the possibilities of different types of particle configurations.
Because of the highly exothermic nature of FSP liquid precursors, flame temperatures of up to 2600 K65,70 or 2800 K71 have been measured. At the same time, the high gas velocities of the FSP induce radial entrainment of surrounding gas.72 Coupled with the radiation heat loss, this gives rise to extremely short residence times (milliseconds) with high temperature gradients (170 K cm−1) along the flame axis.70 The interplay between high temperature and large temperature gradient is one of the most important features in FSP: the high local temperature promotes the formation of homogeneous and highly crystalline materials and also promotes particle growth by sintering and coalescence,65 while the large temperature gradient (and short residence time) preserves the nanoscale feature of the particles. In fact, the rapid thermal quenching during FSP has resulted in the formation of many metastable materials such as amorphous Ta2O5 up to 45 wt.% in silica composites,20 Y3Al5O12,73 monoclinic BaCO3,74 cubic and tetragonal γ-Fe2O3.22,23 On an industrial scale, although the FSP process for ceramic materials is yet to see an equally large-scale implementation as VAFS with productions of tons h−1, it is making its way into the market. Scalability and reproducibility have been demonstrated at pilot scale production rates of 300 to 1100 g h−1 (SiO2,69 ZrO271,75,76), although FSP industrial units might already run at higher rates.
Among the extensions to the basic FSP synthesis of ceramic oxide nanoparticles, direct deposition of particulate films is perhaps one of the most extensively explored. This technique was demonstrated by Karthikeyan et al.64 by the natural deposition of flame aerosols (i.e. absence of phoresis) onto grit-blasted stainless steel substrate. Thermophoretic deposition was later developed for the direct fabrication of functional porous layers as Au/TiO2 catalytic microreactors77 and SnO2-based gas sensors17 (Fig. 2, VII). Here, deposition is driven by the temperature difference between the cooled masked substrate78 and the gas temperature at which the preformed open aerosol agglomerates are suspended. One of the processing advantages of this in situ technique is the independent nanoparticle synthesis and the film thickness control, where the former depends on the flame synthesis properties while the latter is a function of deposition time. Strictly speaking, this is different conceptually from the combustion chemical vapour deposition (CCVD) where particles are formed upon nucleation of metal vapour onto a temperature-controlled substrate, much alike a conventional CVD process, except the metal vapour was generated through the combustion of liquid or gaseous precursors.79 In CCVD, the substrate properties i.e. temperature and material affect strongly the nucleation and subsequent particle growth, while particles are preformed in the in situ FSP deposition. Typically, the FSP-deposited particulate films consist of agglomerates with Df ∼ 1.7–1.8 (as a result of Brownian coagulation) and are highly porous (>95% porosity). It has been shown theoretically that it is possible to manipulate the film profile, which is an interdependency of film thickness and porosity, by varying primary particle size, aggregate size, morphology and thermophoretic temperature difference.80 The reasons are the depth of penetration into the film of newly arriving particles during deposition, which is a function of the migration velocity and the particle diffusion coefficient as well as the particle/aggregate structure.
Sequential deposition of different aerosol materials gives rise to multilayer films,81,82 each of which can be in principle designed with controlled thickness and morphologies (Fig. 3). The direct immobilisation technique has also been extended to vacuum-assisted flame coating of aerosols onto the external and internal surfaces of porous substrates e.g. ceramic foams.83 Impinging flame-annealing is a film modification technique. Depending on the thermal stability of the inter-particulate bridge, direct annealing by the flame sinters the morphology of the lacy-like porous film to one consisting of dense cauliflower-like aggregates (porosity 62%).78 The wider gaps between cauliflowers were especially beneficial for the enhancement of analyte diffusion in gas sensor devices.78 Similar films morphology could also be achieved using high temperature CCVD84 and impinging VAFS techniques,85 with similar theoretical concepts.
 | ||
| Fig. 3 Directly fabricated single- (A,C) and multi-layer sensing films (B,D) on ceramic substrate by FSP. The technique offers high flexibility in terms of layers composition as well as film thickness. Figure adapted with permission from ref. 82. | ||
While most of FSP particles are synthesised in the form of metal oxides, the synthesis of metallic nanoparticles is possible under restrictive oxygen flame conditions.24,86 To achieve this, the amount of O2 present must be kept minimal (in inert gas-filled glove box), but sufficient for the complete combustion of hydrocarbon fuels (Fig. 2, IX). Further restricting the O2 partial pressure can result in the formation of carbonaceous coating.25 To date, the synthesis of metallic nanoparticles have only been restricted to highly electronegative metallic particles such as Bi (but unstable in air)86 or particles which are protected by surface oxide or carbonaceous layer against further oxidations, such as C/Co,24,25 C/Cu,87 Co3O4/Co24 or even Ni/Mo alloy with oxide shell.88
Flame spraying of multicomponent elements in a single nozzle FSP configuration often limits the flexibility in obtaining a certain desirable particle configuration. For instance, uniformly dispersed Ba species within an Al2O3 matrix, rather than discrete oxide particles, were obtained when spraying both elements simultaneously in a single nozzle.89 To achieve phase separation (i.e. reduced solubility), these aerosol particles (e.g. Ba and Al) have to be co-precipitated in separate flames and interfaced only at a certain angle for even interparticle aerosol mixing (Fig. 2, X). The flame temperature and remaining residence time at the point of mixing is critical to either induce delayed solid-state reactions or suppress any reactions between these discrete phases altogether. A key advantage of the concept is the independent control over particle synthesis (in terms of elements as well as their physical characteristics) at each nozzle. Particle systems that have been synthesised using the multi-nozzle, besides BaCO3/Al2O3,89 include Pt/Ba/CexZr1−xO290 and carbon-supported Pt nanoparticles.91
In other circumstances, induced delay and sequential precipitation (rather than co-precipitation) of one of the metal components is required, which in this sense would render the multi-nozzle unsuitable. To do so, it is required that the secondary metal component be introduced into the flame, for example as vapour phase precursor – similar to VAFS, only after the precipitation of the primary phase particles (Fig. 2, XI).92–94 The technique circumvents the high temperature solid-state reaction dependencies of multiple components in a co-oxidation configuration. More importantly, it allows the design of thin coating and even encapsulation by the secondary metal oxides phase, e.g. SiO2-coated TiO292 and SiO2-coated γ-Fe2O3.95 As in the case of multi-nozzle configuration, the flame temperature and remaining residence time at which mixing of secondary component (i.e. coating precursor) is introduced determines the fate of this phase. Point of mixing at excessively high temperature and long residence time may induce solid-state reaction and uneven segregation of the two components,94 while an excessively low temperature and insufficient residence time may result in incomplete formation of the oxide coating layer. In principle, the technique could also be applied to multi-layer coating by introducing more sequential stages.
3. Designing of liquid precursors
Formulation of liquid precursors is one of the more important steps to FSP particle synthesis. Selection of metal precursors and solvents with suitable combustion enthalpies, melting/decomposition temperatures, miscibility and chemical stability are intrinsic to the overall particles formation in the flame which in turn determine the resultant particle properties. Early formulations of FSP precursors were based on solid nitrates,64,96,97 acetates64,66 and acetylacetonates98,99 as these were natural choices in direct extensions of FASP. While many of these precursors were economical and readily available commercially, they do not always yield the homogeneous morphology comprising of fine and dense particles. The low combustion enthalpy of some of these precursors coupled with their high melting/decomposition points can be disadvantageous, but as discussed below, some of these disadvantages can be circumvented by implementing the right processing conditions.The combination of low combustion enthalpy density (<4.7 kJ g−1gas) and higher metal precursor melting/decomposition point relative to the solvent boiling point i.e. Tbp (solvent)/Td/mp(precursor) < 1 during FSP results in the formation of inhomogeneous particles (Fig. 4).54 Under these conditions, both micron- and nanoparticles co-exist (Fig. 4a,b) due to particle formation through the droplet-to-particle100 and gas-to-particle routes,5,101–104 respectively. The former arises from incompletely evaporated droplets as in classic spray pyrolysis,48 while the latter from the supersaturation of metal vapour.96 Hollow particles can even be formed if the metal precursor precipitates at the droplet surface during solvent evaporation. In the case where an impermeable shell is formed, internal pressure build-up during the evaporation of trapped solvent leads to fragmentation of the spherical shells.96 Subsequent solid-state reactions and densification take place forming micron-sized metal oxides. In the case when FSP is operated at high combustion enthalpy densities, sufficient heat is provided to evaporate the precursor intermediate products to yield homogeneous fine particles, even at Tbp (solvent)/Td/mp(precursor) < 1 (Fig. 4d).
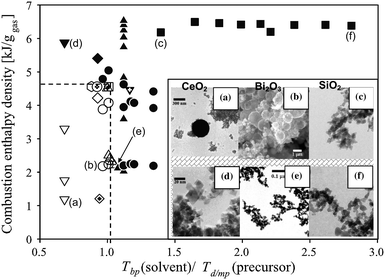 | ||
| Fig. 4 Morphology map of particles made by spray flames adapted and modified from ref. 47. The hollow symbols, within the broken line box, represent inhomogeneous and hollow particles: (a) CeO2;73 (b) Bi2O3,96 while solid symbols represent solid and homogeneous particles: (c,f) SiO2,47 (d) CeO2,73 (e) Bi2O3.96 All images are adapted with permissions from the respective references. | ||
By definition, the criteria for obtaining dense and homogenous fine particles can be achieved as long as the metal precursor, in its pure form i.e. not dissolved in solvent, exists in liquid phase at ambient temperature i.e. Tmp(precursor) < Tbp(solvent) (Fig. 4c,f). Most metal alkoxides fall in this category, with the exception of some precursors e.g. aluminium propoxide, which exists as solid crystals. In general, metal alkoxides in the form of methoxides to pentoxides, are commonly used in FSP for a number of reasons: high volatilities, high combustion enthalpies, miscibility in many organic solvents, low viscosity and commercial availability (see Table 1). One drawback, besides price, is the general moisture sensitivity of metal alkoxides, rendering their handling and preparation in a humidity-free environment necessary. Very recently, a facile conversion of metal chloride (as WCl4 and WCl6) to moisture tolerant alkoxide was demonstrated by reacting with benzyl alcohol at room temperature and pressure.179 Gaseous hydrogen chloride evolves during the alkoxylation reaction, thereby removing the chloride content.
| Element | Metal precursor | Solvent | Particles | Additional remark | Reference |
|---|---|---|---|---|---|
| Li | Lithium–sodium–alumatrane–glycolate | Ethanol | Li-doped Na2O·xAl2O3 | Refluxing of alumatrane + sodium hydroxide + lithium hydroxide hydrate (1 h), followed by N2 distillation to remove water and excess ethylene glycol | Sutorik et al.105 |
| Lithium tert-butoxide | Tetrahydrofuran–toluene | Li–ZnO | Height et al.106 | ||
| Tetrahydrofuran–xylene | LiMn2O4, Li4Ti5O12, LiFe5PO8 | Ernst et al.26 | |||
| B | Tributyl boride | 2-Ethylhexanoic acid | Borosilicate glass | Brunner et al.107 | |
| Vollenweider et al.26 | |||||
| C | Organic solvent | Ethanol | BaCO3 | Strobel et al.74 | |
| Mineral spirit–tetrahydrofuran | C–Co | Restricted oxygen flame, C2H2 | Grass et al.25 | ||
| Tetrahydrofuran | C–Cu | Restricted oxygen flame | Athanassiou et al.87,88 | ||
| Tetrahydrofuran | C–Cu | Restricted oxygen flame, C2H2 | Athanassiou et al.109 | ||
| Xylene | Pt/C and C/Pt | Twin nozzles | Ernst et al.91 | ||
| Xylene–2-ethylhexanoic acid | CaCO3 | Osterwalder et al.110 | |||
| F | Trifluoroacetic acid | Xylene | Ca10(PO4)6(OH)2−xFx | Loher et al.19 | |
| Hexafluorobenzene | 2-Ethylhexanoic acid | Bioglass | Brunner et al.107 | ||
| Xylene | CaF2, SrF2, BaF2 | Grass et al.111 | |||
| Xylene | F-MOx (MOx = TiO2, ZrO2) | Teoh et al.,112 Emeline et al.113 | |||
| Na | Lithium–sodium–alumatrane–glycolate | Ethanol | Li-doped Na2O·xAl2O3 | See Li | Sutorik et al.105 |
| Sodium 2-ethylhexanoate | 2-Ethylhexanoic acid–xylene | NaCl | Sodium hydrogencarbonate + ethylhexanoic acid | Grass and Stark111 | |
| Acetonitrile, HMDSO, tributyl phosphate, tributyl borate, fluorobenzene | Soda-lime glass | Brunner et al.,107 Vollenweider et al.108 | |||
| Mg | Magnesium 2-ethylhexanoate | 2-Ethylhexanoic acid | Mg–Ca3(PO4)2 | Magnesium oxide + 2-ethylhexanoic acid | Loher et al.19 |
| magnesium acetate tetrahydrate | Methanol | Ni:MgO–SiO2 | Suzuki et al.114 | ||
| Methanol–butanol | Mg2SiO4:Cr |
Tani et al.115 | |||
| Methanol–water | MgO | Large and inhomogeneous MgO at high water content | Tani et al.116 | ||
| Magnesium acetylacetonate | Tetrahydrofuran–ethanol | MgO–Al2O3 | Dissolve metal precursor in solvent followed by filtration to remove ∼0.5 wt% of residual solids | Hinklin and Laine33 | |
| Magnesium pentanedionato | Tetrahydrofuran–ethanol | MgO–Fe2O3, MgO–Al2O3 | Hinklin et al.117 | ||
| Al | Alumatrane | Ethanol | 3Al2O3.2SiO2 (mullite) | Aluminium hydroxide hydrate + triethanolamine + ehtylene glycol (N2 distillation, 3–4 h to remove water and ethylene glycol). Product was recovered by vacuum filtration | Baranwal et al.29 |
| Al2O3:(Ce,Pr) | Williams et al.118 | ||||
| CoOx–Al2O3 | Azurdia et al.31 | ||||
| NiO–Al2O3 | Azurdia et al.32 | ||||
| ZrO2–Al2O3 | Kim and Laine119 | ||||
| (CeOx)x(Al2O3)1−x | Kim et al.120 | ||||
| Ethanol–tetrahydrofuran | Al2O3 | Hinklin et al.121 | |||
| MgO–Al2O3 | Hinklin and Laine33 | ||||
| Ethanol, tetrahydrofuran–ethanol, aqueous ethanol | Y3Al5O12 | Marchal et al.122 | |||
| Methanol, butanol | TiO2/Al2O3 | Kim et al.123 | |||
| Aluminium 2-ethylhexanoate | 2-Ethylhexanoate–toluene | Al2O3/ZrO2 | Aluminium 2-ethylhexanoate basic + 2-ethylhexanoic acid followed by N2 distillation to remove water | Jossen et al.76 | |
| Acetic acid anhydride–2-ethylhexanoic acid | Al2O3/CexZr1−xO2 | Aluminium 2-ethylhexanoate (16.5% Al2O3) + acetic acid/2-ethylhexanoic acid (N2 reflux, 2 h) followed by distillation to remove acetic acid | Schulz et al.124 | ||
| Tetrahydrofuran | Hastelloy | Athanassiou et al.88 | |||
| Aluminium acetylacetonate | Ethanol–benzene | Al2O3 | Sokolowski et al.10,98 | ||
| Ethanol–tetrahydrofuran | Al2O3 | Heating to 50 °C for 1 h, and kept at ∼50 °C | Hinklin et al.121 | ||
| Methanol–acetic acid | CuaMgbAlcOx | Haider and Baiker125 | |||
| M/Al2O3 (M = Rh, Pt–Rh–Ru, Rh–Ru or Pt–Rh) | Hannemann et al.,126 van Vegten et al.127 | ||||
| Aluminium chloride | Tetrahydrofuran–ethanol | Al2O3 | Hinklin et al.121 | ||
| Aluminium sec-butoxide | Butanol–2-propanol | Al2O3 | Tani et al.97 | ||
| Diethylene glycol monobutyl ether/acetic anhydride | Pt–Ba/Al2O3 | Strobel et al.,89 Piacentini et al.128 | |||
| Xylene | Pd/Al2O3 | Strobel et al.129 | |||
| Pd/Al2O3 (film) | In situ deposition | Sahm et al.81,82 | |||
| Pd/La2O3/Al2O3 | Strobel et al.130 | ||||
| Pt–Rh–Ru/Al2O3 | Hannemann et al.126 | ||||
| SiO2-coated Al–TiO2, SiO2/Al2O3/TiO2 | In situ sequential flame coating | Teleki et al.92–94 | |||
| Xylene–acetonitrile | Pt–Pd/Al2O3 | Strobel et al.131 | |||
| Aluminium isopropoxide | Isopropanol | Al2O3 | |||
| Xylene–ethylacetate | Pt/Al2O3 | Maintained at 50 °C to prevent precipitation | Strobel et al.132 | ||
| Aluminium nitrate nanohydrate | 2-propanol–methanol | Al2O3 | Tani et al.97 | ||
| Deionised water–kerosene–surfactant (hexa(2-hydroxy-1,3-propylene glycol) diricinoleate) | Al2O3 | Emulsion precursor | Tani et al.97 | ||
| Ethanol, butanol | Y3Al5O12 | Marchal et al.122 | |||
| Isopropanol | Al2O3 | Karthikeyan et al.,64 Tikkanen et al.133 | |||
| Tetrahydrofuran–ethanol | Al2O3 | Hinklin et al.121 | |||
| Lithium–sodium–alumatrane–glycolate | Ethanol | Li-doped Na2O·xAl2O3 | see Li | Sutorik et al.105 | |
| Magna-alumatrane glycolate | Ethanol | MgO–Al2O3 | Aluminium hydroxide hydrate + Magnesium hydroxide hydrate + triethanolamine + ehtylene glycol (200 °C distillation to remove ethylene glycol and water). | Bickmore et al.28 | |
| Al2O3 (predominantly sigma, gamma, theta-phase) | Ethanol | alpha-Al2O3 | Flame spraying of slurries | Laine et al.134 | |
| Si | Hexamathyldisiloxane (HMDSO) | 2-Ethylhexanoic acid | Bioglass | Brunner et al.,107 Vollenweider et al.108 | |
| Acetic acid–methanol | SiO2, SiO2/ZnO | Mädler et al.,135 Tani et al.136,137 | |||
| Deionised water–kerosene–hexa(2-hydroxy-1,3-propylene-gricol) diricinoleate | SiO2, SiO2/ZnO | Emulsion combustion (ECM), mixing at 10 000 rpm for 10 min | Tani et al.67 | ||
| Ethanol | SiO2 | Pilot-scale FSP | Mueller et al.71 | ||
| Ethanol, iso-octane, methanol | SiO2 | Mädler et al.65 | |||
| Methanol | Ni:MgO–SiO2 | Suzuki et al.114 | |||
| Methanol–butanol | Mg2SiO4:Cr |
Tani et al.115 | |||
| Methanol–hydrogen peroxide | SiO2, SiO2/ZnO | Tani et al.67 | |||
| Toluene–2-ethylhexanoic acid | Ag/SiO2 | Loher et al.138 | |||
| Xylene | SiO2/Al2O3/TiO2 | Teleki et al.92–94 | |||
| SiO2/SnO2 | In situ flame deposition-annealing | Tricoli et al.139 | |||
| Xylene, pentane, hexane, octane, decane,dodecane, tetradecane, hexadecane | SiO2 | Jossen et al.47 | |||
| SiO2 sol | Deionised water–kerosene–hexa(2-hydroxy-1,3-propylene-gricol) diricinoleate | SiO2, SiO2/ZnO | Emulsion combustion (ECM), mixing at 10000 rpm for 10 min |
Tani et al.67 | |
| TEA–Si–egH | Ethanol | 3Al2O3.2SiO2 (mullite) | Silica + triethanolamine + ethylene glycol (N2 distillation, 3–4 h). TEA–Si–egH is filtered and washed with acetone. The product is mixed with alumatrane and further distilled (N2, 3–4 h) to remove excess ethylene glycol | Baranwal et al.29 | |
| Tetraethyl siloxane (TEOS) | 2-Ethylhexanoic acid–toluene | SiO2/CexZr1−xO2 | Schulz et al.124 | ||
| 2-Ethylhexanoic acid–toluene | SiO2/BiVO4 | Strobel et al.140 | |||
| Acetonitrile–acetic anhydride, xylene–acetic anhydride/2-ethylhexanoic acid | Yb2O3/SiO2 | Mädler et al.141 | |||
| Ethanol | Y2SiO5:Eu3+ |
Qin et al.142 | |||
| Methanol–AcOH | ZnO–SiO2 | Ramin et al.143 | |||
| Pentane, hexane, dodecane, xylene, 2-ethylhexanoic acid–toluene (all under N2) | Ta2O5/SiO2 | Schulz et al.20 | |||
| Toluene | SiO2–V2O5–WO3–TiO2 | Jossen et al.144 | |||
| SiO2/ZrO2 | Jossen et al.83 | ||||
| Xylene | M/SiO2(M = Au, Ag or Au–Ag) | Hannemann et al.145 | |||
| Xylene–acetonitrile | SiO2/Fe2O3 | Li et al.22,146 | |||
| Tetramethylsilane (TMS), HMDSO, tetramethyl orthosilicate (TEMOS), tetrepropyl orthosilicate (TEPOS) | Xylene | SiO2 | Jossen et al.54 | ||
| P | Tributyl phosphate | 2-Ethylhexanoic acid | Ca10(PO4)6(OH)2−xFx, Ca3(PO4)2, M–Ca3(PO4)2 (M = Mg, Zn) | Loher et al.19,147 | |
| 2-Ethylhexanoic acid–toluene | Ag/Ca3(PO4)2 | Loher et al.138 | |||
| Xylene | FePO4 | Rohner et al.21 | |||
| S | Dimethyl sulfoxide | Xylene | CaSO4 | Osterwalder et al.110 | |
| Cl | Hexachlorobenzene | 2-Ethylhexanoic acid | NaCl | Grass and Stark111 | |
| Ca | Calcium 2-ethylhexanoate | 2-Ethylhexanoic acid | Ca10(PO4)6(OH)2−xFx, Ca3(PO4)2, M–Ca3(PO4)2 (M = Mg, Zn) | Calcium oxide + 2-ethylhexanoic acid | Loher et al.19,146 |
| Calcium hydroxide + 2-ethylhexanoic acid | Brunner et al.147 | ||||
| 2-Ethylhexanoic acid–toluene | Ag/Ca3(PO4)2 | Calcium hydroxide + 2-ethylhexanoic acid | Loher et al.138 | ||
| 2-Ethylhexanoic acid–xylene | CaO, CaCO3, CaSO4 | Calcium hydroxide + 2-ethylhexanoic acid (140 °C, 3 h) | Osterwalder et al.110 | ||
| Glass and bioglass | Brunner et al.,107 Vollenweider et al.108 | ||||
| Ti | Titanatrane | TiO2 | TiO2 in ethylene glycol (EG) + triethanolamine (200 °C, 8 h) followed by distillation to remove EG. Resultant precursor is hydrolytically stable | Bickmore et al.149 | |
| Methanol, butanol | TiO2/Al2O3 | Kim et al.123 | |||
| Titanium tetraisopropoxide (TTIP) | Propionic acid, 2-ethylhexanoic acid, methanol, xylene, pyridine | Au/TiO2 | Chiarello et al.150 | ||
| Toluene | SiO2–V2O5–WO3–TiO2 | Jossen et al.144 | |||
| Xylene | Nb–TiO2, Cu–TiO2 | Teleki et al.151 | |||
| M/TiO2 (M = Au, Ag or Au–Ag) | Hannemann et al.145 | ||||
| SiO2-coated Al–TiO2, SiO2/Al2O3/TiO2 | In situ sequential flame coating | Teleki et al.92–94 | |||
| Xylene–acetonitrile | TiO2 | Teoh et al.,151–154 Teleki et al.155 | |||
| Pt/TiO2 | Schulz et al.,70 Teoh et al.152,154 | ||||
| Fe–TiO2 | Teoh et al.156 | ||||
| V2O5/TiO2 | Schimmoeller et al.83 | ||||
| Ag/TiO2 | Gunawan et al.157 | ||||
| Xylene–tetrahydrofuran | Li4Ti5O12 | Ernst et al.26 | |||
| V | Vanadium oxo-triisopropoxide | Toluene | SiO2–V2O5–WO3–TiO2 | Jossen et al.144 | |
| Xylene–acetonitrile | V2O5/TiO2 | Schimmoeller et al.83 | |||
| Vanadyl naphthenate | Toluene–2-ethylhexanoic acid | BiVO4 and SiO2/BiVO4 | Strobel et al.140 | ||
| Cr | Chromium 2-ethylhexanoate | Mineral spirit–tetrahydrofuran | Hastelloy | Restricted oxygen flame | Athanassiou et al.88 |
| Chromium acetate | Methanol/butanol | Mg2SiO4:Cr |
Tani et al.115 | ||
| Chromium acetylacetonate | Diethylene glycol monbutyl ether/ethanol | Cr–WO3 | Wang et al.158 | ||
| Mn | Manganese 2-ethylhexanoate | Mineral spirit–tetrahydrofuran | Hastelloy | Restricted oxygen flame | Athanassiou et al.88 |
| Xylene–tetrahydrofuran | LiMn2O4 | Ernst et al.26 | |||
| Manganese acetylacetonate | Xylene–tetrahydrofuran | LiMn2O4 | Ernst et al.26 | ||
| Manganese nitrate | Isopropanol | Mn2O3 | Karthikeyan et al.,64 Tikkanen et al.133 | ||
| Fe | Iron 2-ethylhexanoate | Tetrahydrofuran | Hastelloy | Restricted oxygen flame | Athanassiou et al.88 |
| Iron acetylacetonate | Xylene | FePO4 | Rohner et al.21 | ||
| Xylene–acetonitrile | Fe2O3, SiO2/Fe2O3 | Li et al.22,23,146 | |||
| Iron naphthenate | Mineral spirit–xylene | M/Fe2O3/Fe3O4 (M = Au, Ag or Au–Ag) | Hannemann et al.145 | ||
| Mineral sprit–xylene/acetonitrile | Fe–TiO2 | Teoh et al.156 | |||
| Mineral sprit–xylene–tetrahydrofuran | LiFe5PO8 | Ernst et al.26 | |||
| Iron(III) nitrate | Ethanol | Nd:Co:Fe2O3 |
Dosev et al.159 | ||
| Iron propionate | Ethanol | (MgO)x(Fe2O3)1−x | Hinklin et al.117 | ||
| Co | Cobalt 2-ethylhexanoate | 2-Ethylhexanoic acid | Co-soda lime glass | Brunner et al.107 | |
| Mineral spirit–tetrahydrofuran | Co3O4, CoO, fcc–Co | Restricted oxygen flame | Grass and Stark24 | ||
| C–Co | Restricted oxygen flame, C2H2 | Grass et al.25 | |||
| Co nanowire | External magnetic field | Athanassiou et al.109 | |||
| Xylene | Co3O4, Co3O4–ZrO2, Ru–Co3O4–ZrO2 | Teoh et al.160 | |||
| Cobalt acetate tetrahydrate | Carboxylic acids (acetic acid, propionic acid, butyric acid, hexanoic acid, octanoic acid or 2-ethylhexanoic acid) | LaCoO3 | Chiarello et al.161–163 | ||
| Propionic acid–propanol–water | Pd/LaCoO3 | Vigorous stirring at 60 °C | Chiarello et al.164,165 | ||
| Cobalt nitrate tetrahydrate | Ethanol | Nd:Co:Fe2O3 |
Dosev et al.159 | ||
| Propionic acid + alcohol (methanol, ethanol, 1-propanol) | LaCoO3 | Chiarello et al.166 | |||
| Cobalt propionate | Ethanol | Co3O4, NiO–Co3O4 | Reactive distillation of cobalt nitrate hexahydrate + propionic acid under N2 sparging (150 °C, 6 h) to remove water, propionic acid and NOx | Azurdia et al.167 | |
| Ethanol–propionic acid | CoOx–Al2O3 | Cobalt nitrate hydrate + propionic acid under N2 sparging followed by distillation (150 °C, 6 h) to remove water and NOx | Azurdia et al.31 | ||
| Ni | Nickel 2-ethylhexanoate | Tetrahydrofuran | Ni/Mo alloy | Athanassiou et al.88 | |
| Nickel acetate | Methanol | Ni:MgO–SiO2 | Suzuki et al.114 | ||
| Nickel propionate | Ethanol | NiO, NiO–MOx (Mox = Co3O4, MoO3, CuO) | Nickel nitrate hexahydrate + propionic acid under N2 sparging and distillation (150 °C, 4h) to remove water, propionic acid and NOx | Azurdia et al.167 | |
| Ethanol, ethanol–methanol, ethanol–butanol | NiO–Al2O3 | Reactive distillation of nickel nitrate hexahydrate + propionic acid under N2 sparging (150 °C, 6 h) to remove water, propionic acid and NOx | Azurdia et al.32 | ||
| Cu | Copper 2-ethylhexanoate | Tetrahydrofuran | C–Cu | Restricted oxygen flame | Athanassiou et al.87 |
| Xylene | Cu–TiO2 | Teleki et al.151 | |||
| Cu–SiO2 | Height and Pratsinis168 | ||||
| Xylene, xylene–2-ethylhexanoic acid | CuO, Cu–MOx (MOx = SiO2, Al2O3, TiO2, CeO2,ZrO2) | Kydd et al.169,170 | |||
| Copper nitrate trihydrate | Acetic acid–methanol | CuaMgbAlcOx | Haider and Baiker125 | ||
| Copper propionate | Ethanol | CuO, NiO–CuO | Reactive distillation of copper nitrate hydrate + propionic acid under N2 sparging (150 °C, 6 h) to remove water, propionic acid and NOx | Azurdia et al.167 | |
| Zn | Zinc acetate dihydrate | Methanol–water | ZnO | Large and inhomogeneous ZnO at high water content | Tani et al.116 |
| Zinc acetylacetonate | Methanol–AcOH | ZnO–SiO2 | Ramin et al.143 | ||
| Colloidal SiO2 (Ludox)–Methanol–AcOH | ZnO–SiO2 | Flame spraying of slurries | Ramin et al.143 | ||
| Zinc acrylate | Acetic acid–methanol | SiO2/ZnO | Mädler et al.,135 Tani et al.136,137 | ||
| ZnO | Tani et al.171 | ||||
| Zinc napthenate | 2-Ethylhexanoic acid | Zn–Ca3(PO4)2 | Loher et al.19 | ||
| Mineral spirit–ethanol | ZnO, Ag/ZnO | Height et al.172 | |||
| Mineral spirit–toluene | ZnO, M–ZnO (M = In, Sn, Li) | Height et al.106 | |||
| Y | Yttrium 2-ethylhexanoate | 2-Ethylhexanoic acid–ethanol | Y2O3/ZrO2 | Yttrium nitrate hexahydrate + ethanol + 2-ethylhexanoic acid (N2 distilled to remove ethanol, water, NO) | Jossen et al.76 |
| 2-Ethylhexanoic acid–toluene | Y2O3/ZrO2 | Yttrium nitrate hexahydrate + ethanol + 2-ethylhexanoic acid (120 °C distillation to remove ethanol, water, NO) | Jossen et al.75 | ||
| Toluene | Y2O3:Eu3+ |
Yttrium nitrate hexahydrate + aqueous ammonia to form Y(OH)3 followed by refluxing with 2-ethylhexanoic acid + acetic acid (65 °C, 4 h) | Camenzind et al.12,13 | ||
| Tetrahydrofuran–ethanol | Y3Al5O12 | Marchal et al.122 | |||
| Yttrium acetate | Aqeuous acetic acid | Y2O3/ZrO2 | Karthikeyan et al.64 | ||
| Yttrium acetylacetonate | Ethanol | Y3Al5O12 | Marchal et al.122 | ||
| Yttrium butoxide | Toluene | Y2O3/ZrO2 | Jossen et al.76 | ||
| Yttrium methoxyacetate | Aqueous ethanol | Y3Al5O12 | Yttrium trichloride + methoxyacetic acid (reflux under N2,135 °C, 2 h) | Marchal et al.122 | |
| Yttrium nitrate hexahydrate | Ethanol | Y2O3/ZrO2 | Inhomogeneous and segregated Y2O3 | Jossen et al.76 | |
| Y2O3:Eu3+ |
Dosev et al.14 | ||||
| Y2SiO5:Eu3+ |
Qin et al.142 | ||||
| Ethanol, butanol | Y3Al5O12 | Marchal et al.122 | |||
| Ethanol–acetic anhydride | Y2O3/ZrO2 | Slow addition of acetic anhydride under N2. Resulting NOx is bubbled through NaOH. Inhomogeneous and segregated Y2O3 | Jossen et al.76 | ||
| Yttrium propionate | Ethanol, tetrahydrofuran | Y3Al5O12 | Yttrium nitrate hexahydrate + propionic acid (N2,145 °C) | Marchal et al.122 | |
| Zr | Zirconyl 2-ethylhexanoate | 2-Ethylhexanoic acid | Pt/CexZr1−x | Stark et al.173 | |
| 2-Ethylhexanoic acid–toluene | MOx/CexZr1−xO2(MOx = SiO2, Al2O3) | Acetic acid digested zirconium carbonate + 2-ethylhexanoic acid + cerium acetate hydrate (N2 distilled to remove water and acetic acid) | Schulz et al.124 | ||
| Pt/Ba/CexZr1−xO2 | Twin nozzles | Strobel et al.131 | |||
| Ethanol | Y2O3/ZrO2 | Zirconium carbonate hydroxide oxide dissolved in acetic acid (50 °C) + 2-ethylhexanoic acid followed by distillation (120 °C) to remove water and acetic acid | Jossen et al.76 | ||
| Xylene | Rh/CexZr1−xO2 | Hotz et al.174 | |||
| Zirconium acetate | Aqueous acetic acid | Y2O3/ZrO2 | Karthikeyan et al.64 | ||
| Zirconium butoxide | n-Butanol | ZrO2 | Karthikeyan et al.64 | ||
| Zirconium propionate | Ethanol | ZrO2–Al2O3 | Zirconium carbonate + 2-ethylhexanoic acid followed by distillation (120 °C, 2 h) to remove water and propionic acid | KimandLaine119 | |
| Zirconium propoxide | Isopropanol–ethanol | Y2O3/ZrO2 | Jossen et al.76 | ||
| Isopropanol–toluene | MOx/ZrO2 (MOx = SiO2, La2O3, CeO2, Y2O3, Al2O3) | Jossen et al.75 | |||
| Isopropanol–xylene | ZrO2, Co3O4–ZrO2,Ru–Co3O4–ZrO2 | Teoh et al.160 | |||
| Zirconium tetraacetylacetonate | Isooctane/acetic acid/butanol | CeO2/ZrO2 | Low boiling point solvent resulting in segregated ZrO2 and CeO2 phase | Stark et al.175 | |
| lauric acid/acetic acid | CexZr1−xO2 | Heating to full dissolution | Stark et al.175 | ||
| Nb | Niobium 2-ethylhexanoate | Xylene | Nb–TiO2 | Teleki et al.151 | |
| Mo | Ammonium molybdate | Lactic acid | MoO3, NiO–MoO3 | Dissolve ammonium molybdate in aquoeus lactic acid (heating >100 °C, 2h) followed by removal of excess water and acid in rotary evaporator | Azurdia et al.167 |
| Molybdenum 2-ethylhexanoate | Mineral spirit–tetrahydrofuran | Ni/Mo alloy | Athanassiou et al.88 | ||
| Ru | Ruthenium acetylacetonate | Xylene | Ru–Co3O4–ZrO2 | Teoh et al.160 | |
| Ruthenocen | Xylene, methanol–acetic acid | Pt–Rh–Ru/Al2O3, Rh–Ru/Al2O3 | Hannemann et al.126 | ||
| Rh | Rhodium 2-ethylhexanoate | Xylene | Rh/CexZr1−xO2 | Hotz et al.174 | |
| Rhodium acetylacetonate | Xylene, methanol–acetic acid | M/Al2O3 (M = Rh, Pt–Rh–Ru, Rh–Ru or Pt–Rh) | Hannemann et al.,126 van Vegten et al.127 | ||
| Pd | Palladium acetate | Propionic acid–propanol–water | Pd/LaCoO3 | Vigorous stirring at 60 °C | Chiarello et al.164,165 |
| Palladium acetylacetonate | Xylene | M/Al2O3 (M = Pd, Pt–Pd) | Strobel et al.,129,131 Sahm et al.81,82 | ||
| Pd/La2O3/Al2O3 | Strobel et al.130 | ||||
| Ag | Silver acetate | 2-Ethylhexanoate–toluene | Ag/Ca3(PO4)2, Ag/SiO2 | Loher et al.138 | |
| Silver benzoate | Pyridine | Ag/MOx (MOx = SiO2, TiO2, Fe2O3/Fe3O4) | Hannemann et al.145 | ||
| Xylene–TTIP | Ag/TiO2 | Gunawan et al.157 | |||
| Silver nitrate | Ethanol | Ag/ZnO | Height et al.172 | ||
| Ag/TiO2 | Keskinen et al.176,177 | ||||
| In | Indium acetylacetonate | Toluene | In–ZnO | Height et al.106 | |
| Sn | Tin 2-ethylhexanoate | Ethanol | SnO2 (film) | Sahm et al.16 | |
| Toluene | SnO2, Pd/SnO2, Pt/SnO2 (films) | In situ flame deposition | Mädler et al.,17,18 Sahm et al.81,82 | ||
| Sn–ZnO | Height et al.106 | ||||
| Xylene | SnO2, SiO2/SnO2 | In situ flame deposition-annealing | Kuhne et al.,178 Tricoli et al.78,139 | ||
| Ba | Barium(II) 2-ethylhexanoate | Ethanol | BaCO3 | Strobel et al.74 | |
| Pt–Ba/Al2O3 | Twin nozzles | Strobel et al.,89 Piacentini et al.128 | |||
| 2-Ethylhexanoic acid–toluene | Pt/Ba/CexZr1−xO2 | Twin nozzles | Strobel et al.90 | ||
| La | Lanthanum 2-ethylhexanoate | 2-Ethylhexanoic acid–toluene | La2O3/ZrO2 | Lanthanum(III) acetylacetonate hydrate + acetic anhydride + 2-ethylhexanoic acid (distilled under N2 to remove acetic acid, acetylacetonate, water) | Jossen et al.75 |
| Lanthanum acetate dihydrate | Carboxylic acids (Acetic acid, propionic acid, butyric acid, hexanoic acid, octanoic acid, 2-ethylhexanoic acid) | LaCoO3 | Chiarello et al.161–163 | ||
| Propionic acid–propanol–water | Pd/LaCoO3 | Vigorous stirring at 60 °C | Chiarello et al.164,165 | ||
| Lanthanum isopropoxide | Xylene | Pd/La2O3/Al2O3 | Strobel et al.130 | ||
| Ta | Tantalum ethoxide, tantalum butoxide, tantalum tetraethylacetonate | Pentane, hexane, dodecane, xylene, 2-ethylhexanoic acid–toluene (all under N2) | Ta2O5/SiO2 | Schulz et al.20 | |
| W | Ammonium tungstate hydrate | Diethylene glycol monbutyl ether–ethanol | WO3, Cr–WO3 | Wang et al.158 | |
| Tungsten benzyl alkoxide | Benzyl alcohol | WO3 | Vigorous stirring of WCl4 or WCl6 in benzyl alcohol for 8 h at 20 °C until pH 4.5. Gaseous HCl is liberated. | Pokhrel et al.179 | |
| Tungsten carbonyl | Tetrahydrofuran | WO3 | Pokhrel et al.179 | ||
| Tungsten ethoxide | Toluene | SiO2–V2O5–WO3–TiO2 | Jossen et al.144 | ||
| Pt | Platinum acetylacetonate | 2-Ethylhexanoic acid | Pt/CexZr1−xO2 | Stark et al.173 | |
| 2-Ethylhexanoic acid–toluene | Pt/Ba/CexZr1−xO2 | Twin nozzles | Strobel et al.131 | ||
| Ethanol | Pt–Ba/Al2O3 | Twin nozzles | Strobel et al.,89 Piacentini et al.128 | ||
| Toluene | Pt/SnO2 (films) | Mädler et al.17,18 | |||
| Xylene | Pt/C and C/Pt | Twin nozzles | Ernst et al.91 | ||
| Xylene–acetonitrile | Pt/TiO2 | Teoh et al.152,154 | |||
| Xylene–ethyl acetate | Pt/Al2O3 | Strobel et al.132 | |||
| Xylene, methanol/acetic acid | M/Al2O3 (M = Pt–Rh–Ru, Pt–Rh) | Hannemann et al.126 | |||
| Au | Dimethyl gold(III)-acetylacetonate | Xylene | Au/MOx, Au–Ag/MOx (MOx = TiO2, SiO2, Fe2O3/Fe3O4) | Extremely moisture- and thermal-sensitive precursor | Hannemann et al.145 |
| Xylene–pyridine | Au/TiO2 | Chiarello et al.150 | |||
| Gold chloride | Acetonitrile–2-ethylhexanoic acid | Au-soda lime glass | Brunner et al.107 | ||
| Xylene–acetonitrile | Au/MOx (MOx = TiO2, SiO2) | Mädler et al.180 | |||
| Gold-triphenylphosphine-nitrate | Tetrahydrofuran/isooctane | Au/TiO2 (film) | Thybo et al.77 | ||
| Bi | Bismuth 2-ethylhaxanoate | 2-Ethylhexanoic acid–toluene | BiVO4 and SiO2/BiVO4 | Strobel et al.140 | |
| Mineral spirit–tetrahydrofuran | Bi | Restricted oxygen flame | Grass and Stark86 | ||
| CeO2/Bi | Grass et al.181 | ||||
| Bismuth acetate | Acetic acid, nitric acid and ethanol | Bi2O3 | Bismuth trinitrate pentahydrate + acetic acid in an ethanol/nitric acid medium | Mädler and Pratsinis96 | |
| Bismuth trinitrate pentahydrate | Nitric acid–alcohol (alcohol = methanol, ethanol, methoxy propanol, ethoxy ethanol, propylene glycol propylether, diethylene glycol-monoethylether | Bi2O3 | Mädler and Pratsinis,96 Jossen et al.47 | ||
| Ce | Cerium 2-ethylhexanoate | 2-Ethylhexanoic acid | Pt/CexZr1−xO2 | Stark et al.173 | |
| 2-Ethylhexanoic acid–toluene | Pt/Ba/CexZr1−xO2 | Twin nozzles | Strobel et al.131 | ||
| Xylene | Rh/CexZr1−xO2 | Hotz et al.174 | |||
| Cerium acetate hydrate | 2-Ethylhexanoic acid–toluene | MOx/CexZr1−xO2 (MOx = SiO2, Al2O3) | Acetic acid digested zirconium carbonate + 2-ethylhexanoic acid + cerium acetate hydrate (N2 distilled to remove water and acetic acid) | Schulz et al.124 | |
| Acetic acid | CeO2 | Inhomogeneous particle size | Mädler et al.66 | ||
| Acetic acid–isooctane–butanol | CeO2 | Homogeneous particle size | Mädler et al.66 | ||
| CeO2/ZrO2 | Low boiling point solvent resulting in segregated ZrO2 and CeO2 phase | Stark et al.175 | |||
| Lauric acid–acetic acid | CexZr1−xO2 | Heating to full dissolution | Stark et al.175 | ||
| Cerium(III) nitrate | Ethanol | Al2O3:Ce |
Williams et al.118 | ||
| Cerium(III) octoate | 2-Ethylhexanoic acid–toluene | CexZr1−xO2 | Jossen et al.75 | ||
| Tetrahydrofuran | CeO2/Bi | Restricted oxygen flame | Grass et al.181 | ||
| Cerium propionate | Ethanol | (CeOx)x(Al2O3)1−x | Reaction distillation of cerium carbonate with propionic acid (120 °C, 2 h, flowing N2) | Kim et al.120 | |
| Pr | Praseodymium(III) nitrate | Ethanol | Al2O3:Pr |
Williams et al.118 | |
| Nd | Neodymium(III) nitrate | Ethanol | Nd:Co:Fe2O3 |
Dosev et al.159 | |
| Eu | Europium 2-ethylhexanoate | Toluene | Y2O3:Eu3+ |
Camenzind et al.12,13 | |
| Europium(III) nitrate | Ethanol | Y2O3:Eu3+ |
Dosev et al.14 | ||
| Eu:Gd2O3 |
Goldys et al.182 | ||||
| Y2SiO5:Eu3+ |
Qin et al.142 | ||||
| Nd:Co:Fe2O3 in ethanol slurry |
Eu:Gd2O3/Nd:Co:Fe2O3 |
Flame spraying of slurry | Dosev et al.159 | ||
| Gd | Gadolinium(III) nitrate | Ethanol | Eu:Gd2O3 |
Goldys et al.182 | |
| Nd:Co:Fe2O3 in ethanol slurry |
Eu:Gd2O3/Nd:Co:Fe2O3 |
Flame spraying of slurry | Dosev et al.159 | ||
| Ho | Holmium 2-ethylhexanoate | 2-Ethylhexanoic acid | Ho–BaF2 | Holmium oxide in 2-ethylhexanoic acid | Grass and Stark111 |
| Yb | Ytterbium 2-ethylhexanoate | Xylene | Yb2O3/SiO2 | Reactive distillation of yttrium nitrate + 2-ethylhexanoic acid (N2, 107 °C) | Mädler et al.141 |
| Ytterbium nitrate pentahydrate | Acetonitrile–acetic anhydride–acetic acid | Yb2O3/SiO2 | Mädler et al.141 |
Low cost chemical conversion of metal oxides and hydroxides to soluble “trane” complexes is a robust alternative to preparing moisture insensitive metal precursors. Introduced by Laine and co-workers, the procedure involves moderate heating-distillation of solid metal oxides and/or hydroxides in triethanolamine (trane)/ethylene glycol under inert environment.28 In the past, a range of trane complexes ranging from Ti,149 Li, Na,105 Ca, Ba, Sr,183 Si,29 Ce, Zr, Y, Yb, Er, Tm to Pr,11,118 or even multi-metal tranes such as Sr–Si–Al, Ba–Si–Al and Al–Mg28 were synthesised. A similar polymerisable complex or Pechini method is another promising technique for the conversions of metal oxide, carbonates or salts to liquid precursors.184–188 It involves dissolving of single or multi-metal components in an acetic acid (as chelating agent)–ethylene glycol–methanol mixture followed by heating to polymerise the acetic acid and ethylene glycol. Both the trane and polymerised complex can be readily dissolved in alcohols.
Metal carboxylates189 are another group of metal precursors with proven versatility and ease in handling (hydrolytically insensitive). The simplicity of carboxylic acids in forming favourable ligand exchange, especially with metal nitrates, is attractive.96 Although short chain carboxylates such as acetates have low combustion enthalpy, its calorific value can generally be increased by increasing the carbon chain length. This prompted the synthesis of Co, Cu, Ni,167 Y122 propionates or Y12,13,75 and Yb141 2-ethylhexanoates by reaction of the long chain alkanoic acids with metal nitrates (Table 1). Heating or refluxing of the metal nitrate–alkanoic acid mixture promotes ligand exchange. Besides reacting with metal nitrates, the reaction with 2-ethylhexanoic acid can also be extended to the conversion of other low cost materials such as acetates (Na, Ba, Sr, Bi),111 oxides (Ca,19,147 Mg,19 Ho111), hydroxides (Ca,110,148 Al76,124) and even carbonates (Na,111 Zr76). The higher viscosity of 2-ethylhexanoic acid compared to water or ethanol has to be accounted for in nozzle design and liquid pump selection or dilution with e.g. xylene.
4. Particles formation and configurations by FSP
In the previous section, we identified the relevance of liquid precursor design to the formation of particles through the droplet-to-particle or gas-to-particle routes, the latter ensures homogeneous morphologies and sizes. In this section, we further review the different categories of particle configurations that may form in the gas-to-particle route in FSP. These particles form in sequential stages of: (1) precursor spray evaporation/decomposition forming metal vapour; (2) nucleation as a result of supersaturation; (3) growth by coalescence and sintering; and (4) particles aggregation (forming hard agglomerates by chemical bonds) and agglomeration (forming soft agglomerates by mainly physical bonds) (Fig. 5). Since the general gas-to-particle mechanism has been well-studied and reviewed in the past, it will not be repeated here, but readers are referred to previous literature for a more detailed understanding.5,101–103 Despite the common route to particle formation under the classification of gas-to-particle, the uniqueness of every material system prohibits generalisation of each particle configuration predictably. Prior insights of the materials properties and thermodynamic need to be taken into account in relevance to the flame conditions. Fig. 5 summarises all the particle configurations (and combinations) obtained by FSP to date. It must be mentioned that the details of particles formation on most of these routes were based only on the most current conceptual understanding, rather than proven experimentally.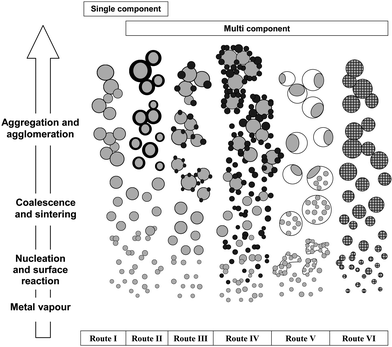 | ||
| Fig. 5 The formation of different particle configurations via the gas-to-particle mechanism for single and multicomponent systems. Please refer to the text for details. Although not shown in the Figure, the stage of droplet spray should in the context of FSP, precede the metal vapour stage. | ||
Flame spraying of single metal component precursors in open ambient (air) condition produces of simple oxides or carbonates in the case of alkaline metals through reaction with the CO2-laden flame (Fig. 5, Route I). A wide range of elements across the periodic table, from MgO to Yb2O3, have been synthesised via this route (see Fig. 6 and Table 1). The high entrainment of ambient air72 during particle synthesis provides a continuous and excess supply of O2 to the flame (oxidizing). Coupled with high flame temperatures, these are the reasons that minimal amount of carbon soot formed on the metal oxide surfaces in this process. The high temperature formation in the flame renders high thermal stability of the as-prepared particles as compared to other lower temperature wet techniques.66,190,191
 | ||
| Fig. 6 Examples of as-prepared metal oxides of different morphologies made by FSP, varying from: (a) hexagonal/octagonal platelet γ-Fe2O3; (b) slightly oblonged ZnO; (c) spherical TiO2; and (d) rhomboid-shaped CeO2 with sharp edges. | ||
Additionally, highly crystalline nanoparticles are often produced by FSP, as induced by the high flame temperature crystallisation. The polymorphic structures of some oxides, such as TiO2,152,192 Y2O312 and WO3,158,179 can be designed by capitalising the inter-relationship of flame synthesis and materials characteristics. Only in a few instances are amorphous materials such as that in SiO265,135 and C91 obtained due to insufficient residence time and/or flame temperatures. Extension of the high temperature flame regime is achievable by enclosing the FSP flame in a quartz tube, where external entrainment of ambient air is prevented while conserving the convection heat within the tube.155 Doing so extends the residence time at which aerosol formation and growth takes place. Besides obtaining larger particle sizes, the technique is unique in the reduction of crystal defects such as those achieved in enhanced magnetic moment of γ-Fe2O323,146 as well as the decreased electron capacitance of TiO2 nanoparticles in dye-sensitised solar cells due to elimination of charge trapping sites.191
Oxygen containing anions such as phosphates19,21 and sulfates (currently limited only to group II metal i.e. Ca2+)110 can also form in the presence of P and S, respectively. The high reactivity of group II metals is particularly interesting as it allows the formation of fluorides (BaF2, CaF2, SrF2) even in the presence of air,111 due to the higher affinity of F− towards this group of metals compared to oxygen anionic species (i.e. oxides and carbonates). Likewise, NaCl salts can be synthesised in the presence of Cl−.111
Enclosure of FSP in an inert environment in practice suppresses the amount of external O2 being entrained (limiting O2 solely from internally delivered sources i.e. dispersant and sheath gases), and depending on the oxidising affinity of the carbon precursor and that of the metal compound, could yield metallic nanoparticles (Fig. 2 IX).24 To date however, only the FSP synthesis of metallic Co,24,109 Bi86 and Cu87,193 have been reported, where metallic Bi must be kept under anaerobic conditions to prevent rapid oxidation to Bi2O3, while metallic Co and Cu nanoparticles are protected either by a surface oxide or carbon layer (Fig. 5, Route II). The latter is formed when the total amount of O2 provided is less than needed for total combustion of all carbon sources, allowing deposition of carbonaceous layers in the form of soot, graphene or graphite. In some cases C2H2, CO or H2 can be provided externally to the spray flame to induce higher degree of reduction and yield highly ordered carbonaceous deposition layers.25,193
The particle formation paths in multicomponent systems are far more complex than single components. A wide range of particle configurations and their permutations are possible, dictated by the kinetics and thermodynamics of the component mixture. Hereby, we present the most general classes of particle configurations for multicomponents (Fig. 5, Route II–VI). In highly complex systems, particularly those comprising a large number of elements with different properties, combinations of two or more configurations from Route I–IV are indeed possible.
Direct encapsulation of a secondary component metal or metal oxide shell or even carbonaceous coating is a demanding process due to the difficult entropic requirements. The possibilities exist for the formation of composite materials (e.g. complex oxides, solid solutions) as well as interparticle phase segregations. To maintain an even coating morphology, favourable and strong interfacial interaction between the two components, besides higher amount of the coating component than the solid-state solubility limit, are required at the stage where the secondary component is precipitated. Phase segregation could result if the temperature-dependent interfacial interaction is weaker than that required to maintain homogeneous coating. To date, only a small number of such systems prepared by direct flame spraying of the components mixture have been achieved i.e. SiO2124 and Al2O3124,194 coatings on CexZr1−xO2, as well as TiO2 on CeO2.195In situ sequential coating technique (Fig. 2, XI and Section 2) capitalizes on the introduction of the secondary component at a stage where the primary component is fully formed, and which temperature and remaining residence time is sufficiently low to avoid segregation of the homogeneous encapsulation layer.92–95
In the case where full encapsulation is not attainable, either due to insufficient amount of the secondary component or occurrence of phase segregation (as small deposits), a supported metal/metal oxide on another metal oxide component can result (Fig. 5, Route III). Most notably, deposition of the secondary component as fine noble metals (Ru, Rh, Pd, Ag, Pt to Au) or even alloys on various metal oxide supports such as Al2O3, TiO2, ZnO, SiO2, CexZr1−xO2, etc. have been reported by FSP (see Fig. 7, Table 1). Such morphologies have been largely explored for catalytic applications,7,196 while others such as gas sensors18,81,82,197 and antimicrobial138,168,157 applications also exist. In many cases, the low boiling/sublimation points of the noble metals relative to that of the oxide supports allow sequential nucleation and growth along the concentration gradient of the spray flame.70,129,132,152 The support metal oxide is first nucleated at high temperature followed by that of noble metals at cooler downstream gas temperature. The rapid quenching and short residence time of the spray flame is advantageous here as it prevents significant sintering of the noble metals upon deposition on support and therefore maintains high surface dispersion of deposits (Fig. 7). Strong metal (or deposit)–support interaction is another factor governing its dispersion, as the strong interaction limits the mobility (and hence sintering) of the deposits at high flame temperatures. See for example ref. 130–132 on the effect of Pt–Pd alloy deposits on Al2O3, as compared to deposits in their pure forms.
 | ||
| Fig. 7 Examples of FSP-made supported noble metals on various metal oxide supports prepared in a single step, as illustrated in Route III – (a) Au/CeO2, (b) Pt/TiO2, (c) Au/SiO2/γ-Fe2O3 and (d) Ag/TiO2. | ||
Despite the elegance of Route III synthesis, it is somewhat limited by the thermodynamic properties of the deposit-support, where besides the difference in boiling points, solid-state phase miscibility of deposit-support materials should also be taken into consideration. This is particularly important when the deposits are nucleated at high temperature where solid-state reaction, e.g. phase miscibility, formation of complex oxides, with support materials is non-negligible. To circumvent this limitation, the support and deposit materials can be precipitated separately in a twin-nozzle configuration (Fig. 2 X), interfacing both flames at a point with desirable extent of solid-state reactions.91 Rapid quenching of the flame at the stage of post-precipitation of noble metal deposits is another effective technique in limiting the deposit/support sintering70 (Fig. 2, VIII).
Because the deposit/support particles are formed at high temperature, they (i.e. dispersion of metal deposits, Fig. 5, Route III) are often presumed to exhibit high thermal stability. While this may be true,132 but depending on the metal-support interactions, they may be subjected to significant loss in metal dispersion when overly exposed to very high temperatures. For instance, the initially highly-dispersed Pd deposits (on Al2O3) sinter significantly from 5 nm to 50–150 nm when exposed to 1000 °C,130 rendering them indifferent from other lower temperature preparations. Besides surface dispersion, the oxidation state of the as-prepared metal deposits is an important characteristic that should not be overlooked. It has direct influence on some targeted end applications, including, catalytic hydrogenation,132 photocatalysis154 and antimicrobial.157 Other non-precious metals deposit/support structures that have been synthesised via Route III are V2O5/TiO283,197 and CuO/CeO2.169,198
In some multicomponent systems, the different particle phases tend to segregate completely with little miscibility allowing only physical inter-particle mixing (Fig. 5, Route IV). Achieving such configuration during co-precipitation in a single flame is usually rare as multicomponent solid state reaction or mixing at an atomic level is usually inevitable at high temperature. CeO2/Bi prepared in a restrictive oxygen flame is the only such system reported so far.181 By adopting a more engineering approach such as that in the twin nozzle synthesis of Pt/Ba/Al2O3, the interaction of Pt/BaCO3 and Al2O3 was deliberately kept apart by separate precipitations in independent spray flames and only mixed at a point where the flame temperature was low enough to avoid solid-state reactions between the two phases.89 Formation of BaAl2O4 is detrimental to the NOx storage capacity during catalytic deNOx applications.89,128 The same concept was adopted for the flame mixing of Pt/BaCO3 and CexZr1−xO2.90
Segregation of phases could also take place within a single particle host matrix (Fig. 5, Route V) such as that of SiO2/ZnO135–137 and SiO2/γ-Fe2O322,146 (Fig. 8). In these systems, molten SiO2 is thought to envelope the precipitated particles i.e. ZnO or γ-Fe2O3 producing a multicore structure (see Fig. 8b,c), while the encapsulation layer acts as diffusion barrier, retarding the agglomeration and sintering of the core particles.22,199 One of the features of such configuration is the phase immiscibility or limited solid-state reactions between the core particles and the encapsulation layer, despite being in intimate contact. This could possibly arise from the limited thermodynamic miscibility of the two phases at the encapsulation temperature, or the formation of mixed oxide layers at the phase boundary that further suppressed the miscibility.
 | ||
| Fig. 8 Examples of embedded core particle systems prepared by a single-step FSP as depicted in Route V – (a) single core and (b) multicore γ-Fe2O3 in a single SiO2 matrix and (c) ZnO cores in amorphous SiO2 matrix. Figure (c) adapted with permission from ref. 136. | ||
For systems where general homogeneous mixing between multicomponents at intra-particle level are preferred and depending on the states as well as their nature of mixing, a wide variety of configurations could result, ranging from substitutionally-doped systems, solid solutions, dispersed mixed oxides, complex metal oxides (perovskite, spinel, etc.) to metal alloys. Such particle configuration in route VI (Fig. 5) is exclusive to highly miscible multi-elements. The bottom-up synthesis and high synthesis temperature in FSP can be advantageous in enhancing substitutional dopant concentrations, while its steep temperature gradient and short residence time prevent segregation of the dopants.156 A classic example of perfect solid solutions by FSP is that of CexZr1−xO2, which was obtainable when using high boiling point solvents (i.e. gas-to-particle route) to ensure good distribution of precursor in the flame.175 Unlike solid solutions, Ta2O5/SiO220 and Co3O4/ZrO2160 retain the individual crystallite structures, while still achieving physical dispersity within a single particle matrix. A large family of complex metal oxides involving the oxide of two or more metals also fall in this route VI configuration: MgAl2O4,28,33 3Al2O3 · 2SiO2,29 Y3Al5O12,122 LaCoO3,161–163 CoOx–Al2O3,31 NiO–Al2O3,32 LiMn2O4, Li4Ti5O12, LiFe5O8,26 BiVO4.140,200,201 In the event of multicomponent synthesis in restrictive oxygen flames, even alloys such as Ni/Mo could be achieved.88
5. Concluding remarks and perspectives
The advancement of FSP currently lies at the intersection of combustion science, aerosols technology and materials chemistry. As such, the interdisciplinary knowledge of all intersecting fields is not only inevitable but is becoming increasingly important in the design of flame-made nanoparticles. As in the past decade, innovations in FSP technology, in terms of reactor engineering and particles design, will continue to be applications-driven. In terms of industrial applications, FSP is a proven scalable and high throughput process unmatched by other wet chemical techniques.67,71,75 Formulation of liquid precursors, which was previously perceived to be a “stumbling block” requiring expensive and moisture-sensitive chemicals, is now reasonably well-understood. In fact, high quality metal precursors can now be synthesised from cheap raw materials through various routes, even though more work is still required in the area of precious metals.In terms of particle homogeneity, it is often limited by the so-called self preserving size distributions (SPSD), which forms the asymptotic limit of polydispersity for particles growth by Brownian coagulation, giving number geometric standard deviation of ∼1.46.5 However, there is increasing demand especially in bioapplications for particles with much narrower size distribution (high monodispersity), whilst maintaining high particle purity and crystallinity. Exclusive particle growth by surface condensation rather than coagulation is one possibility102 but the implementation of such restrictive system is highly challenging, especially within the harsh spray flame conditions. Manipulations of flame combustion, fluid and aerosol dynamics is necessary to achieve this task.
As for the production of non-oxygen containing ceramics, only group II metal fluorides have been reported by FSP to date.111 The synthesis of metal carbides, nitrides, phosphides and sulfides are still elusive in FSP, while the synthesis of SiC and Si3N4 has been shown to be possible by VAFS, using SiH4 precursors.202,203 A requirement for such systems would be to restrict the presence of O2 which would otherwise result in the formation of metal oxides, carbonates, phosphates or sulfates. Creation of anisotropic materials or particle shape control by FSP is also lacking. Besides CeO2 rhomboids,66 BaF2 cubes,111 (In, Sn)-doped ZnO nanorods106 and γ-Fe2O3 hexagonal/octagonal platelets,22,23 most other FSP-derived particles are spherical. Moreover, most of these non-spherical shapes are naturally occurring in the flame thereby providing little flexibility in terms of shape manipulation. Magnetically-assisted formation of metallic Co nanowires by FSP reported recently marks the beginning of externally-induced shape control by FSP.109 More work in this direction is required, for example, template-assisted, thermophoretically- or electrophoretically-induced flame aerosols alignment and assemblies.
In the last few years, we have seen successful combinations of FSP-derived inorganic particles with organic materials particularly for bioapplications, namely dental fillers,20,141 bioactive polymer films147 and magnetic particles with drug delivery204,205 as well as magnetic resonance imaging205,206 functions. This requires the coupling of predesigned flame particles with functional organic molecules such as DNA, protein, polymers and surfactants. More developments in this regard, encompassing various areas of applications from functional pigments, sensors to bioelectronics are expected to take place in the near future. Of equal importance is the fabrication of flame particles-based devices as readily demonstrated for gas sensors.17,85,178 Other potential devices such as solar cells, fuel cells, batteries, lab-on-a-chip etc. are also possible. The ability to create short-range and long-range ordering of these particles as-prepared via FSP, with or without templating, and their further assemblies in devices would also be of tremendous benefit.
Acknowledgements
The authors gratefully acknowledge Prof. Sotiris E. Pratsinis (ETH Zürich) and Prof. Jan-Dierk Grunwaldt (Technical University of Denmark) for their insightful comments and discussion on the manuscript. The authors also thank Dr Dan Li, Dr Richard Kydd and Yung Kent Kho (UNSW) and Dr Frank Krumeich (ETH Zürich) for many of the electron microscopy images presented in this review. W. Y. T. and R. A. thank the Australian Research Council (ARC) for financial support on various FSP-related projects.References
- K. Wegner and S. E. Pratsinis, Chem. Eng. Sci., 2003, 58, 4581 CrossRef CAS.
- R. Strobel and S. E. Pratsinis, J. Mater. Chem., 2007, 17, 4743 RSC.
- G. D. Ulrich, Chem. Eng. News, 1984, 62, 22 CAS.
- G. Kühner, M. VollManufacture of Carbon Black, in Carbon Black ed. Donnet, J.-B., Bansal, R. C., Wang, M.-J., Marcel Dekker, New York, 1993 Search PubMed.
- S. E. Pratsinis, Prog. Energy Combust. Sci., 1998, 24, 197 CrossRef CAS.
- L. Mädler, KONA, 2004, 22, 107 Search PubMed.
- R. Strobel, A. Baiker and S. E. Pratsinis, Adv. Powder Technol., 2006, 17, 457 Search PubMed.
- G. D. Ulrich, Combust. Sci. Technol., 1971, 4, 47 CrossRef CAS.
- B. S. Marshall, I. Telford and R. Wood, Analyst, 1971, 96, 569 RSC.
- M. Sokolowski, A. Sokolowska, A. Michalski and B. Gokieli, J. Aerosol Sci., 1977, 8, 219 CrossRef.
- R. M. Laine, T. Hinklin, G. Williams and S. C. Rand, Mater. Sci. Forum, 2000, 343–346, 500 CrossRef CAS.
- A. Camenzind, R. Strobel and S. E. Pratsinis, Chem. Phys. Lett., 2005, 415, 193 CrossRef CAS.
- A. Camenzind, R. Strobel, F. Krumeich and S. E. Pratsinis, Adv. Powder Technol., 2007, 18, 5 Search PubMed.
- D. Dosev, B. Guo and I. M. Kennedy, J. Aerosol Sci., 2006, 37, 402 CrossRef CAS.
- T. R. Hinklin, J. Azurdia, M. Kim, J. C. Marchal, S. Kumar and R. M. Laine, Adv. Mater., 2008, 20, 1270 CrossRef CAS.
- T. Sahm, L. Mädler, A. Gurlo, N. Bârsan, S. E. Pratsinis and U. Weimar, Sens. Actuators, B, 2004, 98, 148 CrossRef.
- L. Mädler, A. Roessler, S. E. Pratsinis, T. Sahm, A. Gurlo, N. Barsan and U. Weimar, Sens. Actuators, B, 2006, 114, 283 CrossRef.
- L. Mädler, T. Sahm, A. Gurlo, J. D. Grunwaldt, N. Barsan, U. Weimar and S. E. Pratsinis, J. Nanopart. Res., 2006, 8, 783 CrossRef.
- S. Loher, W. J. Stark, M. Maciejewski, A. Baiker, S. E. Pratsinis, D. Reichardt, F. Maspero, F. Krumeich and D. Gunther, Chem. Mater., 2005, 17, 36 CrossRef CAS.
- H. Schulz, L. Mädler, S. E. Pratsinis, Burtscher and N. Moszner, Adv. Funct. Mater., 2005, 15, 830 CrossRef CAS.
- F. Rohner, F. O. Ernst, M. Arnold, M. Hilbe, R. Biebinger, F. Ehrensperger, S. E. Pratsinis, W. Langhans, R. F. Hurrell and M. B. Zimmermann, J. Nutr., 2007, 137, 614 CAS.
- D. Li, W. Y. Teoh, C. Selomulya, R. C. Woodward, R. Amal and B. Rosche, Chem. Mater., 2006, 18, 6403 CrossRef CAS.
- D. Li, W. Y. Teoh, C. Selomulya, R. C. Woodward, P. Munroe and R. Amal, J. Mater. Chem., 2007, 17, 4876 RSC.
- R. N. Grass and W. J. Stark, J. Mater. Chem., 2006, 16, 1825 RSC.
- R. N. Grass, E. K. Athanassiou and W. J. Stark, Angew. Chem., Int. Ed., 2007, 46, 4909 CrossRef CAS.
- F. O. Ernst, H. K. Kammler, A. Roessler, S. E. Pratsinis, W. J. Stark, U. Ufheil and P. Novák, Mater. Chem. Phys., 2007, 101, 372 CrossRef CAS.
- T. J. Patey, R. Büchel, S. H. Ng, F. Krumeich, S. E. Pratsinis and P. Novák, J. Power Sources, 2009, 189, 149 CrossRef CAS.
- C. R. Bickmore, K. F. Waldner, D. R. Treadwell and R. M. Laine, J. Am. Ceram. Soc., 1996, 79, 1419 CAS.
- R. Baranwal, M. P. Villar, R. Garcia and R. M. Laine, J. Am. Ceram. Soc., 2001, 84, 951 CAS.
- A. C. Sutorik and M. S. Baliat, Mater. Sci. Forum, 2002, 386–388, 371 CrossRef.
- J. Azurdia, J. Marchal and R. M. Laine, J. Am. Ceram. Soc., 2006, 89, 2749 CAS.
- J. A. Azurdia, J. Marchal, P. Shea, H. Sun, X. Q. Pan and R. M. Laine, Chem. Mater., 2006, 18, 731 CrossRef CAS.
- T. R. Hinklin and R. M. Laine, Chem. Mater., 2008, 20, 553 CrossRef CAS.
- Nanomaterials08, http://www.nanomaterials08.com/sponsor_exhibit.php, accessed 20 October 2008 Search PubMed.
- F. Klopfer German Patent DE 762,723, 1942.
- Degussa, Aerosil® brochure, 2004 Search PubMed.
- J. M. Rowell, Sci. Am., 1986, 255, 146 CrossRef.
- M. Formenti, F. Juiliet, P. Meriaudeau, S. J. Teichner and P. Vergnon, J. Colloid Interface Sci., 1972, 39, 79 CrossRef CAS.
- P. T. Spicer, C. Artelt, S. Sanders and S. E. Pratsinis, J. Aerosol Sci., 1998, 29, 647 CrossRef CAS.
- H. K. Kammler and S. E. Pratsinis, J. Mater. Res., 2003, 18, 2670 CrossRef CAS.
- T. Johannessen and S. Koutsopoulos, J. Catal., 2002, 205, 404 CrossRef CAS.
- R. A. Dobbins and C. M. Megaridis, Langmuir, 1987, 3, 254 CrossRef CAS.
- W. Koch and S. K. Friedlander, J. Colloid Interface Sci., 1990, 140, 419 CrossRef CAS.
- S. Tsantilis, H. K. Kammler and S. E. Pratsinis, Chem. Eng. Sci., 2002, 57, 2139 CrossRef CAS.
- T. Johannessen, S. E. Pratsinis and H. Livbjerg, Chem. Eng. Sci., 2000, 55, 177 CrossRef CAS.
- S. E. Pratsinis, W. Zhu and S. Vemury, Powder Technol., 1996, 86, 87 CrossRef CAS.
- R. Jossen, S. E. Pratsinis, W. J. Stark and L. Mädler, J. Am. Ceram. Soc., 2005, 88, 1388 CrossRef CAS.
- G. L. Messing, S. C. Zhang and G. V. Jayanthi, J. Am. Ceram. Soc., 1993, 76, 2707 CAS.
- J. H. Brewster and T. T. Kodas, AIChE J., 1997, 43, 2665 CrossRef CAS.
- Y. C. Kang, D. J. Seo, S. B. Park and H. D. Park, Jpn. J. Appl. Phys., 2001, 40, 4083 CrossRef CAS.
- K. Okuyama and I. W. Lenggoro, Chem. Eng. Sci., 2003, 58, 537 CrossRef CAS.
- H. Chang, I. W. Lenggoro, K. Okuyama and T.-O. Kim, Jpn. J. Appl. Phys., 2004, 43, 3535 CrossRef CAS.
- W. Lee, Y. M. Gao, K. Dwight and A. Wold, Mater. Res. Bull., 1992, 27, 685 CrossRef CAS.
- Y. J. Jang, C. Simer and T. Ohm, Mater. Res. Bull., 2006, 41, 67 CrossRef CAS.
- R. Kriegel, J. Töpfer, N. Preuss, S. Grimm and J. Böer, J. Mater. Sci. Lett., 1994, 13, 1111 CrossRef CAS.
- R. A. M. Giacomuzzi, M. Portinari, I. Rosetti and L. Forni, Stud. Surf. Sci. Catal., 2000, 130, 197.
- I. Rosetti and L. Forni, Appl. Catal., B, 2001, 33, 345 CrossRef.
- E. Campagnoli, A. Tavares, L. Fabbrini, I. Rosetti, Y. A. Dubitsky, A. Zaopo and L. Forni, Appl. Catal., B, 2005, 55, 133 CrossRef CAS.
- P. A. Tanner and K. L. Wong, J. Phys. Chem. B, 2004, 108, 136 CrossRef CAS.
- X. Qin, Y. Ju, S. Bernhard, N. YaoProc. in NSTI Nanotech 2005, NSTI Nanotechnology Conference and Trade Show, Anaheim, CA, United States, May 8–12, 2005, 9, 12.
- A. Purwanto, I. W. Lenggoro, H. Chang and K. Okuyama, J. Chem. Eng. Jpn., 2006, 39, 68 CrossRef CAS.
- K. Y. Jung and Y. C. Kang, Mater. Lett., 2004, 58, 2161 CrossRef CAS.
- H. Chang, I. W. Lenggoro, T. Ogi and K. Okuyama, Mater. Lett., 2005, 59, 1183 CrossRef CAS.
- J. Karthikeyan, C. C. Berndt, J. Tikkanen, J. Y. Wang, A. H. King and H. Herman, Nanostruct. Mater., 1997, 8, 61 CrossRef CAS.
- L. Mädler, H. K. Kammler, R. Mueller and S. E. Pratsinis, J. Aerosol Sci., 2002, 33, 369 CrossRef CAS.
- L. Mädler, W. J. Stark and S. E. Pratsinis, J. Mater. Res., 2002, 17, 1356 CrossRef CAS.
- T. Tani, N. Watanabe and K. Takatori, J. Nanopart. Res., 2003, 5, 39 CrossRef CAS.
- T. Xia, M. Kovochich, M. Liong, L. Mädler, B. Gilbert, H. Shi, J. I. Yeh, J. I. Zink and A. E. Nel, ACS Nano, 2008, 2, 2121 CrossRef CAS.
- R. Mueller, L. Mädler and S. E. Pratsinis, Chem. Eng. Sci., 2003, 58, 1969 CrossRef.
- H. Schulz, L. Mädler, R. Strobel, R. Jossen, S. E. Pratsinis and T. Johannessen, J. Mater. Res., 2005, 20, 2568 CrossRef CAS.
- R. Mueller, R. Jossen, S. E. Pratsinis, M. Watson and M. K. Akhtar, J. Am. Ceram. Soc., 2004, 87, 197 CrossRef CAS.
- M. C. Heine, L. Mädler, R. Jossen and S. E. Pratsinis, Combust. Flame, 2006, 144, 809 CrossRef CAS.
- R. M. Laine, J. Marchal, H. Sun and X. Q. Pan, Adv. Mater., 2005, 17, 830 CrossRef CAS.
- R. Strobel, M. Maciejewski, A. Baiker and S. E. Pratsinis, Thermochim. Acta, 2006, 445, 23 CrossRef CAS.
- R. Jossen, R. Mueller, S. E. Pratsinis, M. Watson and M. K. Akhtar, Nanotechnology, 2005, 16, S609 CrossRef.
- R. Jossen, M. C. Heine, S. E. Pratsinis and M. K. Akhtar, Chem. Vap. Deposition, 2006, 12, 614 CrossRef CAS.
- S. Thybo, S. Jensen, J. Johansen, T. Johannessen, O. Hansen and U. J. Quaade, J. Catal., 2004, 223, 271 CrossRef CAS.
- A. Tricoli, M. Graf, F. Mayer, S. Kühne, A. Hierlemann and S. E. Pratsinis, Adv. Mater., 2008, 20, 3005 CrossRef CAS.
- A. T. Hunt, W. B. Carter and J. K. Cochran, Jr., Appl. Phys. Lett., 1993, 63, 266 CrossRef CAS.
- L. Mädler, A. A. Lall and S. K. Friedlander, Nanotechnology, 2006, 17, 4783 CrossRef CAS.
- T. Sahm, W. Rong, N. Bârsan, L. Mädler, S. K. Friedlander and U. Weimar, J. Mater. Res., 2007, 22, 850 CrossRef CAS.
- T. Sahm, W. Rong, N. Bârsan, L. Mädler and U. Weimar, Sens. Actuators, B, 2007, 127, 63 CrossRef.
- B. Schimmoeller, H. Schulz, S. E. Pratsinis, A. Bareiss, A. Reitzmann and B. Kraushaar-Czarnetzki, J. Catal., 2006, 243, 82 CrossRef CAS.
- Y. Liu, E. Koep and M. Liu, Chem. Mater., 2005, 17, 3997 CrossRef CAS.
- E. Thimsen, N. Rastgar and P. Biswas, J. Phys. Chem. C, 2008, 112, 4134 CrossRef CAS.
- R. N. Grass and W. J. Stark, J. Nanopart. Res., 2006, 8, 729 CrossRef CAS.
- E. K. Athanassiou, R. N. Grass and W. J. Stark, Nanotechnology, 2006, 17, 1668 CrossRef.
- E. K. Athanassiou, R. N. Grass, N. Osterwalder and W. J. Stark, Chem. Mater., 2007, 19, 4847 CrossRef CAS.
- R. Strobel, M. Piacentini, L. Mädler, M. Maciejewski, A. Baiker and S. E. Pratsinis, Chem. Mater., 2006, 18, 2532 CrossRef CAS.
- R. Strobel, F. Krumeich, S. E. Pratsinis and A. Baiker, J. Catal., 2006, 243, 229 CrossRef CAS.
- F. O. Ernst, R. Büchel, R. Strobel and S. E. Pratsinis, Chem. Mater., 2008, 20, 2117 CrossRef CAS.
- A. Teleki, M. K. Akhtar and S. E. Pratsinis, J. Mater. Chem., 2008, 18, 3547 RSC.
- A. Teleki, B. Buesser, M. C. Heine, F. Krumeich, M. K. Akhtar and S. E. Pratsinis, Ind. Eng. Chem. Res., 2009, 48, 85 CrossRef CAS.
- A. Teleki, M. C. Heine, F. Krumeich, M. K. Akhtar and S. E. Pratsinis, Langmuir, 2008, 24, 12553 CrossRef CAS.
- A. Teleki, M. Suter, P. R. Kidambi, O. Ergeneman, F. Krumeich, B. J. Nelson and S. E. Pratsinis, Chem. Mater., 2009, 21, 2094 CrossRef CAS.
- L. Mädler and S. E. Pratsinis, J. Am. Ceram. Soc., 2002, 85, 1713 CAS.
- T. Tani, K. Takatori and S. E. Pratsinis, J. Am. Ceram. Soc., 2004, 87, 523 CrossRef CAS.
- M. Sokolowski, A. Sokolowska, A. Michalski and Z. Ziolkowski, J. Cryst. Growth, 1981, 52, 274 CrossRef CAS.
- S. Grimm, M. Schultz, S. Barth and R. Müller, J. Mater. Sci., 1997, 32, 1083 CrossRef CAS.
- T. T. Kodas, M. J. Hampden-SmithAerosol Processing of Materials, Wiley-VCH, New York, 1999 Search PubMed.
- M. S. Wooldridge, Prog. Energy Combust. Sci., 1998, 24, 63 CrossRef CAS.
- S. K. Friedlander Smoke, Dust and Haze: Fundamentals of Aerosol Dynamics, Oxford University Press, New York, 2nd edn, 2000 Search PubMed.
- A. Gutsch, H. Mühlenweg and M. Krämer, Small, 2005, 1, 30 CrossRef CAS.
- P. Roth, Proc. Combust. Inst., 2007, 31, 1773 CrossRef.
- A. C. Sutorik, S. S. Neo, D. R. Treadwell and R. M. Laine, J. Am. Ceram. Soc., 1998, 81, 1477.
- M. J. Height, L. Mädler, S. E. Pratsinis and F. Krumeich, Chem. Mater., 2006, 18, 572 CrossRef CAS.
- T. J. Brunner, R. N. Grass and W. J. Stark, Chem. Commun., 2006, 1384 RSC.
- M. Vollenweider, T. J. Brunner, S. Knecht, R. N. Grass, M. Zehnder, T. Imfeld and W. J. Stark, Acta Biomater., 2007, 3, 936 CrossRef CAS.
- E. K. Athanassiou, P. Grossmann, R. N. Grass and W. J. Stark, Nanotechnology, 2007, 18, 165606 CrossRef.
- S. Osterwalder, S. Loher, R. N. Grass, T. J. Brunner, L. K. Limbach, S. C. Halim and W. J. Stark, J. Nanopart. Res., 2007, 9, 275 CrossRef CAS.
- R. N. Grass and W. J. Stark, Chem. Commun., 2005, 1767 RSC.
- W. Y. Teoh, T. Fukahori, T. Kamai, T. Ohno, R. AmalProc. in International Conference on Photochemical Conversion and Storage of Solar Energy (IPS-17), Sydney, 27 July–1 August, 2008.
- A. V. Emeline, N. V. Sheremetyeva, N. V. Khomchenko, G. N. Kuzmin, V. K. Ryabchuk, W. Y. Teoh and R. Amal, J. Phys. Chem. C, 2009, 113, 4566 CrossRef CAS.
- T. Suzuki, Y. Ohishi and T. Tani, Mater. Sci. Eng., B, 2006, 128, 151 CrossRef CAS.
- T. Tani, S. Saeki, T. Suzuki and Y. Ohishi, J. Am. Ceram. Soc., 2007, 90, 805 CrossRef CAS.
- T. Tani, A. Kato and H. Morisaka, J. Ceram. Soc. Jpn., 2005, 113, 255 CrossRef CAS.
- T. R. Hinklin, J. Azurdia, M. Kim, J. C. Marchal, S. Kumar and R. M. Laine, Adv. Mater., 2008, 20, 1373 CrossRef CAS.
- G. R. Williams, S. B. Bayram, S. C. Rand, T. Hinklin and R. M. Laine, Phys. Rev. A: At., Mol., Opt. Phys., 2001, 65, 013807 CrossRef.
- M. Kim and R. M. Laine, J. Ceram. Process. Res., 2007, 8, 129 Search PubMed.
- M. Kim, T. R. Hinklin and R. M. Laine, Chem. Mater., 2008, 20, 5154 CrossRef CAS.
- T. Hinklin, B. Toury, C. Gervais, F. Babonneau, J. J. Gislason, R. W. Morton and R. M. Laine, Chem. Mater., 2004, 16, 21 CrossRef CAS.
- J. Marchal, T. John, R. Baranwal, T. Hinklin and R. M. Laine, Chem. Mater., 2004, 16, 822 CrossRef CAS.
- S. Kim, J. J. Gislason, R. W. Morton, X. Q. Pan, H. P. Sun and R. M. Laine, Chem. Mater., 2004, 16, 2336 CrossRef CAS.
- H. Schulz, W. J. Stark, M. Maciejewski, S. E. Pratsinis and A. Baiker, J. Mater. Chem., 2003, 13, 2979 RSC.
- P. Haider and A. Baiker, J. Catal., 2007, 248, 175 CrossRef CAS.
- S. Hannemann, J.-D. Grunwaldt, P. Lienemann, D. Günther, F. Krumeich, S. E. Pratsinis and A. Baiker, Appl. Catal., A, 2007, 316, 226 CrossRef CAS.
- N. van Vegten, D. Ferri, M. Maciejewski, F. Krumeich and A. Baiker, J. Catal., 2007, 249, 269 CrossRef CAS.
- M. Piacentini, R. Strobel, M. Maciejewski, S. E. Pratsinis and A. Baiker, J. Catal., 2006, 243, 43 CrossRef CAS.
- R. Strobel, F. Krumeich, W. J. Stark, S. E. Pratsinis and A. Baiker, J. Catal., 2004, 222, 307 CrossRef CAS.
- R. Strobel, S. E. Pratsinis and A. Baiker, J. Mater. Chem., 2005, 15, 605 RSC.
- R. Strobel, J.-D. Grunwaldt, A. Camenzind, S. E. Pratsinis and A. Baiker, Catal. Lett., 2005, 104, 9 CrossRef CAS.
- R. Strobel, W. J. Stark, L. Mädler, S. E. Pratsinis and A. Baiker, J. Catal., 2003, 213, 296 CrossRef CAS.
- J. Tikkanen, K. A. Gross, C. C. Berndt, V. Pitkänen, J. Keskinen, S. Raghu, M. Rajala and J. Karthikeyan, Surf. Coat. Technol., 1997, 90, 210 CrossRef CAS.
- R. M. Laine, J. C. Marchal, H. P. Sun and X. Q. Pan, Nat. Mater., 2006, 5, 710 CrossRef CAS.
- L. Mädler, W. J. Stark and S. E. Pratsinis, J. Appl. Phys., 2002, 92, 6537 CrossRef CAS.
- T. Tani, L. Mädler and S. E. Pratsinis, J. Mater. Sci., 2002, 37, 4627 CrossRef CAS.
- T. Tani, L. Mädler and S. E. Pratsinis, Part. Part. Syst. Charact., 2002, 19, 354 CrossRef CAS.
- S. Loher, O. D. Schneider, T. Maienfisch, S. Bokorny and W. J. Stark, Small, 2008, 4, 824 CrossRef CAS.
- A. Tricoli, M. Graf and S. E. Pratsinis, Adv. Funct. Mater., 2008, 18, 1969 CrossRef CAS.
- R. Strobel, H. J. Metz and S. E. Pratsinis, Chem. Mater., 2008, 20, 6346 CrossRef CAS.
- L. Mädler, F. Krumeich, P. Burtscher and N. Moszner, J. Nanopart. Res., 2006, 8, 323 CrossRef.
- X. Qin, Y. Ju, S. Bernhard and N. Yao, Mater. Res. Bull., 2007, 42, 1440 CrossRef CAS.
- M. Ramin, V. van Vegten, J.-D. Grunwaldt and A. Baiker, J. Mol. Catal. A: Chem., 2006, 258, 165 CrossRef CAS.
- R. Jossen, M. C. Heine, S. E. Pratsinis, S. Augustine and M. K. Akhtar, Appl. Catal., B, 2007, 69, 181 CrossRef CAS.
- S. Hannemann, J.-D. Grunwaldt, F. Krumeich, P. Kappen and A. Baiker, Appl. Surf. Sci., 2006, 252, 7862 CrossRef CAS.
- D. Li, W. Y. Teoh, R. C. Woodward, J. D. Cashion, C. Selomulya and R. Amal, J. Phys. Chem. C, 2009, 113, 12040 CrossRef CAS.
- S. Loher, V. Reboul, T. J. Brunner, M. Simonet, C. Dora, P. Neuenschwander and W. J. Stark, Nanotechnology, 2006, 17, 2054 CrossRef CAS.
- T. J. Brunner, R. N. Grass, M. Bohner and W. J. Stark, J. Mater. Chem., 2007, 17, 4072 RSC.
- C. R. Bickmore, K. F. Waldner, R. Baranwal, T. Hinklin, D. R. Treadwell and R. M. Laine, J. Eur. Ceram. Soc., 1998, 18, 287 CrossRef CAS.
- G. L. Chiarello, E. Selli and L. Forni, Appl. Catal., B, 2008, 84, 332 CrossRef CAS.
- A. Teleki, N. Bjelobrk and S. E. Pratsinis, Sens. Actuators, B, 2008, 130, 449 CrossRef.
- W. Y. Teoh, L. Mädler, D. Beydoun, S. E. Pratsinis and R. Amal, Chem. Eng. Sci., 2005, 60, 5852 CrossRef CAS.
- W. Y. Teoh, F. Denny, R. Amal, D. Friedmann, L. Mädler and S. E. Pratsinis, Top. Catal., 2007, 44, 489 CrossRef CAS.
- W. Y. Teoh, L. Mädler and R. Amal, J. Catal., 2007, 251, 271 CrossRef CAS.
- A. Teleki, S. E. Pratsinis, K. Kalyanasundaram and P. I. Gouma, Sens. Actuators, B, 2006, 119, 683 CrossRef.
- W. Y. Teoh, R. Amal, L. Mädler and S. E. Pratsinis, Catal. Today, 2007, 120, 203 CrossRef CAS.
- C. Gunawan, W. Y. Teoh, C. P. Marquis, Juniahani Lifia and R. Amal, Small, 2009, 5, 341 CrossRef CAS.
- L. Wang, A. Teleki, S. E. Pratsinis and P. I. Gouma, Chem. Mater., 2008, 20, 4794 CrossRef CAS.
- D. Dosev, M. Nichkova, R. K. Dumas, S. J. Gee, B. D. Hammock, M. Liu and I. M. Kennedy, Nanotechnology, 2007, 18, 055102 CrossRef.
- W. Y. Teoh, R. Setiawan, L. Mädler, J.-D. Grunwaldt, R. Amal and S. E. Pratsinis, Chem. Mater., 2008, 20, 4069 CrossRef CAS.
- G. L. Chiarello, I. Rosetti and L. Forni, J. Catal., 2005, 236, 251 CrossRef CAS.
- G. L. Chiarello, I. Rosetti, P. Lopinto, G. Migliavacca and L. Forni, Catal. Today, 2006, 117, 549 CrossRef CAS.
- G. L. Chiarello, I. Rosetti, L. Forni, P. Lopinto and G. Migliavacca, Appl. Catal., B, 2006, 72, 219.
- G. L. Chiarello, J.-D. Grunwaldt, D. Ferri, F. Krumeich, C. Oliva, L. Forni and A. Baiker, J. Catal., 2007, 252, 127 CrossRef CAS.
- G. L. Chiarello, D. Ferri, J.-D. Grunwaldt, L. Forni and A. Baiker, J. Catal., 2007, 252, 137 CrossRef CAS.
- G. L. Chiarello, I. Rosetti, L. Forni, P. Lopinto and G. Migliavacca, Appl. Catal., B, 2006, 72, 228.
- J. A. Azurdia, A. McCrum and R. M. Laine, J. Mater. Chem., 2008, 18, 3249 RSC.
- M. J. Height, S. E. Pratsinis International Patent WO 2006/084411 A1, 2006.
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, A. B. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369 CrossRef CAS.
- R. Kydd, W. Y. Teoh, J. Scott, D. Ferri and R. Amal, ChemCatChem, 2009, 1, 286 Search PubMed.
- T. Tani, L. Mädler and S. E. Pratsinis, J. Nanopart. Res., 2002, 4, 337 CrossRef CAS.
- M. J. Height, S. E. Pratsinis, O. Mekasuwandumrong and P. Praserthdam, Appl. Catal., B, 2006, 63, 305 CrossRef CAS.
- W. J. Stark, J. D. Grunwaldt, M. Maciejewski, S. E. Pratsinis and A. Baiker, Chem. Mater., 2005, 17, 3352 CrossRef CAS.
- N. Hotz, M. J. Stutz, S. Loher, W. J. Stark and D. Poulikakos, Appl. Catal., B, 2007, 73, 336 CrossRef CAS.
- W. J. Stark, L. Mädler, M. Maciejewski, S. E. Pratsinis and A. Baiker, Chem. Commun., 2003, 588 RSC.
- H. Keskinen, J. M. Mäkelä, M. Aromaa, J. Keskinen, S. Areva, C. V. Teixeira, J. B. Rosenholm, V. Pore, M. Ritala, M. Leskelä, M. Raulio, M. S. Salkinoja-Salonen, E. Levänen and T. Mäntylä, Catal. Lett., 2006, 111, 127 CrossRef CAS.
- H. Keskinen, J. M. Mäkelä, Aromaa, J. Ristimäki, T. Kanerva, E. Levänen, T. Mäntyla and J. Keskinen, J. Nanopart. Res., 2007, 9, 569 CrossRef CAS.
- S. Kühne, M. Graf, A. Tricoli, F. Mayer, S. E. Pratsinis and A. Hierlemann, J. Micromech. Microeng., 2008, 18, 035040 CrossRef.
- S. Pokhrel, J. Birkenstock, M. Schowalter, A. Rosenauer and L. Mädler, Cryst. Growth Des., 2006, 10, 632.
- L. Mädler, W. J. Stark and S. E. Pratsinis, J. Mater. Res., 2003, 18, 115 CrossRef CAS.
- R. N. Grass, T. F. Albrecht, F. Krumeich and W. J. Stark, J. Mater. Chem., 2007, 17, 1485 RSC.
- E. M. Goldys, K. Drozdowicz-Tomsia, S. Jinjun, D. Dosev, I. M. Kennedy, S. Yatsunenko and M. Godlewski, J. Am. Chem. Soc., 2006, 128, 14498 CrossRef CAS.
- R. Narayanan and R. M. Laine, Appl. Organomet. Chem., 1997, 11, 919 CrossRef.
- M. Kakihana and K. Domen, MRS Bull., 2000, 25, 27 CAS.
- M. Yoshino, M. Kakihana, W. S. Cho, H. Kato and A. Kudo, Chem. Mater., 2002, 14, 3369 CrossRef CAS.
- S. Ikeda, M. Hara, J. N. Kondo, K. Domen, H. Takahashi, T. Okubo and M. Kakihana, Chem. Mater., 1998, 10, 72 CrossRef CAS.
- Y. Miseki, H. Kato and A. Kudo, Chem. Lett., 2006, 35, 1052 CrossRef CAS.
- K. Yoshioka, V. Petrykin, M. Kakihana, H. Kato and A. Kudo, J. Catal., 2005, 232, 102 CrossRef CAS.
- S. Mishra, S. Daniele and L. G. Hubert-Pfalzgraf, Chem. Soc. Rev., 2007, 36, 1770 RSC.
- W. J. Stark, L. Mädler, M. Maciejewski, S. E. Pratsinis and A. Baiker, J. Catal., 2003, 220, 35 CrossRef CAS.
- G. Tsekuoras, M. Miyashita, Y. K. Kho, W. Y. Teoh, A. J. Mozer, R. Amal, S. Mori and G. G. Wallace, IEEE J. Sel. Top. Quantum Electron. Search PubMed , in press.
- Y. K. Kho, A. Iwase, W. Y. Teoh, L. Mädler, A. Kudo and R. Amal, J. Phys. Chem. C, 2010, 114, 2821 CrossRef CAS.
- E. K. Athanassiou, C. Mensing and W. J. Stark, Sens. Actuators, A, 2007, 138, 120 CrossRef.
- M. Kim and R. M. Laine, J. Am. Chem. Soc., 2009, 131, 9220 CrossRef CAS.
- X. Feng, D. C. Sayle, Z. L. Wang, M. S. Paras, B. Santora, A. C. Sutorik, T. X. T. Sayle, Y. Yang, Y. Ding, X. Wang and Y.-S. Her, Science, 2006, 312, 1504 CrossRef CAS.
- R. Strobel and S. E. Pratsinis, Platinum Met. Rev., 2009, 53, 11 CrossRef CAS.
- B. Schimmoeller, H. Schulz, A. Ritter, A. Reitzmann, B. Kraushaar-Czarnetzki, A. Baiker and S. E. Pratsinis, J. Catal., 2008, 256, 74 CrossRef CAS.
- R. Zhang, W. Y. Teoh, R. Amal, B. Chen and S. Kaliaguine, J. Catal. Search PubMed , in press.
- S. H. Ehrman, S. K. Friedlander and M. R. Zachariah, J. Mater. Res., 1999, 14, 4551 CrossRef CAS.
- H. J. Metz, R. Strobel, S. E. Pratsinis. European Patent, EP1829825A1, 2007.
- Y. K. Kho, W. Y. Teoh, L. Mädler, A. Iwase, A. Kudo, R. AmalProc. in International Conference on Photochemical Conversion and Storage of Solar Energy (IPS-17), Sydney, 27 July–1 August, 2008.
- H. F. Calcote, W. Felder, D. G. Keil, D. B. Olson23rd Symposium (International) on Combustion, p. 1739. The Combustion Institute, Pittsburgh, 1990 Search PubMed.
- D. G. Keil, H. F. Calcote and R. J. Gill, Mater. Res. Soc. Symp. Proc., 1996, 410, 167 CAS.
- C. Boyer, V. Bulmus, P. Priyanto, W. Y. Teoh, R. Amal and T. P. Davis, J. Mater. Chem., 2009, 19, 111 RSC.
- C. Boyer, P. Priyanto, T. P. Davis, D. Pissuwan, V. Bulmus, M. Kavallaris, W. Y. Teoh, R. Amal, M. Caroll, R. C. Woodward and T. St. Pierre, J. Mater. Chem., 2010, 20, 255 RSC.
- M. R. J. Carroll, R. C. Woodward, M. J. House, W. Y. Teoh, R. Amal, T. L. Hanley and T. G. St. Pierre, Nanotechnology, 2010, 21, 035103 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |