Preparation and applications of functional nanofibers based on the combination of electrospinning, controlled radical polymerization and ‘Click Chemistry’
Fang
Yao
,
Liqun
Xu
,
Baoping
Lin
and
Guo-Dong
Fu
*
School of Chemistry and Chemical Engineering, Southeast University, Nanjing, P. R. China 211189. E-mail: fu7352@seu.edu.cn; Tel: +86-25-52090625
First published on 1st June 2010
Abstract
This feature article provides an overview of the preparation of functional nanofibers by combined electrospinning, controlled radical polymerization and ‘Click Chemistry’. A combination of the powerful capability of controlled radical polymerization and ‘Click Chemistry’ for the synthesis of functional macromolecules and on surface modification as well as their wide applicability to electrospinning materials, functional nanofibers with a crosslinked structure, core–shell structures, and switchable surface properties etc. were prepared. In addition, the applications of the functional nanofibers in antibacterial fields and controlled release are also explored.
 Fang Yao | Fang Yao, received her joint Master’s degree in industrial chemistry from Technische Universität München (TUM) and National University of Singapore (NUS) in 2006. Now she is working for her PhD degree at Southeast University. Her current interest includes the preparation of dendrimer-like polymers and functional Interpenetrating Networks (IPN) by simultaneous ATRP and ‘Click chemistry’ reactions. |
 Liqun Xu | Liqun Xu obtained his Bachelor’s degree in chemical engineering from Southeast University in 2008. Now he studies for his PhD degree at the National University of Singapore (NUS). His research interests involve material surface modification and preparing polymeric brush by simultaneous ATRP and ‘Click Chemistry’ reactions. |
 Guo-Dong Fu | Guodong Fu received his BSc and MSc degrees in polymer science from Beijing University of Chemical Technology in 1996 and 1999, respectively. In 2005, he received a PhD degree in polymer science from the National University of Singapore. From 2009 to 2010, he worked at Bayreuth University with Prof. Mukundan Thelakkat. Currently he is a professor in the school of chemistry and chemical engineering, Southeast University. His research centers on the preparation of nanostructured, smart and crosslinked polymeric materials by living radical polymerization, ‘Click Chemistry’ and electrospinning, as well as exploring their new functions and applications. |
1. Introduction
Nanofibers are expected to play an important role as both interconnecting wires and functional units in fabricating electronic, optoelectronic, electrochemical and electromechanical devices with nanoscale dimensions.1 The preparation of nanofibers, especially polymeric nanofibers, has attracted much attention for their diverse applications, including energy conversion, energy storage, filtration, adsorption of heavy metal ion or organic compounds and environmental detection.2,3 The high surface-to-volume ratio, desired properties as well as the tunable functionality of polymeric nanofibers extend their applications in biomedical fields,4 such as tissue engineering,5,6 wound dressing,7 enzyme immobilization8 and drug delivery.9 Polymeric nanofibers were prepared by various methods, such as template-directed techniques,10 vapor-phase approaches,11 solution–liquid–solid methods12 and self-assembly.13 Electrospinning has attracted much attention both of research laboratories and industry, because: (1) electrospinning can fabricate continuous fibers with diameters down to a few nanometres; (2) electrospinning is applicable to a wide range of materials such as synthetic and natural polymers, metals as well as ceramics and composite systems; (3) and electrospinning can prepare nanofibers with low cost and high yield.14–16Recent development in polymer synthesis techniques such as controlled radical polymerization (CRP) and ‘Click Chemistry’ provides a feasible approach to prepare well-defined macromolecules with various functionalities. CRP, especially atom transfer radical polymerization (ATRP) and reversible addition–fragmentation chain transfer (RAFT) polymerization, has been demonstrated a convincing tool for synthesizing macromolecules with a well-defined molecular weight and desired architecture.17–25 ‘Click Chemistry’, such as copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), can also prepare various complex macromolecular architectures due to the complete specificity, quantitative yields and high functional group tolerance.26–30
However, at present, most polymeric nanofibers from electrospinning were fabricated using commercial polymers. Thus, the functionality and applications of nanofibers were largely restricted due to the lack of functionality of the original polymers. The combination of macromolecular design and synthesis with CRP, ‘Click Chemistry’ and electrospinning provides a novel and applicable approach to the preparation of nanofibers with desired functionalities and applications. Here, we summarize our recent works on the preparation of nanofibers with desired nanostructures, various functionalities and tunable surface properties, based on a combined technology of electrospinning, CPR and ‘Click Chemistry’. The applications of these nanofibers, such as in antibacterial fields and controlled release are also explored.
2. Nanofibers from surface-initiated ATRP (SI-ATRP) and electrospinning
2.1 Core–sheath nanofibers
Recently, surface-functionalized polymeric nanofibers, especially those with core–sheath structures are of growing interest for their potential applications such as in tissue engineering,31,32 biosensor technology,33 drug delivery34 and fabrication of nanotubes.35 Core–sheath polymeric nanofibers have been prepared by template-directed approaches35,36 and coaxial electrospinning.37–42 However, core–sheath nanofibers with a sheath covalently attached are still preferred. Surface-initiated graft polymerization is a common approach to prepare core–shell structures,43,44 as well as to modify the surface properties of materials covalently without affecting the properties of matrices.45 However, general methods, including chemical treatment, plasma deposition, UV-induced surface graft polymerization, high energy irradiation, and ozone-induced surface graft polymerization, are restricted in applications of polymeric nanofibers because of the destruction of the nano- and chemical structures of the fibers during processing.46A very simple method to prepare nanofibers with surface-grafted polymer brushes, or core–sheath microfiber via combined ATRP and electrospinning has been developed recently.46 The procedure for preparing surface functionalized nanofibers is shown in Fig. 1. Initially, macromolecules of polystyrene (PS) with a molecular weight in the range of 6 × 103 g mol−1 to 3.3 × 104 g mol−1 were synthesized. Electrospinning of synthesized PS allows the preparation of nanofibers at 400–800 nm. The mechanism of atom transfer radical polymerization (ATRP) involves a rapid dynamic equilibrium between a minute amount of growing free radicals and a large amount of dormant species.18–20 Thus, the resulting polymers with ‘active’ or ‘living’ end groups can be used as macroinitiators to synthesize block copolymers. In order to study the effect of electrospinning on the dormant alkyl halide chain ends of polymers, α-methylbenzyl bromide (MBB) was used to initiate ATRP of styrene. Thus, there are no other functional groups except alkyl bromide in the polystyrene (PS) macromolecule.
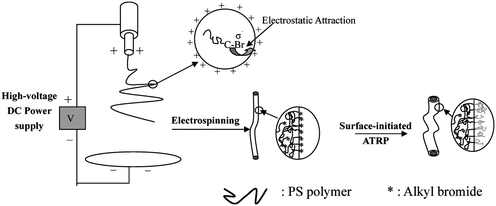 | ||
| Fig. 1 Schematic illustration of the preparation of core–shell nanofibers from combined electrospinning and surface-initiated atom transfer radical polymerization. The inset shows schematically migration of the alkyl bromide groups of PS to the liquid surface under the electrostatic attraction. Reprinted from ref. 46. | ||
The surface compositions of nanofibers were studied by X-ray photoelectron spectroscopy (XPS). Surface elemental stoichiometries were determined from the spectral area ratios, after correcting with the experimentally determined sensitivity factors, and were reliable to within ±10%. XPS results suggest that the dormant alkyl halide species at the PS chain ends is well preserved on the resulting fiber surfaces, when electrospinning of PS is carried out with the anode positioned at the spinneret (PSn-a). The XPS results also indicate that the surface Br concentration of PS nanofibers electrospun from cathode positioned at the spinneret (PSn-c) is much higher than that of the original PS. Also, the surface Br concentration of PSn-c nanofibers electrospun from PS polymer is about 0.56 atom%, which is much higher than that (0.28 atom%) of PSn-a nanofibers. Surface-initiated ATRP of acrylamide (AAM) and N-(hydroxymethyl)acrylamide (HMAM) from PS films, and PSn-c and PSn-a nanofibers were carried out in aqueous solutions at ambient temperature. Since PS is hydrophobic, the nanofibers will remain intact as a heterogeneous phase during ATRP in an aqueous medium. Most importantly, only those alkyl bromide groups on the fiber surface can initiate the ATRP of hydrophilic monomers. Since ATRP reactions were carried out under the same reaction condition, the lengths of PAAM and PHMAM brushes on the PSn-a-g-PAAM and PSn-c-g-PAAM nanofibers should be comparable. The XPS results reveal that the PAAM brushes on the PSn-c-g-PAAM nanofiber surface are denser than those on the PSn-a-g-PAAM nanofiber and PS film surfaces (Fig. 2). The [C]/[O] molar ratio of PSn-a-g-PAAM fibers derived from XPS result is about 10.3 which is comparable to the theoretical value (9.4) of the PS-g-PAAM film. The [C]/[O] molar ratio of PSn-c-g-PAAM fibers is about 3.2, which is close to the theoretical value (3.0) of PAAM. In Fig. 2(c), the substantial increase in the intensity of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species and the disappearance of π–π* shake-up satellite signal of the aromatic rings at BE of 291.4 eV, suggest that the surfaces of PSn-c-g-PAAM nanofibers are completely covered by the PAAM brushes to a thickness greater than the probing depth of the XPS techniques (∼8 nm in an organic matrix47). XPS results also indicate that a core–sheath structure of nanofibers must be formed.
O species and the disappearance of π–π* shake-up satellite signal of the aromatic rings at BE of 291.4 eV, suggest that the surfaces of PSn-c-g-PAAM nanofibers are completely covered by the PAAM brushes to a thickness greater than the probing depth of the XPS techniques (∼8 nm in an organic matrix47). XPS results also indicate that a core–sheath structure of nanofibers must be formed.
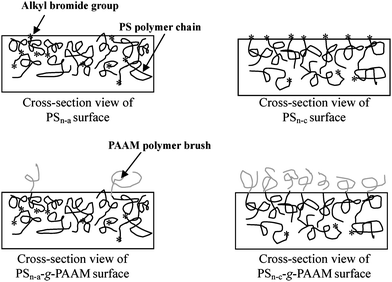 | ||
| Fig. 2 Schematic illustration of the difference in density of polymer brushes via surface-initiated ATRP from a PSn-a and PSn-c nanofiber. Reprinted from ref. 46. | ||
Due to applicability of ATRP to a wide selection of functional monomers, core–sheath fibers with diameter ranging from the nano-scale to the micro-scale and with various functionalities can be readily prepared. The present method also provides a possibility for preparing nanotubes by the preparation of core–sheath nanofibers and subsequent selective removal of the core. Arising from the narrow polydispersity index of the macromolecules from ATRP, the sheath thickness of the nanofibers or the wall thickness of the nanotubes could be defined and controlled by simply regulating the polymerization time or the molar ratio. The core–sheath nanofibers could be used as the precursor for the preparation of nanotubes.
2.2 Yardlong-bean-shaped nanofibers
Preparation of nanoparticle array in a controlled manner is always interesting in nanoscience and nanoengineering due to the wide applications of nano-arrays in biodiagnotics, nanomachine fabrication, electronics, optics and catalytic chemistry.48 Methods including topographical templates and biological macromolecules, surfacants, as well as block copolymer assisted self-assembly have been developed to prepare ordered 1-dimensional (1 D) or 2-dimensional (2 D) arrays of nanoparticles. An alternative approach to the preparation of crosslinked nanofibers by electrospinning-directed self-assembly of SiO2 nanoparticles with surface grafted poly(glycidyl methacrylate) (PGMA) brushes (core–shell SiO2-g-PGMA) has been developed.49The procedure of preparation of yardlong-bean-shaped crosslinked nanofibers (or nanofibers with a linear single-particle-array structure) is shown in Fig. 3. Initially, the initiator for atom-transfer radical polymerization (ATRP), trichloro(4-chloromethylphenyl)silane, was immobilized on the surface of SiO2 nanoparticles (SiO2-initiator) of about 25 nm in diameter. Subsequent surface-initiated ATRP of glycidyl methacrylate (GMA) gave rise to SiO2-g-PGMA nanoparticles of 60–100 nm in diameters (Fig. 4(a)). Electrospinning-directed self-assembly of SiO2-g-PGMA nanoparticles was carried out on a coaxial electrospinning setup with SiO2-g-PGMA nanoparticles solution in THF loaded into the core capillary and with 1,2-diaminohexane (DAH) solution in THF loaded into the sheath capillary. Nanofibers electrospun from a 20 wt% of SiO2-g-PGMA63 solution in THF have an average diameter of about 180 nm and are uniform. Fig. 4(c) shows the SEM image of nanofibers electrospun from SiO2-g-PGMA148 at a concentration of about 11 wt%. The nanofibers are well-defined and have a diameter of about 90 nm on average, which is comparable to the size of a SiO2-g-PGMA148 nanoparticle. The TEM image (inset of Fig. 4(c)) indicates that the nanofibers have a yardlong-bean-shaped nanostructure.
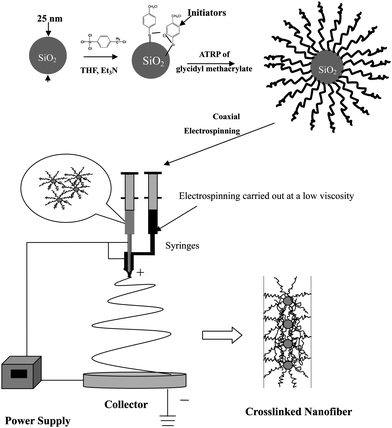 | ||
| Fig. 3 Schematic illustration of the preparation of a linear single nanoparticle array nanofiber via electrospinning directed self-assembly of core–shell nanoparticles and preparation of core–shell nanoparticles via surface-initiated ATRP. | ||
 | ||
| Fig. 4 (a) TEM image of core–shell SiO2-g-PGMA98 nanoparticles; (b) SEM image of nanofibers electrospun from a 20 wt% of SiO2-g-PGMA63 solution in THF, inset: TEM image of fiber; (c) SEM image of nanofibers electrospun from a 11 wt% of SiO2-g-PGMA148 solution in THF, inset: TEM image of fiber; (d) TEM image of nanofibers electrospun from a 20 wt% of SiO2-g-PGMA63 solution in THF after 10% of HF acid treating for 10 h. | ||
The TEM micrograph suggests that there is an array of multi-nanoparticles along the nanofibers (inset of Fig. 4(b)). TEM image also reveals that the SiO2 nanoparticles are dispersed in the nanofibers in a orderly manner. In Fig. 4(b) and 4(c), the appearance of some of the SiO2 core with sizes larger than 25 nm is attributable to the agglomeration of SiO2 nanoparticles in the process of surface-initiated ATRP. A crosslinked structure of SiO2-g-PGMA nanofibers was firstly ascertained by the well-preserved nanostructure of fibers after immersing in THF solvent for 1 h. Nanofibers with a linear single-particle-array structure were obtained via electrospinning-directed self-assembly of core–shell SiO2-g-PGMA nanoparticles. Removal of the silica cores by HF etching gave rise to crosslinked porous nanofibers with uniformly dispersed closed-cell structure.
Yardlong-bean-shaped nanofibers with different inorganic cores, such as magnetic Fe3O4, gold, CdSe and silver, and different functional polymer shells could be prepared due to the wide applicability of surface-initiated ATRP to various functional monomers and inorganic particle substrates, as well as the wide applicability of electrospinning to materials. These kind of nanofibers could find applications in biodiagnotics, nanomachine fabrication, electronics, optics and catalytic chemistry. Removal of the inorganic cores gave rise to nanofibers with uniformly dispersed closed pores, which could be good candidates for carriers in drug delivery.
3. Antibacterial polymeric nanofibers from CRP and electrospinning
Nanofibers with antibacterial activities are of global interest for their potential applications in various areas, particularly in medical devices, health care, hygienic applications, water-treatment, and food packaging and storage.50 The applications of antibacterial polymers have attracted much attentions because they minimize the environmental problems accompanying conventional disinfectants or antimicrobial agents with a low molecular weight. The conventional disinfectants or antimicrobial agents show toxicity after leaching into the environment.51 Solvent-resistant antibacterial nanofibers of self-quaternized block copolymers were prepared by combined ATRP and electrospinning.52A procedure for the preparation of antibacterial nanofibers is schematically shown in Fig. 5. Initially, the copolymers of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and glycidyl methacrylate (GMA) (P(DMAEMA-c-GMA)) were synthesized via ATRP and used as macro-initiators to prepare the block copolymer of P(DMAEMA-c-GMA) and pentachlorophenyl acrylate polymer (PPCPA) (P(DMAEMA-c-GMA)-b-PPCPA). GMA was introduced into the copolymer to prepare crosslinked nanofibers via the reaction between the epoxy groups and diamines. Electrospinnning of P(DMAEMA-c-GMA)-b-PPCPAP and subsequent treatment with diamine gave rise to crosslinked nanofibers with diameters in the range of 300 nm to 1.3 μm. For P(DMAEMA-c-GMA)-b-PPCPA, quaternary ammonium salts (QASs) were generated via N-alkylation of tertiary alkylamine groups of PDMAEMA blocks by the chloro-aromatic compounds of PPCPA block (self-quaternization of P(DMAEMA-c-GMA)-b-PPCPA). The formation of the QASs via self-quaterization of the P(DMAEMA-c-GMA)-b-PPCPA polymers is schematically shown in Fig. 6. Combination of the hydrophobic interaction of the PPCPA block and electrostatic interaction of quaternary ammonium salts generated from the N-alkylation between the chloro-aromatic compounds of the PPCPA and tertiary amine groups of DMAEMA, the cross-linked nanofibers exhibit good antibacterial activities to E. coli and S. aureus. The antimicrobial assays were first conducted with suspensions of Gram-positive bacteria (S. aureus), containing 1 × 105 cells ml−1. For cross-linked nanofibers electrospun from P(DMAEMA-c-GMA)-b-PPCPA polymer (Mn = 2.1 × 104 g mol−1, [GMA]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[DMAEMA]:[PPCPA] = 8:110:20, average diameter = 300 nm), the number decreases by 3 orders of magnitude in 30 min (Fig. 7(a)). Thus, an antibacterial efficiency of 99.9% was obtained. The antibacterial assays were also carried out with suspensions of Gram-negative bacteria, E. coli, containing 1 × 105 cells ml−1. All nanofibers from P(DMAEMA-c-GMA)-b-PPCPA polymer exhibit a higher antibacterial efficiency than that of PPCPA. The PPCPA polymers have a poor solubility in common solvents and are difficult to be prepared into nanofiber via electrospinning. Thus, PPCPA polymer powders were used for the antibacterial assays. For nanofibers from P(DMAEMA-c-GMA)-b-PPCPA (Mn = 2.1 × 104 g mol−1, [GMA]:[DMAEMA]:[PPCPA] = 8:110:20, average diameter = 300 nm), the viable cell number decreases by 2.5 orders of magnitude in 30 min (Fig. 7(b)). It is well-known that the cell surface of bacteria is negatively charged.53 When cells contact with P(DMAEMA-c-GMA)-b-PPCPA nanofibers having QASs on surface, the normal function of cell membrane was disrupted due to the electrostatic interaction.54 The increase in PPCPA content of nanofibers can improve the antibacterial efficiency. However, the content of PPCPA is not more than 50 mol% due to the poor solubility and the difficulty in electrospinning of the resulting copolymers. The permanence of the antibacterial activity of P(DMAEMA-c-GMA)-b-PPCPA nanofibers were demonstrated by five batches of antibacterial assays.
:[DMAEMA]:[PPCPA] = 8:110:20, average diameter = 300 nm), the number decreases by 3 orders of magnitude in 30 min (Fig. 7(a)). Thus, an antibacterial efficiency of 99.9% was obtained. The antibacterial assays were also carried out with suspensions of Gram-negative bacteria, E. coli, containing 1 × 105 cells ml−1. All nanofibers from P(DMAEMA-c-GMA)-b-PPCPA polymer exhibit a higher antibacterial efficiency than that of PPCPA. The PPCPA polymers have a poor solubility in common solvents and are difficult to be prepared into nanofiber via electrospinning. Thus, PPCPA polymer powders were used for the antibacterial assays. For nanofibers from P(DMAEMA-c-GMA)-b-PPCPA (Mn = 2.1 × 104 g mol−1, [GMA]:[DMAEMA]:[PPCPA] = 8:110:20, average diameter = 300 nm), the viable cell number decreases by 2.5 orders of magnitude in 30 min (Fig. 7(b)). It is well-known that the cell surface of bacteria is negatively charged.53 When cells contact with P(DMAEMA-c-GMA)-b-PPCPA nanofibers having QASs on surface, the normal function of cell membrane was disrupted due to the electrostatic interaction.54 The increase in PPCPA content of nanofibers can improve the antibacterial efficiency. However, the content of PPCPA is not more than 50 mol% due to the poor solubility and the difficulty in electrospinning of the resulting copolymers. The permanence of the antibacterial activity of P(DMAEMA-c-GMA)-b-PPCPA nanofibers were demonstrated by five batches of antibacterial assays.
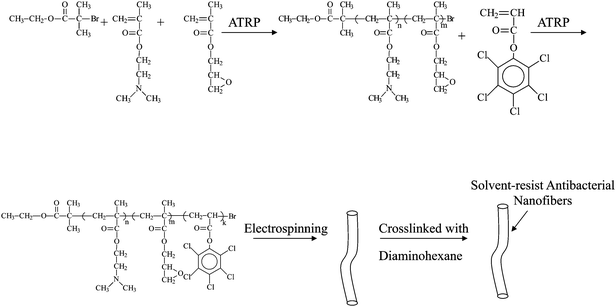 | ||
| Fig. 5 Scheme illustration of the preparation of P(DMAEMA-c-GMA)-b-PPCPA nanofibers via ATRP and electrospinning. Reprinted from ref. 52. | ||
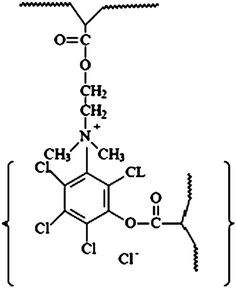 | ||
| Fig. 6 Scheme illustration of the self-quaternized P(DMAEMA-c-GMA)-b-PPCPA. Reprinted from ref. 52. | ||
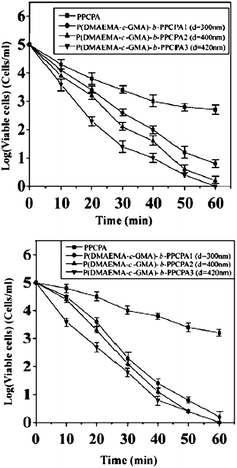 | ||
| Fig. 7 Antibacterial efficacy of 50 mg of PPCPA polymers and self-quaternized nanofibers electrospun from P(DMAEMA-c-GMA)-b-PPCPA copolymers in contact with (a) 50 ml of S. aureus suspension (105 CFU ml−1) and (b) 50 ml of of E. coli suspension (105 CFU ml−1). Reprinted from ref. 52. | ||
Antibacterial nanofibers of self-quaternized block copolymers of 4-vinyl pyridine and pentachlorophenyl acrylate were also prepared by RAFT and electrospinning.55 Well-defined antibacterial block copolymers of 4-vinyl pyridine (4VP) and pentachlorophenyl acrylate (PCPA) (P(4VP-b-PCPA)) were prepared via RAFT (Fig. 8). Electrospinning of the P(4VP-b-PCPA) from a solution in mixed THF and DMF gave rise to fibers with diameters in the range of 0.5–4.0 μm. The QASs were generated by N-alkylation of pyridine groups of P4VP block and chloro-aromatic compounds of PPCPA block (or self-quaternization of P(4VP-b-PCPA)). The self-quaternization of P(4VP-b-PCPA) nanofibers was studied by XPS. Attributable to the hydrophobicity of the PPCPA blocks and the electrostatic interaction of QASs generated from the self-quaternization of P(4VP-b-PCPA), the resulting nanofibers exhibit a high antibacterial efficiency. The antibacterial effect of the P(4VP-b-PCPA) nanofibers was assayed using Escherichia coli and Staphylococcus aureus cultures. 99.6% of E. coli and 99.1% S. aureus were killed after being contacted with 50 mg nanofibers in 10 min. The permanence of antibacterial activity of the self-quaternized P(4VP-b-PCPA) nanofibers was also demonstrated in repeat applications. In comparison to the other QASs containing polymers, which were prepared by N-alkylation of the tertiary amine groups by alkyl halides, the QASs of the nanofiber are formed from the self-quaternization of P(DMAEMA-c-GMA)-b-PPCPA. Thus, the loss of the antibacterial activity of the nanofibers due to slowly leaching of the small molecules is largely prevented. In summer, the combination of design and synthesis of functional macromolecules via CRP and electrospinning is a simple and feasible approach to the preparation of nanofibers with desired functionalities and applications.
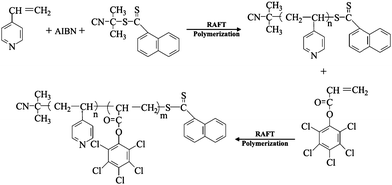 | ||
| Fig. 8 Scheme illustration of the preparation of the P4VP-b-PPCPA via RAFT polymerization. Reprinted from ref. 55. | ||
4. Smart nanofibers from the combination of CRP, ‘Click Chemistry’ and electrospinning
4.1. Smart nanofibers with thermal sensitive surface
Nanofibers with environmentally-responsive (or smart) surfaces and crosslinked structures (or solvent-resistant) are especially interesting for applications to sensors and bioengineering.32,33 Poly(N-isopropyl acrylamide) (PNIPAM) is one of the most studied synthetic responsive polymers, which undergoes a transition from a hydrophilic state to a hydrophobic state in water at 32 °C.56 A simple method for preparing solvent-resistant nanofibers with a thermally sensitive surface has been developed by the combined technology of RAFT, ATRP, electrospinning, and ‘Click Chemistry’.57 The experimental procedure is shown in Fig. 9. Initially, PVBC was synthesized in a controlled manner via RAFT polymerization using 2-cyanoprop-2-yl 1-dithionaphthalate (CPDN) as the chain transfer agents. Another step of RAFT polymerization of GMA using the prepared PVBC as the macro-CTAs allows the synthesis of PVBC-b-PGMA block copolymers. Well-defined PVBC50-b-PGMA15 (PVBC-b-PGMA copolymers with, on average, 50 VBC repeat units and 15 GMA repeat units), PVBC50-b-PGMA42, PVBC74-b-PGMA32 and PVBC74-b-PGMA46 were obtained by regulating the polymerization times. Electrospinning of PVBC-b-PGMA polymers gave rise to nanofibers with a diameter in the range of 400 nm–1.5 μm (Fig. 10). It is interesting to find that a crosslinked gel was obtained when PVBC-b-PGMA reacted with sodium azide. PVBC-b-PGMA-N3 nanofibers were obtained when PVBC-b-PGMA nanofibers were exposed to NaN3 solution in mixed DMF and water. Most importantly, the resulting nanofibers have a crosslinked structure. Thermally responsive polymers of alkyne-terminated PNIPAM (PNIPAMAT) were synthesized via ATRP of NIPAM. Cross-linked PVBC-b-PGMA nanofibers with thermal sensitive PNIPAM brushes on surface (PVBC-b-PGMA-g-PNIPAM) were obtained by ‘Click Chemistry’ between PVBC-b-PGMA-N3 nanofibers and PNIPAMAT polymers.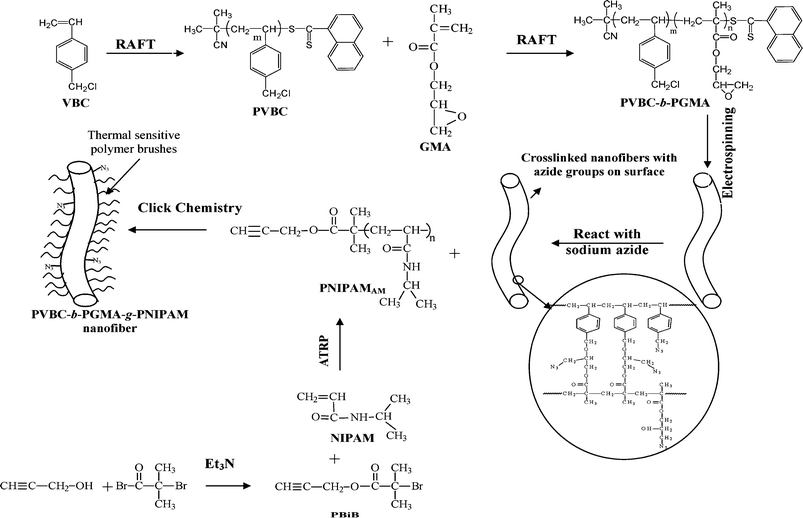 | ||
| Fig. 9 Preparation of solvent resistant nanofibers with a thermally sensitive surface by combined atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT) polymerization, electrospinning and ‘Click Chemistry’. (VBC = 4-vinylbenzyl chloride; PVBC = poly(4-vinylbenzyl chloride); GMA = glycidyl methacrylate; PVBC-b-PGMA = poly(4-vinylbenzyl chloride)-b-poly(glycidyl methacrylate); PNIPAMAM = alkyne-terminated PNIPAM; PBiB = propargyl-2-bromoisobutyrate). Reprinted from ref. 57. | ||
 | ||
| Fig. 10 Scanning electron microscope (SEM) surface images of the (a) nanofibers electrospun from PVBC74-b-PGMA46 (having 74 VBC repeat units and 46 GMA repeat units in each macromolecule) at a concentration of about 20 wt% in THF and (b) prepared nanofibers after reacting with sodium azide in mixed DMF–H2O solution (1:1 in volume ratio) and immersion in THF for 2 h. Reprinted from ref. 57. | ||
To explore the thermal sensitivity behavior of a surface, water contact angles were measured at different temperatures. Fig. 11 (a) shows the photographs of the water-drop profiles on nanofibers of PVBC74-b-PGMA46-g-PNIPAM73 at 20 °C and 45 °C, respectively. PVBC74-b-PGMA46-g-PNIPAM73 nanofibers exhibited hydrophobic surface properties at 45 °C, having a water contact angle about 140°. When the temperature was cooled down to 20 °C, the nanofibers changed to a hydrophilic surface, and the water contact angle decreased to less than 30°. The permanence of the thermally sensitive surface of PVBC-b-PGMA-g-PNIPAM nanofibers was also demonstrated by several repeated temperature-changing cycles from 20 °C to 45 °C. A switch of character from hydrophilic to hydrophobic indicates a good reversibility of the surface wettability of PVBC-b-PGMA-g-PNIPAM nanofibers (Fig. 11(b)). The prepared crosslinked nanofibers with smart surfaces can be potentially used as biosensors, in scaffold-supported cell therapies for tissue engineering and as carrying materials for drug and gene delivery.
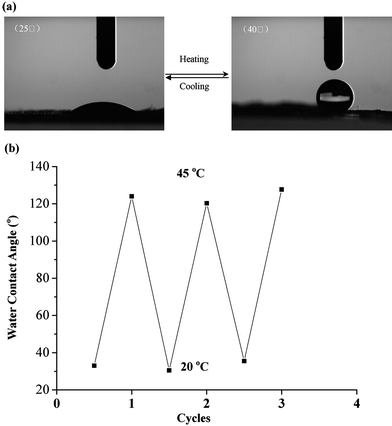 | ||
| Fig. 11 (a) Photographs of a water-droplet on the surface of nanofibers of PVBC74-b-PGMA46-g-PNIPAM73, which shows the thermally responsive surface of the nanofibers that can switch between hydrophilicity at low temperature (20 °C) and hydrophobicity at high temperature (45 °C). (b) Reversibility of water contact angle transition on the nanofibers of PVBC74-b-PGMA46-g-PNIPAM73 at 20 °C and 45 °C, respectively. Half cycles: 20 °C; and integral cycles: 45 °C. Reprinted from ref. 57. | ||
4.2 Smart nanofibers with a photo-responsive surface for controlled release
Stimuli-responsive or ‘smart’ materials have attracted much attention for controlled release under specific conditions. The photo-responsive delivery systems have been of special interest to scientists because of their unique advantage of quick response without the addition of any chemical stimulants or the production of chemical waste in the photo-induced release process.58 Combination of the mechanisms of the host–guest interaction and drug conjugation with cyclodextrins (CDs), a smart delivery system based on crosslinked nanofibers with surface-loaded CD prodrugs has been developed59 (Fig. 12). Initially, crosslinked nanofibers with azido groups on the surface (CNFPVBC-b-PGMA-N3) were prepared via electrospinning of the block copolymer of vinylbenzyl chloride (VBC) and glycidyl methacrylate (GMA) (PVBC-b-PGMA) synthesized by consecutive reversible addition–fragmentation chain transfer (RAFT) polymerization and subsequent reaction with sodium azide. Then, ‘Click Chemistry’ between 4-propargyloxyazobenzene and CNFPVBC-b-PGMA-N3 allows the preparation of nanofibers with photo-sensitive azobenzene (AB) groups on the surface (CNFPVBC-b-PGMA-AB).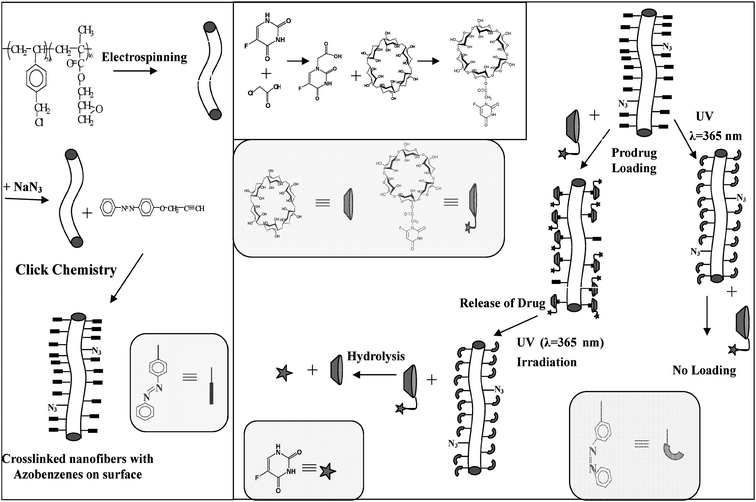 | ||
| Fig. 12 Schematic illustration of the preparation of the crosslinked nanofibers of poly(glycidyl methacrylate)-block-poly(vinylbenzyl chloride) (PVBC-b-PGMA) with azobenzene groups on the surface (CNFPVBC-b-PGMA-AB), the synthesis of cyclodextrin and 5-fluorouracil prodrug (α-CD-5FU) and the photo-responsive loading and release of α-CD-5FU prodrug on the CNFPVBC-b-PGMA-AB surface by host–guest interaction. Reprinted from ref. 59. | ||
Cyclodextrins (CDs) and azobenzene (AB) molecules are a photo-responsive host–guest pair.58 Driven by hydrophobic and van der Waals interactions, trans-AB is well-recognized by α-cyclodextrin (α-CD). The host–guest interaction is hindered when AB transforms from the trans to cis configuration under UV-irradiation. The 5-fluorouracial (5FU) and α-CD conjugate (α-CD-5FU), a prodrug for colon, has been synthesized. Initially, the loading of the α-CD-5FU prodrug on the photosensitive CNFPVBC-b-PGMA-AB was carried out by immersion of the nanofibers in a 1 mmol L−1 aqueous solution of α-CD-5FU for 12 h in the dark. The successful loading of α-CD-5FU prodrug on the nanofiber surface was revealed by XPS analysis. Photo-controlled release of the α-CD-5FU prodrug from the CNFPVBC-b-PGMA-AB surface was conducted by immersing 10 mg of the α-CD-5FU loaded nanofibers in 20 mL of water, followed by UV irradiation at the wavelength of 365 nm. Fig. 13 shows the release profiles of α-CD-5FU from the CNFPVBC-b-PGMA-AB surface as a function of UV irradiation time. In the dark, there was almost no release of α-CD-5FU from the α-CD-5FU prodrug-loaded CNFPVBC-b-PGMA-AB after 1 h of immersion in water (curve a in Fig. 13), suggesting that dissociation of the host–guest interaction between α-CD-5FU and CNFPVBC-b-PGMA-AB did not occur in the absence of UV irradiation. When exposed to UV irradiation, α-CD-5FU was released quickly into the solution, as the azobenzene transformed from the trans to cis configuration. An α-CD-5FU solution concentration of 0.5 μg mL−1 was reached after 5 min of UV irradiation (Curve b in Fig. 13). The quick response and controllable release of α-CD-5FU prodrug in this delivery system are revealed by the multi-step ‘ON–OFF’ release profile under UV irradiation (Curve c in Fig. 13). The release profile shows that there is no release of α-CD-5FU from the nanofiber surface during the initial period (10 min) in the dark. The concentration of α-CD-5FU in solution increases gradually in the next 10 min upon UV exposure. The concentration of α-CD-5FU ceases to increase upon removal of the UV irradiation at t = 20 min. The concentration of α-CD-5FU remains almost constant in the next 20 min, in the absence of UV irradiation. When UV irradiation is resumed at t = 40 min, the concentration of α-CD-5FU in solution increases again.
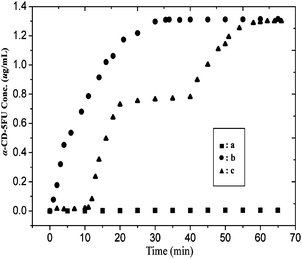 | ||
| Fig. 13 Time release profile of the α-CD-5FU prodrug in 20 mL of water from 10 mg of CNFPVBC-b-PGMA-AB with surface-loaded α-CD-5FU. Curve a is the release profile in the dark. Curve b is the release profile upon continuous exposure to 365 nm UV irradiation, and curve c is the release profile upon intermittent exposure to the 365 nm UV irradiation in the time interval of 10–20 min and 40–70 min. Reprinted from ref. 59. | ||
A novel, photo-responsive, controlled-release system, in which the α-CD-5FU prodrugs are loaded on the photo-responsive and crosslinked nanofiber surface via host–guest interaction has been developed. Differing from the other controlled release systems, in which the drug is loaded inside the carrier and is susceptible to release delays in response to external stimuli, the present system provides a photo-controlled fast ‘ON–OFF’ release characteristic. With the availability of a wide variety of CD prodrugs, stimuli-responsive nanofiber systems with loaded CD prodrugs are expected to provide unique opportunities for the effective and controlled ‘ON–OFF’ release of therapeutic agents.
Conclusion
This feature article has presented the recent development of the preparation of functional polymeric nanofibers based on macromolecular design and synthesis, utilizing the controlled radical polymerization and ‘Click Chemistry’, and electrospinning. Macromolecules with desired functionality and architecture can be synthesized by controlled radical polymerization and ‘Click Chemistry’, attributable to their powerful capability for the design and synthesis of functional macromolecules. Electrospinning of the designed macromolecules allows the preparation of functional nanofibers with extended functionalities, large-scale, low cost and high production rate. It was demonstrated that nanofibers with desired nanostructures and functionalities, such as core–shell structure, high antibacterial capability and switchable surface properties, could be prepared by a combined technology of electrospinning, controlled radical polymerization and ‘Click Chemistry’. The application of prepared functional nanofibers in the antibacterial field, controlled release etc. have been shown. In summary, the combination of controlled radical polymerization, ‘Click Chemistry’ and electrospinning provide a feasible approach to the preparation of nanofibers with desired functionalities and nanostructures.Acknowledgements
This work was supported by the National Natural Science Foundation of China project grant 20804009 and the Key Project of Chinese Ministry of Education grant 108062. This work was also funded by the Program for New Century Excellent Talents of the University grant NCET-08-0117, the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry, Southeast University foundations grants 4007041012 and 4007041023.References
- Y. N. Xia, P. D. Yang, Y. G. Sun, Y. Y. Wu, B. Mayers, B. Gates, Y. D. Yin, F. Kim and Y. Q. Yan, Adv. Mater., 2003, 15(5), 353–389 CrossRef CAS.
- V. Thavasi, G. Singh and R. Ramakrishna, Energy Environ. Sci., 2008, 1, 205–221 RSC.
- B. Ding, M. Wang, J. Yu and G. Sun, Sensors, 2009, 9, 1609–1624 CrossRef CAS.
- D. Liang, B. S. Hsiao and B. Chu, Adv. Drug Delivery Rev., 2007, 59, 1392–1412 CrossRef CAS.
- S. H. Lim and H. Q. Mao, Adv. Drug Delivery Rev., 2009, 61, 1084–1096 CrossRef CAS.
- B. M. Baker, A. M. Handorf, L. C. Ionscu, W. J. Li and R. L. Mauck, Expert Rev. Med. Devices, 2009, 6(5), 515–532 Search PubMed.
- K. S. Rho, L. Jeong and G. Lee, Biomaterials, 2006, 27, 1452–1461 CrossRef CAS.
- J. P. Chen, K. H. Ho, Y. P. Chiang and K. W. Wu, J. Membr. Sci., 2009, 340(1–2), 9–15 CrossRef CAS.
- H. Yoon, Y. Park and G. Kim, Polym. Korea, 2009, 33(3), 219–223 Search PubMed.
- B. B. Lakshmi, P. K. Dorhout and C. R. Martin, Chem. Mater., 1997, 9, 857–862 CrossRef CAS.
- M. Law, J. Goldberger and P. Yang, Annu. Rev. Mater. Res., 2004, 34, 83–122 CrossRef CAS.
- M. S. Gudiksen and C. M. Lieber, J. Am. Chem. Soc., 2000, 122, 8801–8802 CrossRef CAS.
- X. Peng, U. Manna, W. Yang, J. Wickham, E. Scher, A. Kadavanich and A. P. Allvisatos, Nature, 2000, 404, 59–61 CrossRef CAS.
- A. Greiner and J. H. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS.
- G. C. Rutledge and S. V. Fridrikh, Adv. Drug Delivery Rev., 2007, 59(14), 1384–1391 CrossRef CAS.
- S. Ramakrishna, K. Fujihara, W. E. Teo, T. Yong, Z. Ma and R. Ramaseshan, Mater. Today, 2006, 9(3), 40–50 CrossRef CAS.
- J. A. Johnson, M. G. Finn, J. T. Koberstein and N. J. Turro, Macromol. Rapid Commun., 2008, 29, 1052–1072 CrossRef CAS.
- K. Matyjaszewski and J. H. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS.
- T. E. Patten and K. Matyjaszewski, Adv. Mater., 1998, 10, 901–915 CrossRef CAS.
- V. Coessens, T. Pintauer and K. Matyjaszewski, Prog. Polym. Sci., 2001, 26, 337–377 CrossRef CAS.
- B. Lowe and C. L. McCormick, Prog. Polym. Sci., 2007, 32(3), 283–351 CrossRef CAS.
- T. P. T. Le, G. Moad, E. Rizzardo and S. H. Thang, WO Patent 9801478, 1998.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- L. Borner, T. P. Davis, M. H. Stenzel and C. Barner-Kowollik, Macromol. Rapid Commun., 2007, 28, 539–559 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2005, 58, 379–410 CrossRef CAS.
- D. Fournier, R. Hoogenboom and U. S. Schubert, Chem. Soc. Rev., 2007, 36, 1369–1380 RSC.
- P. A. Wu, K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. J. Pyun, M.J. Fre'chet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928–3934 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40(11), 2004 CrossRef CAS.
- W. H. Binder and R. Sachsenhof, Macromol. Rapid Commun., 2007, 28, 15–54 CrossRef CAS.
- J. Lutze, Angew. Chem., Int. Ed., 2007, 46, 1018–1025 CrossRef CAS.
- R. Langer and J. P. Vacanti, Science, 1993, 260, 920–926 CrossRef CAS.
- C. J. Koh and A. Atala, J. Am. Soc. Nephrol., 2004, 15, 1113–1125 CrossRef.
- Y. J. Chang, Y. C. Chen, H. L. Kuo and P. K. Wei, J. Biomed. Opt., 2006, 11(1), 014032 CrossRef.
- K. S. Scoppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS.
- H. Hou, Z. Jun, A. Reuning, A. Schaper, J. H. Wendorff and A. Greiner, Macromolecules, 2002, 35, 2429–2431 CrossRef CAS.
- R. A. Crauso, J. H. Schattka and A. Greiner, Adv. Mater., 2001, 13, 1577 CrossRef CAS.
- V. Kalra, S. Mendez, J. H. Lee, H. Nguyen, M. Marquez and Y. L. Joo, Adv. Mater., 2006, 18, 3299–3303 CrossRef CAS.
- Z. Sun, E. Zussman, A. L. Yarin, J. H. Wendorff and A. Greiner, Adv. Mater., 2003, 15, 1929–1932 CrossRef CAS.
- G. Larsen, R. Velarde-Ortiz, K. Minchow, A. Barrero and I. G. Loscertales, J. Am. Chem. Soc., 2003, 125, 1154–1155 CrossRef CAS.
- D. Li and Y. Xia, Nano Lett., 2004, 4, 933–938 CrossRef CAS.
- M. Ma, V. Krikorian, J. H. Yu, E. L. Thomas and G. C. Rutledge, Nano Lett., 2006, 6, 2969–2972 CrossRef CAS.
- X. Y. Sun, R. Shankar, H. G. Borner, T. K. Chosh and R. Spontak, Adv. Mater., 2007, 19, 87–91 CrossRef CAS.
- M. X. Gu, T. C. A. Yeung and C. M. Tan, J. Appl. Phys., 2006, 100(9), 094304 CrossRef.
- Z. W. Ma, M. Kotaki, R. Inai and S. Ramakrishna, Tissue Eng., 2005, 11(1–2), 101–109 CrossRef CAS.
- K. Kato, E. Uchida, E. T. Kang, Y. Uyama and Y. Ikada, Prog. Polym. Sci., 2003, 28, 209–214 CrossRef CAS.
- G. D. Fu, J. Y. Lei, C. Yao, X. S. Li, F. Yao, S. Z. Nie, E. T. Kang and K. G. Neoh, Macromolecules, 2008, 41(18), 6854–6858 CrossRef CAS.
- G. D. Fu, J. P. Zhao, Y. M. Sun, E. T. Kang and K. G. Noeh, Macromolecules, 2007, 40, 2271–2275 CrossRef CAS.
- S. I. Lim and C. Z. Zhong, Acc. Chem. Res., 2009, 42(6), 798–808 CrossRef CAS.
- H. S. Wang and G. D. Fu, unpublished work.
- C. Burger, B. S. Hsiao and B. Chu, Annu. Rev. Mater. Res., 2006, 36, 333–368 CrossRef CAS.
- K. de Arruda Alerda, A. A. A. de Queiroz, O. Z. Higa, G. A. Abraham and J. S. Roman, J. Biomed. Mater. Res., 2004, 68a, 473–478 Search PubMed.
- G. D. Fu, F. Yao, Z. G. Li and X. S. Li, J. Mater. Chem., 2008, 18, 859–867 RSC.
- T. Tashiro, Macromol. Mater. Eng., 2001, 286, 63–87 CrossRef CAS.
- A. M. Klibanov, J. Mater. Chem., 2007, 17, 2479–2482 RSC.
- L. Q. Xu, F. Yao, S. Yin, G. D. Fu, L. Shen, S. Z. Nie and M. F. Zhu, High Perfm. Polym. DOI:10.1177/0954008309104776.
- E. S. Gil and S. M. Hudson, Prog. Polym. Sci., 2004, 29, 1173–222 CrossRef CAS.
- G. D. Fu, L. Q. Xu, F. Yao, K. Zhang, X. F. Wang, M. F. Zhu and S. Z. Nie, ACS Appl. Mater. Interfaces, 2009, 1(2), 239–243 Search PubMed.
- S. Yagai and A. Kitamura, Chem. Soc. Rev., 2008, 37, 1520–1529 RSC.
- G. D. Fu, L. Q. Xu, F. Yao, G. L. Li and E. T. Kang, ACS Appl. Mater. Interfaces, 2009, 1(11), 2424–2427 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |