Decorating graphene oxide with CuO nanoparticles in a water–isopropanol system
Junwu
Zhu
*a,
Guiyu
Zeng
b,
Fude
Nie
b,
Xiaoming
Xu
a,
Sheng
Chen
a,
Qiaofeng
Han
a and
Xin
Wang
*a
aKey Laboratory of Soft Chemistry and Functional Materials(Nanjing University of Science and Technology), Ministry of Education, Nanjing, 210094, China. E-mail: wxin@public1.ptt.js.cn; zhujw@mail.njust.edu.cn
bInstitute of Chemical Materials, China Academy of Engineering Physics, Mianyang, 621900, China
First published on 27th April 2010
Abstract
A facile chemical procedure capable of aligning CuO nanoparticles on graphene oxide (GO) in a water–isopropanol system has been described. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) observations indicate that the exfoliated GO sheets are decorated randomly by spindly or spherical CuO nanoparticle aggregates, forming well-ordered CuO:GO nanocomposites. A formation mechanism of these interesting nanocomposites is proposed as intercalation and adsorption of Cu2+ ions onto the GO sheets, followed by the nucleation and growth of the CuO crystallites, which in return resulted in the exfoliation of GO sheets. Moreover, the obtained nanocomposites exhibit a high catalytic activity for the thermal decomposition of ammonium perchlorate (AP), due to the concerted effect of CuO and GO.
Introduction
Graphene, monolayers of carbon atoms arranged in a honeycomb lattice, has attracted enormous attention in recent years from both the experimental and theoretical scientific communities.1 Graphene oxide (GO) is one of the most important derivatives of graphene, it is a layered material bearing oxygen functional groups on its basal planes and edges.2 Because of the special surface properties and layered structure, it may provide potential nanoscale building blocks for new materials.3It is well known that carbon nanotubes, owning to their unique structural, optoelectronic properties and ability to improve catalytic properties, have drawn special attention over the past few decades.4,5 Of particular interest is the dispersion of a catalytic-active or redox-active transition metal oxide throughout a high surface area carbon support, which shows enhanced catalytic performance and electrochemical capacitance.5–7 Since graphene may be considered as unrolled carbon nanotubes, it can also provide a higher specific area comparable to carbon nanotubes and has become the focus of considerable interest as a consequence of wide potential applications in the fields of transistors, microwave switches, optoelectronic devices, nanoelectronic and nanoelectromechanical devices.8–12 Meanwhile, GO is receiving increased attention due to its special surface properties, large specific surface area and layered structure for synthesis of GO-containing nanocomposites. The organization of functional semi-conductor nanoparticles (NPs) on GO attracts substantial research efforts motivated by the desire to combine the properties of two functional nanoscale materials to achieve a wider range of applications.7,13–15
CuO has received considerable attention in recent years due to its potential applications in many important fields such as gas sensors, magnetic phase transitions, catalysts and superconductors.16–19 The fabrication of CuO nanocrystals has been most extensively studied in the past two decades, and their morphology as well as size can be well controlled by adjusting the reaction parameters.20–24
GO can be exfoliated to form quasi-two-dimensional carbon nanosheets. These exfoliated GO sheets possess large surface area, and hydroxyl, carboxyl and epoxy groups are introduced on carbon nanosheets due to oxidation procedures,25–27 which can be used as nucleation centers to form NP:GO nanocomposites. Therefore, it is feasible to obtain CuO:GO nanocomposites. The new hybrid material might possess unusual properties, as compared with their individual counterparts, and present some special features, especially in catalysis.
NH4CIO4 (AP) is the most common oxidizer in composite solid propellants, and its thermal decomposition performance has a great influence on the combustion behavior of propellants. The catalytic activities of several metal oxide nanoparticles in the thermal decomposition of AP have been reported.28–31
Herein, we describe a facile procedure in a water–isopropanol reaction system, which is capable of in situ depositing CuO nanoparticles on GO sheets without the need for any chemical additives. Meanwhile, the catalysis of these nanocomposites for the decomposition of AP was also investigated. To the best of our knowledge, little work on synthesis of CuO, especially CuO:GO nanocomposites in water–isopropanol reaction systems, has been reported. Considering the polarity, steric hindrance and electrostatic effect etc., the different coordination capabilities between water species and isopropanol might affect the morphology and texture of as-obtained products. Therefore, the introduction of a double solvent system would provide a wider space for us to manipulate the properties of the products. Furthermore, this synthetic method can, in principle, be tuned to produce modulations in compositions, phases, and microstructures that are designed to enhance material performance in catalytic applications.
Experimental
Preparation
All chemical reagents were analytical grade and used as received without further purification. In a typical procedure, GO was synthesized from natural graphite (400 mesh, Qingdao Zhongtian Company) using a modified Hummers method.32,33 After being purified by washing and centrifugation, the obtained GO was dried at 120 °C to eliminate the water contained in the product. Nanocomposites with different mass ratios of CuO:GO (0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:1, 2:1 and 3:1) were synthesized. Herein, the mass of CuO is determined by Cu(OAc)2 according to the hydrolysis reaction. Considering the yield percentage of CuO in the actual process, the mass of Cu(OAc)2·H2O employed is a little excessive. The typical route, for example, when the mass ratio of CuO:GO is 0.5:1, is as follows: the 0.05 g of dried GO was suspended in 50 mL of isopropanol to give a brown dispersion by sonicating for ∼30 min. The resulting homogeneous dispersion was mixed with 0.07 g of Cu(OAc)2·H2O in a round-bottomed flask equipped with a refluxing device. The mixture was heated to approximately 83 °C with vigorous stirring, and maintained at that temperature for 30 min. Then 5 mL of deionized (DI) water was rapidly introduced into the above boiling solution, and the mixture was heated at 83 °C for another 30 min. During this process, the initial brown color of the dispersion gradually turned into black. After cooling to room temperature, the as-synthesized composite was centrifuged, washed with ethanol several times, and dried at 70 °C in air overnight. The nanocomposites with different mass ratios of CuO:GO, labeled as CuO:GOratio (e.g. CuO:GO0.5, CuO:GO1, CuO:GO2 and CuO:GO3).
:1, 1:1, 2:1 and 3:1) were synthesized. Herein, the mass of CuO is determined by Cu(OAc)2 according to the hydrolysis reaction. Considering the yield percentage of CuO in the actual process, the mass of Cu(OAc)2·H2O employed is a little excessive. The typical route, for example, when the mass ratio of CuO:GO is 0.5:1, is as follows: the 0.05 g of dried GO was suspended in 50 mL of isopropanol to give a brown dispersion by sonicating for ∼30 min. The resulting homogeneous dispersion was mixed with 0.07 g of Cu(OAc)2·H2O in a round-bottomed flask equipped with a refluxing device. The mixture was heated to approximately 83 °C with vigorous stirring, and maintained at that temperature for 30 min. Then 5 mL of deionized (DI) water was rapidly introduced into the above boiling solution, and the mixture was heated at 83 °C for another 30 min. During this process, the initial brown color of the dispersion gradually turned into black. After cooling to room temperature, the as-synthesized composite was centrifuged, washed with ethanol several times, and dried at 70 °C in air overnight. The nanocomposites with different mass ratios of CuO:GO, labeled as CuO:GOratio (e.g. CuO:GO0.5, CuO:GO1, CuO:GO2 and CuO:GO3).
Characterization
Crystal structure identification was determined by X-ray diffraction (XRD) using a Bruker D8 Advance X-ray diffractometer with Cu-Kα (λ = 0.15405nm) radiation. Transmission electron microscopy (TEM) measurements were performed on a JEOL JEM-2100 microscope operating at 200 kV, by depositing a drop of sample dispersion onto 300 mesh Cu grids coated with carbon layer. The shape of samples was determined by field emission scanning electron microscopy (FESEM) using a LEO-1550 microscope. Laser Raman spectroscopy (LRS) studies were carried out using a Renishaw Invia Raman spectrometer at room temperature. Differential scanning calorimetry (DSC) analyses were recorded on a TA-DSC-Q20 from 100 to 500 °C at a heating rate of 10 °C min−1 under nitrogen flow.Results and discussion
Fig. 1 shows the XRD patterns of GO and CuO:GO nanocomposites. The diffraction peak at around 2θ = 10.5° in Fig. 1a belongs to the (001) reflection of GO, and the interlayer spacing (0.84 nm) is much larger than that of pristine graphite (about 0.34 nm) owing to the introduction of oxygen-containing functional groups on the graphite sheet surfaces. As shown in Fig. 1b, no obvious peaks attributable to CuO were found in CuO:GO0.5 nanocomposite owing to the small amount of CuO in the system. When the content of CuO was increased (the mass ratio of CuO:GO was 1:1), the characteristic peaks of CuO could be detected (Fig. 1c). Moreover, the diffraction peaks of CuO intensified significantly with increasing CuO content (Fig. 1d, e). The diffraction peaks of CuO:GO nanocomposite are similar to those of CuO monoclinic phase (JCPDS 80-1268). Nevertheless, the (001) reflection peak of GO disappeared, which may indicate that the regular layered structure of GO has been destroyed, and exfoliated GO sheets due to the growth of CuO nanocrystals are formed.34,35 The XRD patterns demonstrate that the CuO:GO nanocomposite products can be successfully obtained in a water–isopropanol system.
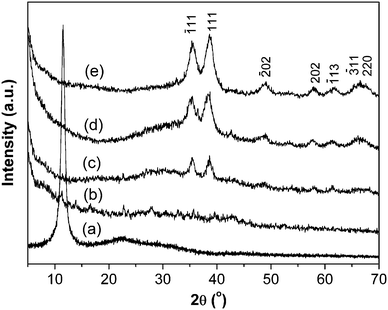 | ||
| Fig. 1 XRD patterns of (a) GO, (b) CuO:GO0.5, (c) CuO:GO1, (d) CuO:GO2 and (e) CuO:GO3. | ||
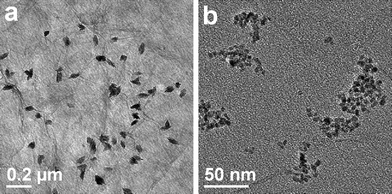
Fig. 2 illustrates the typical TEM images of CuO:GO0.5 nanocomposites. From Fig. 2a, it can be clearly seen that the exfoliated GO sheet was decorated by spindly CuO, with diameters of 40–60 nm and lengths of 80–120 nm. Closer examination (Fig. 2b) reveals that each of these spindly particles is made up of many smaller nanorods, with a diameter of 7 nm and a length of 40 nm. The inset of Fig. 2b shows an enlarged HRTEM image of an individual CuO spindle, which exhibits an interplanar spacing of about 0.252 nm corresponding to the (−111) plane of the monoclinic CuO crystal. Compared with the bright- and dark-field TEM images displayed in Fig. 2c and d, the single layered GO sheet can be clearly distinguished from the background. Obviously, it can be seen that some CuO spindles are brighter than the ones which seem to be enveloped by a thin film. This may be because some of the spindles are located above the GO sheet, while others are situated at the back of the sheet. Moreover, the TEM images demonstrate that most CuO spindles are attached to the GO sheet, and few scattered CuO particles can be found. Remarkably, due to the oxidation, these randomly distributed functional groups (hydroxyl, carbonyl, carboxyl and epoxy groups) on the GO surface could act as anchor sites, and consequently, made in situ formed by CuO particles attaching on the surfaces and edges of GO sheets.36,37 In addition, in situ formation of CuO particles in return caused the exfoliation of the lamellar GO.
 | ||
| Fig. 2 TEM images of the CuO:GO0.5 nanocomposite. (a, b, c) Bright-field TEM images. The inset of (b) is a HRTEM image of CuO particle. (d) Dark-field TEM image. | ||
Furthermore, Fig. 3 reveals the TEM images of CuO:GO1, CuO:GO2 and CuO:GO3 nanocomposites. As displayed in Fig. 3a, the CuO:GO1 system is still composed of spindly CuO attached to the GO sheets. However, when the mass ratio of CuO:GO was increased to 2:1, the exfoliated GO sheets were decorated by spherical particles of 60–80 nm in size (Fig. 3b). In fact, the large spherical particles shown in the TEM image still consist of small nanorods (Fig. 3c). Compared to CuO:GO3, as shown in Fig. 3d, we found that many CuO particles were anchored on the GO sheets, with some particles scattered out of the supports. Such marked increase in the density of CuO on GO sheets with the mass ratio of CuO:GO can be attributable to the nucleation mechanism of CuO nanoparticles at the functional sites of GO.
 | ||
| Fig. 3 TEM images of (a) CuO:GO1, (b, c) CuO:GO2 and (d) CuO:GO3 nanocomposites. | ||
It is well known that GO sheets have their basal planes decorated mostly with epoxy and hydroxyl groups, in addition to carbonyl and carboxyl groups located at the edges. The C:O ratio is ideally 2 when conversion of graphite to graphite oxide is completed;32 thus a lot of oxygen-containing functional groups should exist in as-prepared GO sheets, which can act as anchor sites, and consequently make in situ formed nanoparticles attach on the surfaces and edges of GO sheets . Accordingly, when a small amount of Cu(OAc)2 was introduced into the reaction system, only part of the anchor sites were occupied by CuO crystallites, resulting in the formation of highly dispersed CuO particles on GO surfaces. With the mass ratio of CuO:GO increasing from 0.5 to 2, more Cu2+ of Cu(OAc)2 bound with the negative charged functional groups, thus markedly increasing the density of CuO nanoparticles (Fig. 2 and Fig. 3). However, when the ratio of CuO:GO reached 3, almost all the anchor sites were occupied by CuO crystallites, and a part of Cu(OAc)2 would be outside the supports. Consequently, the GO sheets were decorated with CuO nanocrystals with high density, and some CuO nanoparticles even scatter out of the GO sheet (Fig. 3d).
The representative FESEM images are displayed in Fig. 4, which shows further that the surface of GO sheets were decorated by spindly or spherical CuO nanoparticle aggregates, forming the CuO:GO nanocomposite. The SEM observations are consistent with the TEM results. The nanoparticle aggregates were so stable that they could not be destroyed even after a long ultrasonic treatment at room temperature. The considerable wrinkles shown in Fig. 4 demonstrate the high degree of oxidation for pristine graphite.38
 | ||
| Fig. 4 FESEM images of (a) CuO:GO0.5 and (b) CuO:GO2 nanocomposites. | ||
Based on the above results and analysis, we propose a mechanism to interpret the formation process of CuO:GO nanocomposites (Scheme 1). As mentioned earlier, GO is a layered material bearing oxygen-containing functional groups on their basal planes and edges;38 these functional groups can act as anchor sites, and consequently, make nanoparticles formed in situ attach on the surfaces and edges of GO sheets. Accordingly, in the early stages, the positive Cu2+ ions formed in isopropanol could easily adsorb onto these negative GO sheets via the electrostatic force. With the addition of a small amount of H2O at the relatively high temperature of 83 °C, a large amount of nuclei were formed in a short time owing to the hydrolysis reaction of Cu(OAc)2. The oxygen atoms of the crystal growth units then might form bonds with the functional groups via intermolecular hydrogen bonds or coordination bonds, acting as anchor sites for the crystallites to grow further. Meanwhile, the growth of the CuO crystallites in return led to the exfoliation of GO sheets.
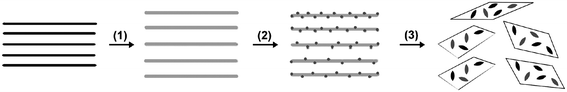 | ||
| Scheme 1 A proposed formation mechanism for CuO:GO nanocomposite. (1) Oxidation of graphite to graphite oxide with larger interlayer spacing. (2) Intercalation and adsorption of Cu2+ ions onto the GO sheets. (3) The nucleation and growth of the CuO crystallites, which in return results in the exfoliation of GO sheets. | ||
Scheme 2 illustrates a proposed growth mechanism of rod-like crystallites based on the phenomenon of different growth rates along the various crystal directions. As pointed out by our previous studies,39 it is considered that the coordination number of Cu2+ generally keeps six in solution. Based on the anionic coordinative polyhedra theoretical model,40–44 the growth units of coordinating octahedron would be formed, in which two water molecules locate at its axis, and other coordinating atoms surround Cu2+ to form a square structure.45,46 Therefore, with the addition of a small amount of water (DI water–isopropanol = 5mL:50mL), H2O molecules might only occupy the axis position of growth units (Scheme 2a). The binding energies of two H2O molecules located at the axis are lower than those of coordinating atoms located at the equator.45,46 So the two H2O molecules located at the axis easily react with Cu2+ to form CuO crystallites owing to the hydrolysis of Cu(OAc)2. That is to say, as a polar crystal, the growth rates of CuO crystals along the axis are higher than those in the plane. Consequently, adding a small amount of water allows CuO crystallites to grow preferentially in the axial direction of the coordinating octahedron. A difference in growth rates along various directions emerges, facilitating the formation of anisotropic CuO nanocrystals. The formation of rod-like crystallites illustrates nicely the growth course of CuO nanocrystals (Fig. 2b). The oxygen atoms in isopropanol can easily form hydrogen bonds by interconnection. The complicated association of hydrogen bonds could result in coarse aggregation of rod-like CuO crystallites. Furthermore, in the process of crystal growth, the Gibbs free energies of crystallite surfaces are usually very high, and the crystallites have the tendency to aggregate together to decrease the Gibbs free energies of the surfaces.47 Regarding the formation of spindly or spherical structure, the geometrical shape of building blocks must have played a key role, since no surfactants/emulsions were used.48 Meanwhile, the aggregation approaches of rod-like CuO crystallites were varied with differing CuO concentrations.
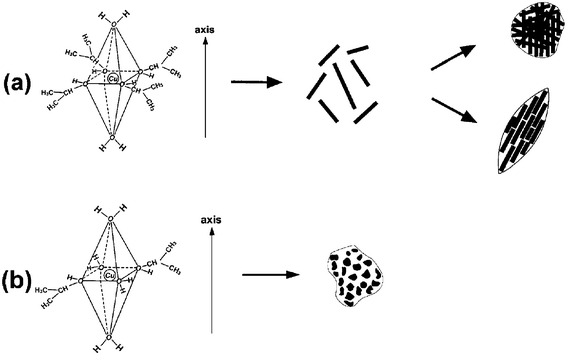 | ||
| Scheme 2 A proposed formation mechanism for CuO nanocrystals. (a) A small amount of water was introduced; (b) a large amount of water was added. | ||
The amount of added water is a key factor for the growth of the CuO crystallites. In fact, similar spindly nanocrystals were formed when 20 mL of water was added (Fig. 5a). However, the interaction of H2O molecules with Cu2+ in other directions became possible when more water was introduced (DI water–isopropanol = 50 mL:50 mL). With increasing amounts of water, the added water and isopropanol molecules might compete against each other. Consequently, some water molecules would occupy coordination sites in the equatorial direction of the coordinating octahedron (Scheme 2b), resulting in the crystal growth which was much less selective in direction and hence produced spherical CuO nanocrystals (Fig. 5b). Compared with the aggregated rod-like CuO, the spherical product disperses relatively well. The result indicates that aggregation growth of CuO nanocrystals is effectively inhibited, which might be mainly caused by the various interactions between isopropanol and water.49
 | ||
| Fig. 5 TEM images of CuO:GO0.5 nanocomposites when different volume ratios of the added water and isopropanol are used: (a) 20mL:50 mL, (b) 50 mL:50 mL. | ||
The use of isopropanol and the presence of a small amount of water were central to the formation of a well-ordered CuO:GO nanocomposite. Without water, no obvious CuO particles were formed in the solution phase. This result is primarily due to the lack of water, which is essential to the hydrolysis reaction of Cu(OAc)2. Similarly, few CuO particles can be found on GO sheets when only water was used as the solvent (Fig. 6), indicating that the isopropanol molecules play an active role in interacting between Cu2+ and GO sheets. Usually, considering the effect of steric hindrance and electrostatic effects in the water–isopropanol system, because the coordination capability of water species should be larger than that of the isopropanol,50 there is a competition reaction of the coordination with Cu2+ and GO between water molecules and isopropanol. Specifically, isopropanol was used as a solvent for dispersing dried GO in the initial stage, where the isopropanol molecules formed bonds with the oxygen-containing groups of GO via a weak hydrogen bond, giving a homogeneous black dispersion. Afterwards, when Cu2+ was introduced into the dispersion system with ultrasonication, part of the isopropanol molecules were replaced by Cu2+, which would interact with the oxygen-containing groups of GO via electrostatic forces. Because the coordination number of Cu2+ is kept at six in solution, based on the anionic coordinative polyhedra theoretical model, several isopropanol molecules would interact with Cu2+ to form a distorted octahedron. After the addition of water into the reaction system, Cu2+ were easily coordinated with H2O; with the introduction of a small amount of water, H2O molecules might occupy the axial positions of the CuO growth units, leaving iospropanol in the equatorial positions. Moreover, the interaction of H2O molecules with Cu2+ in other directions became possible when more water was introduced, resulting in CuO nanocrystals with other shapes.

Remarkably, due to the higher tendency of H2O molecules to form bonds with Cu2+ and GO, when H2O was employed as the only solvent, interactions of Cu2+ with GO would be effectively inhibited, thus CuO nanocrystals are seldom attached to the GO sheets.
Raman spectra of GO and CuO:GO1 nanocomposite are illustrated in Fig. 7. In Fig. 7a, two prominent peaks of GO appearing at around 1354 and 1592 cm−1 are attributed to the D and G modes, respectively. In the spectrum of CuO:GO1, the D and G modes of GO still exist. For the monoclinic CuO, this belongs to the C62h space group with two molecules per unit cell. There are nine zone-center optical phonon modes with symmetries 4Au + 5Bu + Ag + 2Bg. Only three Ag + 2Bg modes are Raman active.51,52 As can be seen from Fig. 7b, there are three Raman peaks at 280, 322 and 614 cm−1, and these peaks can be assigned to one Ag and two Bg modes, respectively. Compared with vibrational spectra of CuO macrocrystalline powder and the single crystal, the Raman peaks shown in Fig. 7b are lower than these reported previously due to size effects.39,53
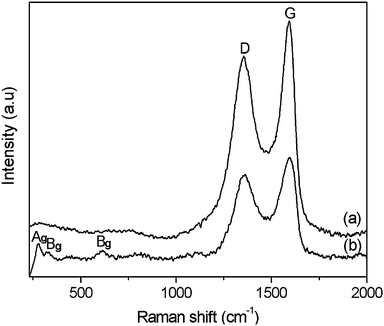 | ||
| Fig. 7 Raman spectra of (a) GO and (b) the CuO:GO1 nanocomposite. | ||
The catalysis of CuO:GO nanocomposites and each individual component for the thermal decomposition of AP (ammonium perchlorate) was investigated by DSC. Fig. 8 presents the DSC curves of the decomposition of AP and AP with 2% GO, CuO and CuO:GO2. As shown in Fig. 8a, the thermal decomposition of AP includes two stages: the low-temperature decomposition (LTD) at around 313 °C and the high-temperature decomposition (HTD) at around 414 °C. The endothermic peak at 247 °C in Fig. 8a is attributed to a crystal transition from orthorhombic to cubic AP.54 The exothermic quantity of decomposition of pure AP is 590 J g−1. Fig. 8b–d illustrate the DSC curves for the decomposition of AP mixed with GO, CuO and CuO:GO2. The additives have no obvious effects on the crystal transition temperature of AP. However, the DSC results show a noticeable change in HTD and exothermic quantity in the presence of additives. When only GO was added, the exothermic peak of HTD occurs at 396 °C with a exothermic quantity of 691 J g−1. The addition of CuO and CuO:GO2 further decreased the temperatures of LTD and HTD (Fig. 8c and d). The HTD of AP mixed with 2% CuO:GO2 (315 °C) was much lower than that of CuO (334 °C); moreover, the former exothermic quantity (1347 J g−1) was also more than that of the latter (1093 J g−1), suggesting the catalytic properties of the nanocomposites are enhanced by the concerted effect between GO and CuO. According to previous studies, CuO may promote the heterogeneous decomposition of deprotonized HClO4 gas on the solid surface in the HTD, which reduces the temperature of high temperature exothermic decomposition process, but without changing the low-temperature exothermic decomposition process much.55–57 Since nanoscale CuO possess a large specific area and more active sites, they exhibit a high catalytic activity upon the decomposition of AP. But aggregation would occur when they are dried in air, resulting in a decrease in the catalytic capability. In our procedure, when CuO nanocrystals are attached onto GO sheets, the aggregation would be effectively inhibited to a certain extent, giving a higher catalytic performance.
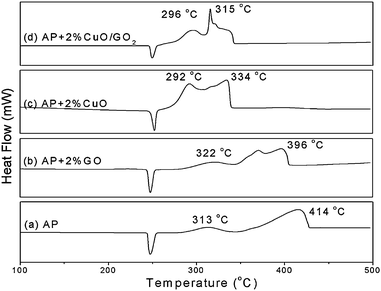 | ||
| Fig. 8 DSC curves for the decomposition of (a) AP, (b) AP with 2% GO, (c) AP with 2% CuO and (d) AP with 2% CuO:GO2 nanocomposite. | ||
Remarkably, the mass ratios of CuO:GO nanocomposites have an important influence on their catalytic properties. The HTD (exothermic quantity) of AP mixed with 2% CuO:GO0.5, CuO:GO1, CuO:GO2 and CuO:GO3 are 330 °C (1056 J g−1), 321 °C (1297 J g−1), 315 °C (1347 J g−1) and 329 °C (1151 J g−1), respectively (not shown). The catalytic activity was enhanced with the increase of CuO in nanocomposites except for CuO:GO3. Actually, this phenomenon is in agreement with the TEM result (Fig. 3d), in which some CuO particles are scattered out of the GO supports; meanwhile, some particles anchored on the GO sheets are aggregated. As a result, CuO:GO3 demonstrates a lower catalytic activity, close to that of CuO. Accordingly, the decoration of CuO on GO sheets, or the formation of chemical bonds between CuO and GO, is speculated to play an important role in the concerted effect.
Conclusions
In summary, we have succeeded in preparing CuO:GO nanocomposites by a facile chemical method without the use of any templates or surfactants. In this procedure, the water/isopropanol mixture was employed as the reaction system, which is essential for in situ forming of CuO particles on the surfaces of GO sheets. SEM and TEM observations indicate that the exfoliated GO sheets are decorated randomly by spindly or spherical CuO nanoparticle aggregates, forming well-ordered CuO:GO nanocomposites. The morphology and size of CuO nanocrystals on the GO sheets could be altered by manipulating the ratio of reactants. The obtained CuO:GO nanocomposites have a high catalytic activity for the thermal decomposition of AP. The various interesting structures of CuO:GO nanocomposites are also expected to have wide applications in catalysts, gas sensors, magnetic phase transitions, and nanoelectronic devices.Acknowledgements
This investigation was supported by the Natural Science Foundation of China and China Academy of Engineering Physics (No.10776014, 50902070), the Natural Science Foundation of Jiangsu province (No.BK2009391), and the Research Fund for the Doctoral Program of Higher Education of China (No. 20093219120011).Notes and references
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS.
- H. He, J. Klinowski, M. Forster and A. Lerf, Chem. Phys. Lett., 1998, 287, 53 CrossRef CAS.
- S. Stankovich, D. A. Dikin, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- B. Yue, Y. W. Ma, H. S. Tao, L. S. Yu, G. Q. Jian, X. Z. Wang, X. S. Wang, Y. N. Lu and Z. Hu, J. Mater. Chem., 2008, 18, 1747 RSC.
- N. Mackiewicz, G. Surendran, H. Remita, B. Keita, G. Zhang, L. Nadjo, A. Hagege, E. Doris and C. Mioskowsk, J. Am. Chem. Soc., 2008, 130, 8110 CrossRef CAS.
- Y. W. Ju, G. R. Choi, H. R. Jung, C. Kim, K. S. Yang and W. J. Lee, J. Electrochem. Soc., 2007, 154, A192 CrossRef CAS.
- R. Muszynski, B. Seger and P. V. Kamat, J. Phys. Chem. C, 2008, 112, 5263 CrossRef CAS.
- X. R. Wang, Y. J. Ouyang, X. L. Li, H. L. Wang, J. Guo and H. J. Dai, Phys. Rev. Lett., 2008, 100, 206803 CrossRef.
- M. Dragoman, D. Dragoman, F. Coccetti, R. Plana and A. A. Muller, J. Appl. Phys., 2009, 105(5), 054309 CrossRef.
- Y. M. Lin and P. Avouris, Nano Lett., 2008, 8, 2119 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197 CrossRef.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170 CrossRef CAS.
- B. Liu and J. Y. Lee, J. Phys. Chem. B, 2005, 109, 23783 CrossRef CAS.
- C. Xu, X. Wang, J. W. Zhu, X. J. Yang and L. D. Lu, J. Mater. Chem., 2008, 18, 5625 RSC.
- C. Xu, X. Wang and J. W. Zhu, J. Phys. Chem. C, 2008, 112, 19841 CrossRef CAS.
- Y. P. Sukhorukov, N. N. Loshkareva, A. A. Samokhvalov, S. V. Naumov, A. S. Moskvin and A. S. Ovchinnikov, J. Magn. Magn. Mater., 1998, 183, 356 CrossRef CAS.
- P. C. Dai, H. A. Mook, G. Aeppli, S. M. Hayden and F. Dogan, Nature, 2000, 406, 965 CrossRef CAS.
- M. Frietsch, F. Zudock, J. Goschnick and M. Bruns, Sens. Actuators, B, 2000, 65, 379 CrossRef.
- C. L. Carnes and K. J. Klabunde, J. Mol. Catal. A: Chem., 2003, 194, 227 CrossRef CAS.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2004, 126, 8124 CrossRef CAS.
- G. H. Du and G. V. Tendeloo, Chem. Phys. Lett., 2004, 393, 64 CrossRef CAS.
- Y. Chang, J. J. Teo and H. C. Zeng, Langmuir, 2005, 21, 1074 CrossRef CAS.
- R. Yang and L. Gao, Solid State Commun., 2005, 134, 729 CrossRef CAS.
- J. W. Zhu, H. Q. Chen, H. B. Liu, X. J. Yang, L. D. Lu and X. Wang, Mater. Sci. Eng., A, 2004, 384, 172 CrossRef.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef CAS.
- J. T. Paci, T. Belytschko and G. C. Schatz, J. Phys. Chem. C, 2007, 111, 18099 CrossRef CAS.
- Y. Matsuo, S. Higashika, K. Kimura, Y. Miyamoto, T. Fukutsuka and Y. Sugie, J. Mater. Chem., 2002, 12, 1592 RSC.
- D. V. Survase, M. Gupta and S. N. Asthana, Prog. Cryst. Growth Charact. Mater., 2002, 45, 161 CrossRef CAS.
- J. W. Zhu, W. G. Zhang, H. Z. Wang, X. J. Yang, L. D. Lu and X. Wang, Chin. J. Inorg. Chem., 2004, 20, 863 CAS.
- Y. P. Wang, J. W. Zhu, X. J. Yang, L. D. Lu and X. Wang, Thermochim. Acta, 2005, 437, 106 CrossRef CAS.
- Y. P. Wang, X. J. Yang, L. D. Lu and X. Wang, Thermochim. Acta, 2006, 443, 225 CrossRef CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- D. Cai and M. Song, J. Mater. Chem., 2007, 17, 3678 RSC.
- A. B. Bourlinos, D. Gournis, D. Petridis, T. Szabo, A. Szeri and I. Dekany, Langmuir, 2003, 19, 6050 CrossRef CAS.
- M. S. Raghuveer, S. Agrawal, N. Bishop and G. Ramanath, Chem. Mater., 2006, 18, 1390 CrossRef CAS.
- J. Li, S. B. Tang, L. Lu and H. C. Zeng, J. Am. Chem. Soc., 2007, 129, 9401 CrossRef CAS.
- T. Szabo, O. Berkesi, P. Forgo, K. Josepovits, Y. Sanakis, D. Petridis and I. Dekany, Chem. Mater., 2006, 18, 2740 CrossRef CAS.
- J. W. Zhu, H. P. Bi, Y. P. Wang, X. Wang, X. J. Yang and L. D. Lu, Mater. Chem. Phys., 2008, 109, 34 CAS.
- W. J. Li, E. W. Shi, W. Z. Zhong and Z. W. Yin, J. Cryst. Growth, 1999, 203, 186 CrossRef CAS.
- W. J. Li, E. W. Shi, W. Z. Zhong, Y. Q. Zheng and Z. W. Yin, J. Chin Ceram. Soc., 1999, 27, 164 Search PubMed.
- W. Z. Zhong, G. Z. Liu, E. W. Shi, S. K. Hua, D. Y. Tang and Q. L. Zhao, Sci. China Ser. B, 1994, 24, 349.
- E. W. Shi, W. Z. Zhong, S. K. Hua, R. L. Yuan, B. G. Wang, C. T. Xia and W. J. Li, Sci. China Ser. E, 1998, 28, 37.
- X. Zhang, Y. Xie, F. Xu, X. H. Liu and D. Xu, Inorg. Chem. Commun., 2003, 6, 1390 CrossRef CAS.
- S. L. Xu, Q. H. Xu, Y. C. Tian, S. Y. Liu, Inorganic Chemistry Series, China, Beijing, 1995, Vol. 6 Search PubMed.
- W. Z. Zhong, S. K. Hua, Crystal Morphology, China, Beijing, 1999 Edit. 1 Search PubMed.
- H. Wang, J. Z. Xu, J. J. Zhu and H. Y. Chen, J. Cryst. Growth, 2002, 244, 88 CrossRef CAS.
- B. Liu and H. C. Zeng, J. Am. Chem. Soc., 2004, 126, 8124 CrossRef CAS; M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877 CrossRef CAS.
- M. J. Kamlet, J. L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877 CrossRef CAS.
- M. J. Kamlet, J.-L. M. Abboud, M. H. Abraham and R. W. Taft, J. Org. Chem., 1983, 48, 2877 CrossRef CAS.
- Z. Wang, V. Pischedda, S. K. Saxena and P. Lazor, Solid State Commun., 2002, 121, 275 CrossRef CAS.
- J. F. Xu, W. Ji, Z. X. Shen, S. H. Tang, X. R. Ye, D. Z. Jia and X. Q. Xin, J. Solid State Chem., 1999, 147, 516 CrossRef CAS.
- K. Reimann and K. Syassen, Solid State Commun., 1990, 76, 137 CrossRef CAS.
- T. Ganga Devi, M. P. Kannan and B. Hema, Thermochim. Acta, 1996, 285, 269 CrossRef.
- Y. Xu, D. Chen, X. Jiao and K. Xue, Mater. Res. Bull., 2007, 42, 1723 CrossRef CAS.
- H. Xu, X. Wang and L. Zhang, Powder Technol., 2008, 185, 176 CrossRef CAS.
- V. V. Boldyrev, Thermochim. Acta, 2006, 443, 1 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |