Microwave chemistry for inorganic nanomaterials synthesis
Idalia
Bilecka
and
Markus
Niederberger
*
Laboratory for Multifunctional Materials, Department of Materials, ETH Zürich, Wolfgang-Pauli-Strasse 10, 8093, Zürich, Switzerland. E-mail: markus.niederberger@mat.ethz.ch
First published on 25th February 2010
Abstract
This Feature Article gives an overview of microwave-assisted liquid phase routes to inorganic nanomaterials. Whereas microwave chemistry is a well-established technique in organic synthesis, its use in inorganic nanomaterials' synthesis is still at the beginning and far away from having reached its full potential. However, the rapidly growing number of publications in this field suggests that microwave chemistry will play an outstanding role in the broad field of Nanoscience and Nanotechnology. This article is not meant to give an exhaustive overview of all nanomaterials synthesized by the microwave technique, but to discuss the new opportunities that arise as a result of the unique features of microwave chemistry. Principles, advantages and limitations of microwave chemistry are introduced, its application in the synthesis of different classes of functional nanomaterials is discussed, and finally expected benefits for nanomaterials' synthesis are elaborated.
 Idalia Bilecka | Idalia Bilecka earned a master's degree in chemistry from the University of Geneva where she gained experience in photocatalysis of nanoparticles (Prof. Augustynski, University of Geneva) and dye sensitized solar cells (Prof. Grätzel, Swiss Federal Institute of Technology Lausanne, EPFL). Since 2007 she has been a doctoral student in the Department of Materials at the Swiss Federal Institute of Technology (ETH) Zürich under the supervision of Prof. Niederberger, working on the microwave synthesis and processing of inorganic nanomaterials in organic solvents. Her research interests include the synthesis and application of inorganic nanoparticles, and the study of formation mechanisms and reaction kinetics. |
 Markus Niederberger | Markus Niederberger studied chemistry at the Swiss Federal Institute of Technology (ETH) Zürich, where he also received his Ph.D. degree in 2000. After a postdoctoral stay at the University of California Santa Barbara, in 2002 he became group leader at the Max Planck Institute of Colloids and Interfaces at Potsdam. Since 2007 he has been Assistant Professor in the Department of Materials at ETH Zürich. His research interests include the liquid-phase synthesis of inorganic functional materials, investigation of formation and crystallization mechanisms, particle assembly and organic–inorganic hybrids. Targeted applications are in the field of lithium ion batteries, chemoresitive gas sensors, photocatalysis, and transparent conducting oxides. |
1. Introduction
Time is valuable – this quote is certainly also true for Science. Especially in the field of synthetic chemistry often a large number of time-consuming trial-and-error experiments are required either to develop a completely new synthesis route to a targeted material, or to optimize an existing synthesis protocol with respect to yield, product quality and sustainability. Any new synthesis technique that helps to save time would be extremely beneficial, because it would allow the scientists to perform more experiments within the same time, or, in other words, testing of new theories and processes could be done much faster and more time would remain to expand scientific creativity.1 Microwave chemistry definitively fulfils the promise of being a fast synthesis technique. Since the first reports in 1986,2,3 the use of microwave ovens became an indispensable tool in modern organic synthesis.1,4–11 However, there is no doubt that the microwave technique has the potential to greatly contribute to all areas of synthetic chemistry. In particular, the synthesis of nanoparticles and nanostructures, whose growth is highly sensitive to the reaction conditions, could benefit from the efficient and controlled heating as provided by microwave irradiation. As a matter of fact, the number of papers dedicated to microwave-assisted synthesis of inorganic nanomaterials is growing rapidly, and almost all classes of functional materials have been targeted, including metals, oxides, sulfides, phosphates, and halides.12–19 But microwave heating does not only reduce the chemical reaction times by several orders of magnitude, but also suppresses side reactions, and thus improves the yield and reproducibility of a specific synthesis protocol. All these features represent important parameters that have to be considered during the development of “greener” synthesis methodologies, and therefore it can be expected that the microwave technique will also play a fundamental role in a future environmentally friendlier “synthetic nanotechnology”. Accordingly, the main question addressed in this review is how liquid-phase synthesis of inorganic nanomaterials can efficiently take advantage of the unique features of microwave chemistry.We start with a short overview of the experimental set-up and a brief introduction of the basic principles and some peculiarities of dielectric heating, followed by a general discussion of the advantages and limitations of this technique. Even though most of these pros and cons are based on experiences made during microwave-assisted organic synthesis, many of them also apply for nanoparticle preparation. Then we present selected applications of dielectric heating in inorganic nanomaterials' synthesis summarized in subchapters according to their composition. We did not intend to provide an exhaustive overview. Instead, we selected a few examples from every family of functional materials to underline the versatility of the microwave technique for accessing a large compositional diversity of compounds. Additionally, the most important synthesis parameters determining the morphology of the particles are mentioned. The final part of the review elaborates the main benefits of microwave-assisted routes for the preparation of nanomaterials. Based on illustrative examples from literature and from our work we propose new opportunities for nanoparticle synthesis offered by microwave heating.
2. Principles, advantages and limitations of microwave chemistry
2.1 Microwave equipment
Whereas the early pioneering work on microwave-assisted organic synthesis was carried out in domestic microwave ovens, the use of dedicated instruments is strongly recommended nowadays. Experimental parameters like irradiation power, reaction temperature and pressure inside the vessel are not known precisely enough in domestic microwave ovens. These uncertainties represent a serious safety issue and also lead to a lack of reproducibility. Modern commercially available dedicated microwave reactors for chemical synthesis, on the other hand, feature built-in magnetic stirrers, and direct temperature and pressure monitoring by various probes and sensors. In fact, one has to be very careful with the pressure inside a sealed reaction tube when working with high absorbing solvents with low boiling points like e.g. ethanol.1 In addition, dedicated microwave reactors are generally connected to a computer, which regulates the temperature and pressure by adjusting the power output. An exemplary reaction profile recorded for the synthesis of ZnO nanoparticles in benzyl alcohol is given in Fig. 1a, together with a photograph of the microwave oven used in our laboratory (Fig. 1b) and the corresponding pressurized reaction vessels of 10 (Fig. 1c) or 50 ml. Nowadays, several manufacturers offer a large variety of reactor systems with different degrees of sophistication with respect to automation, database capabilities, safety features, temperature and pressure monitoring, as well as vessel design.9 An exhaustive review of available microwave equipment is given in a recently published book by Kappe, Dallinger and Murphee.11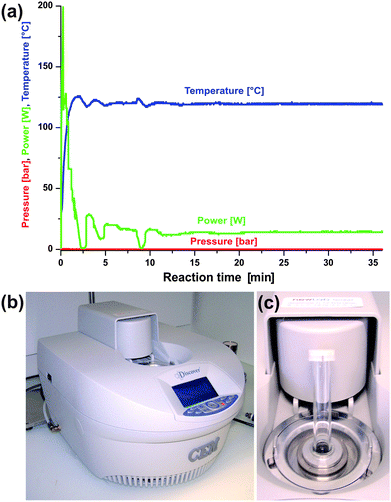 | ||
| Fig. 1 (a) Typical temperature, pressure and power profile for the synthesis of ZnO nanoparticles in benzyl alcohol at 120 °C; (b) single-mode CEM Discover microwave system; (c) 10 ml pressurized reaction tube and reactor cavity. | ||
2.2 Principles and peculiarities of microwave chemistry
Microwave irradiation is electromagnetic radiation in the frequency range 0.3 to 300 GHz, which corresponds to wavelengths of 1 mm to 1 m. A large fraction of the microwave spectrum is reserved for applications in telecommunication and Radar technology. All kitchen microwave ovens and the great majority of commercially available dedicated microwave reactors operate at a frequency of 2.45 GHz (corresponding to a wavelength of 12.25 cm). The choice of this frequency is the result of a compromise to fulfil several requirements. First of all, any interference with telecommunication, wireless networks and cellular phones has to be avoided. Second, this frequency is perfect for kitchen microwaves, because the corresponding magnetrons can be produced at low cost, the wavelength of 12.25 cm is smaller than the cooking chamber, and the typical penetration depth in food is in the range of a few centimetres. On the other hand one has to know that this frequency is not optimized for the heating of water, because liquid water has a much higher resonance frequency of 18 GHz, and therefore the most effective conversion of microwave energy into thermal energy would occur in this frequency region.20Microwave chemistry is based on the efficient heating of matter by microwave dielectric heating, i.e., on the ability of a specific material (e.g. solvent and/or reagents) to absorb microwave energy and to convert it into heat.7,11 The energy of a microwave photon at a frequency of 2.45 GHz is only 1.0 × 10−5 eV (or about 1 J mol−1),21 which is much too low to cleave chemical bonds.22,23 As a matter of fact, it is even lower than Brownian motion and only affects molecular rotations.7 Accordingly, microwave-enhanced chemistry is based on the efficient heating of materials, rather than on inducing chemical reactions by direct absorption of high energy electromagnetic radiation, as for example applied in photochemistry processes.
The heating mechanism involves two main processes, namely dipolar polarization and ionic conduction (Fig. 2).7 Irradiation of a sample with microwaves results in the alignment of the dipoles (Fig. 2a) or ions (Fig. 2b) in the electric field. Due to the fact that the electromagnetic radiation produces an oscillating field, the dipoles or ions continuously attempt to realign themselves in the electric field. Depending on the time scales of the orientation and disorientation phenomena relative to the frequency of the irradiation, different amounts of heat are produced through molecular friction and dielectric loss.7,24 If the dipole does not have enough time to realign (i.e., the frequency of the field is much higher than the response time of the dipoles), or easily keeps pace with the field changes (i.e., time to change the direction of the electric field is longer than the response time of the dipoles), then no heating occurs.7,20,24 The frequency of 2.45 GHz lies in between these two extremes. The rotations of many polar molecules in the liquid begin to lag behind the electric field oscillations at this frequency, producing resistive heating in the medium. This is described as dielectric loss, which is the amount of input microwave energy that is lost to the sample by being dissipated as heat.1 In the case of ionic conduction the dissolved charged particles oscillate back and forth under the influence of the microwave irradiation, collide with neighbouring molecules, and thus create heat. It is important to note that the ionic conduction mechanism represents a much stronger effect than the dipolar polarization with respect to the heat-generating capacity, and this has of course great consequences for the synthesis of nanoparticles in ionic liquids.11
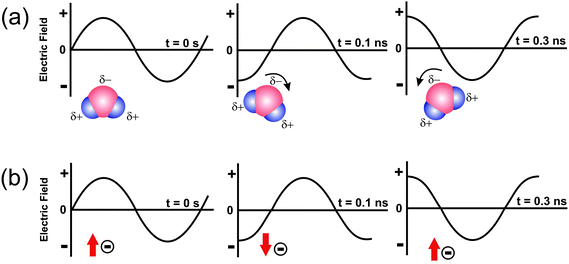 | ||
| Fig. 2 Two main heating mechanisms under microwave irradiation: (a) dipolar polarization; (b) ionic conduction mechanism.11 | ||
Two parameters define the dielectric properties of a substance: (i) the dielectric constant ε′, describing the ability to be polarized by the electric field, and (ii) the dielectric loss ε′′, indicating the efficiency with which electromagnetic radiation is converted into heat.24 The ratio of these two parameters defines the dielectric loss tangent tanδ = ε′′/ε′. This loss factor provides a measure for the ability of a material to convert electromagnetic energy into heat at a given frequency and temperature. A reaction medium with a high loss factor, i.e., with a high tanδ value, is required for efficient absorption and rapid heating.
Organic solvents are generally categorized in three different groups according to their high, medium, and low absorbing properties. High microwave absorbing solvents typically have a tanδ > 0.5, whereas medium and low absorbing solvents have values of 0.1–0.5 and < 0.1, respectively.7 Solvents with high loss factors (measured at room temperature and 2.45 GHz) include ethylene glycol (1.350), ethanol (0.941) or dimethyl sulfoxide (0.825),1 whereas solvents without a permanent dipole moment are basically microwave transparent. In this context it is interesting to point out that some of the high absorbing solvents have been used in the fabrication of inorganic nanoparticles by conventional heating in an oil bath for a long time. One example is the so-called polyol route, involving the reaction between metal salts and glycols.25–27 Polyalcohols like ethylene glycol, 1,3-propanediol, 1,4-butanediol or glycerol20 have high loss tangents, and therefore they are suitable solvents for microwave chemistry. As a matter of fact, the polyol route is nowadays frequently used for the microwave-assisted synthesis of metal oxide and metal chalcogenide nanoparticles (see Section 3). Also benzyl alcohol with tanδ = 0.66720 belongs to the family of strongly microwave absorbing solvents. It has been widely employed for the preparation of a large family of metal oxide nanoparticles by conventional heating,28–32 and also in this case the approach was recently extended to microwave chemistry.33 Benzyl alcohol is a relatively bulky molecule with a large molecular volume. Therefore, it could be expected that it needs a relatively long time to realign in the microwave field, which should restrict its absorption properties. However, its relaxation time, i.e., the time needed to go from the aligned state to the randomised state, is not much longer than that of ethanol.20 It was proposed that the relaxation process of large molecules in the microwave region is not associated with rotation of the whole molecule, but with localized rotational processes. In the case of molecules containing CH2OH groups attached to a phenyl ring, like e.g. in benzyl alcohol, the short relaxation time refers to a localized rotation of the CH2OH group.20
At this point it is worth mentioning that some solvents with very high dielectric constants, like water or acetonitrile, are surprisingly only medium absorbers with tanδ = 0.123 and 0.062, respectively. However, solvents with very low loss factors can also be used in microwave-assisted synthesis procedures, because the substrates, reagents or catalysts present might be polar enough to allow sufficient heating, or the addition of polar additives such as alcohols or ionic liquids increases the absorbance level of the medium.7
The loss tangent values depend on the frequency and the temperature. Because the penetration depth is inversely proportional to the loss tangent, these two parameters also strongly affect the penetration depth, which is defined as the point where 37% of the initially irradiated microwave power is still present.21 As a consequence, solvents and materials with relatively high loss tangents are characterized by short penetration depths and thus only the outer layer is efficiently heated. In the case of water, the penetration depth at room temperature is just a few centimetres,11 and this has of course far-ranging implications for the scale-up of microwave procedures. For pure water and most of the organic solvents the loss tangent decreases with temperature, i.e., the absorption of microwave irradiation decreases with temperature. From the practical point of view this means that microwave heating at higher temperatures might become somewhat difficult.
Almost from the very beginning of the groundbreaking work of Gedye2 and Giguere3 a continuous and controversial discussion started about the question why microwave irradiation can cause such an immense acceleration of reaction rates reaching up to several orders of magnitude.5,6,34 Basically two effects were claimed to be responsible for this observation: Thermal and non-thermal effects. Thermal effects are responsible for the fast heating of the reaction mixture, whereas the role of non-thermal effects, having their origin in the inherent characteristics of microwaves, is still rather speculative and ranges from lowering the Gibbs activation energy to increase of the collision efficiency by mutual orientation of polar molecules and possible excitement of rotational or vibrational transitions.6,8,34 In most of the cases, reports on non-thermal effects were based on inaccurate comparisons of microwave experiments with conventional heating, i.e., the syntheses were not carried out under exactly similar conditions.5,6,35 For example, ionic liquids can be heated in the microwave up to 200 °C within a few seconds, and of course such a temperature profile cannot be reproduced by conventional heating.7 Therefore, no clear and unequivocal evidence for the existence of a non-thermal effect has been reported so far. As a matter of fact, Kappe et al. recently reported a clever experiment that made it possible to discriminate whether or not an observed reaction enhancement is the result of a purely thermal phenomenon.36 The trick is to use reaction vessels made out of silicon carbide SiC together with a single-mode microwave reactor equipped with an external infrared and an internal fiber-optics for the temperature monitoring. SiC is a strongly microwave-absorbing material, and thus can rapidly be heated by microwaves. At the same time it was found that SiC can effectively shield the reaction mixture from the electromagnetic field. In combination with the high thermal conductivity and effusivity of SiC, such an experimental set-up provides a means to heat a reaction mixture by a conventional heat transfer mechanism and not by dielectric heating effects, although using microwave irradiation. Applied to eighteen previously published microwave-assisted organic chemical reactions, for which enhancements have been observed in comparison to conventional heating, the authors did not find any significant differences in terms of conversion, purity and product yields between experiments performed in microwave-transparent Pyrex and SiC vials.36 This observation confirms that for the studied cases only bulk temperature effects are responsible for the observed enhancements and that the electromagnetic field has no direct influence on the reaction pathway.
Nevertheless, microwaves provide some peculiar features that contribute to the acceleration of the reaction rates. Although these specific microwave effects are basically still thermal effects, they are caused by the uniqueness of the microwave dielectric heating mechanism and cannot be achieved by conventional heating. Examples include the superheating effect, the selective heating of polar species in a microwave transparent (or at least less absorbing) medium, the formation of “molecular radiators” by direct coupling of microwave energy to specific reagents in homogeneous solution, and the elimination of wall effects caused by inverted temperature gradients (i.e., from the “inside” to the exterior).7,8 The superheating effect is related to the boiling point of a liquid under microwave irradiation and represents a basic problem of microwave heating. Microwave-heated liquids can have boiling points of 10–20 °C above the conventional boiling point at atmospheric pressure, and of course this effect makes a comparison of the reaction rates under conventional and microwave conditions rather difficult. However, superheating is clearly related to stirring, and therefore in microwave reactors with a stirring device this effect is strongly suppressed.6 Probably the most important effect specific for microwaves results from the selective heating of strongly microwave-absorbing heterogeneous catalysts or reagents in a less polar medium.5,6,8,35 It can reasonably be assumed that such an effect plays a crucial role in the synthesis of inorganic nanoparticles.
At this point we don't want to go into more detail about the working principle and effects of microwave irradiation. The interested reader is referred to some excellent reviews7,20,22,23 and books,1,11 which, however, discuss these topics mainly in the context of organic chemistry.
2.3 General advantages and limitations of microwave-assisted chemical synthesis
In the liquid phase preparation of inorganic nanoparticles most of the syntheses are carried out by conductive heating with an external heat source like an oil bath, a heating mantle or a laboratory furnace as in the case of hydro- and solvothermal reactions. The limitations for such heating procedures are obvious: They are slow and rather inefficient, because they depend on convection currents and on the thermal conductivity of the various materials and substances that have to be penetrated, and often the temperature of the reaction vessel is considerably higher than that of the reaction mixture. Microwave irradiation, on the other hand, produces efficient internal heating, increasing the temperature of the whole volume simultaneously and uniformly. Accordingly, microwave dielectric heating and conventional thermal heating are completely different processes and it is therefore rather problematic to compare results obtained from these two techniques.Microwave heating can provide the following advantages in comparison to conventional heating for chemical synthesis (Fig. 3):9,20 (i) high heating rates, thus increasing the reaction rates; (ii) no direct contact between the heating source and the reactants and/or solvents; (iii) excellent control of the reaction parameters, which is not only important with respect to the quality of the product, but also addresses a serious safety issue; (iv) selective heating, if the reaction mixture contains compounds with different microwave absorbing properties; (v) higher yields; (vi) better selectivity due to reduced side reactions; (vii) improved reproducibility; and (viii) automatization and high throughput synthesis.
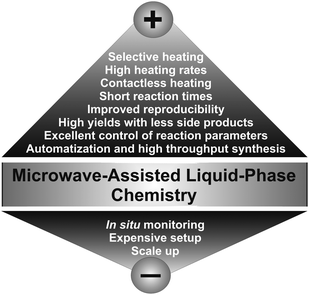 | ||
| Fig. 3 Current pros and cons of microwave-assisted synthesis. | ||
Most of these advantages were established in the context of microwave-assisted organic synthesis. Nevertheless, many of them also apply to the synthesis of inorganic nanoparticles, as discussed in Section 4.
In addition to these numerous advantages, microwave chemistry also has some significant limitations (Fig. 3). One of the major drawbacks are the high costs for dedicated microwave reactors, which can easily be in the range of several ten thousands of Euro. The short penetration depth of microwave irradiation into the liquid medium limits the size of the reactors, which is a serious problem for scale-up. The currently largest microwave systems used on the pilot plant-scale comprise approximately 100 L.37 Another problem, which is however not only specific for microwave chemistry, but for nanoparticle synthesis in general, is the difficulty to monitor nanoparticle formation in situ under reaction conditions. In the case of microwave synthesis, this problem is particularly pronounced due to the field containment and the interaction with metallic objects in the field.38 Nevertheless, in the last few years several combinations of microwave reactors with characterization tools such as X-ray techniques,38–40 neutron scattering,41,42 and Raman spectroscopy38,43–45 have been proposed to study organic as well as inorganic reactions under the respective experimental conditions.
3. Application of microwave heating in inorganic nanomaterials' synthesis
3.1 Introduction
The possibility to tune the physical and chemical properties of nanoscale materials through varying the crystal size and shape is a major driving force in nanoparticle research.46–48 However, this fascinating effect also brings an additional challenge to the synthetic chemist, namely the need to control, not only the composition of a product, but also its morphological characteristics. Although gas-phase processes are successfully employed for the low-cost production of large quantities of nanopowders,49–51 it seems that liquid-phase syntheses,52–57 including coprecipitation, hydrolytic58,59 as well as nonhydrolytic sol–gel processes,29,31,48,60,61 hydrothermal13,62 or solvothermal methods,13 sonochemistry,63,64 template synthesis,65 biomimetic66–70 and environmentally friendly approaches,71 are more flexible with regard to the subtle control of crystal size and shape. Although microwave irradiation has been used for the sintering of nanopowders72 and materials processing,73 it is, in the context of nanoparticles, mainly applied to liquid-phase syntheses, which means that it is basically possible to combine microwave chemistry with the full spectrum of available liquid-phase synthesis techniques (Fig. 4). Without any doubts, the broad variety of possible combinations opens up unique and exciting opportunities for the preparation of inorganic nanoparticles and nanostructures. This great flexibility and versatility is also the reason why it is impossible to give an exhaustive overview of all the inorganic nanoparticles prepared by microwave-assisted liquid phase routes.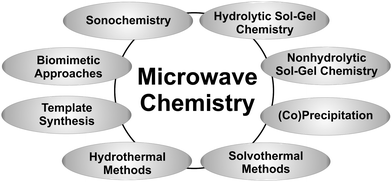 | ||
| Fig. 4 Possible combinations of microwave chemistry with well-established liquid-phase synthesis routes. | ||
3.2 Metallic nanostructures
Microwave-assisted routes have been applied for the one-pot synthesis of a large variety of mono- and bimetallic nanoparticles and nanostructures such as Au, Ag, Pd, Pt, Cu, In, and combinations thereof in solution.18 The irradiation of metal particles by microwaves immediately rises questions about safety issues related to destructive electrical arcing.74 It was suggested, that particle sizes in the submicron range significantly reduce the level of arcing.75Generally, protocols for metal nanoparticles involve the chemical reduction of metal salts either in organic solvents such as methanol, ethanol, N,N-dimethyl formamide or in aqueous medium.
Luo reported the colloidal synthesis of Pt nanoparticles by microwave heating of an aqueous solution of H2PtCl6 and 3-thiophenemalonic acid.76 The size can be controlled by adjusting the molar ratio of the reactants. Kundu et al. proposed the use of 2,7-dihydroxy naphthalene as the reducing agent for the preparation of multi-shaped gold nanoparticles.77 Different particle shapes such as spheres, rods, or triangles (Fig. 5a) were achieved by varying the metal ion to cetyltrimethylammonium bromide ratio and the concentration of the reducing agent. The morphology of gold nanoparticles can also be strongly influenced by physical parameters as provided by the microwave reactor. Liu et al. have shown that with increasing temperature, longer heating time, and fast ramping the gold nanoparticles become smaller and their size distribution narrower.78
 | ||
| Fig. 5 TEM overview images of various metal nanoparticles. (a) Au nanoparticles with different shapes (reproduced from ref. 77 with permission of the American Chemical Society); (b) Ag nanoparticles (inset: aqueous dispersion) (reproduced from ref. 81 with permission of the American Chemical Society); and (c) Ni nanoparticles (reproduced from ref. 82 with permission of Wiley-VCH). | ||
An overview of green syntheses of silver nanoparticles can be found in a recent review by Sharma and Lin.79 An interesting example along these lines is the use of carboxymethyl cellulose sodium as a green reducing and stabilizing agent, allowing the preparation of uniform and stable silver nanoparticles.80 Another “green” synthesis was presented by Hu et al., employing basic amino acids as the reducing and soluble starch as the protecting agent.81Fig. 5b shows a TEM image of such Ag nanoparticles obtained at 150 °C irradiated for just 10 s with a narrow size distribution of 26 ± 2 nm. The nanoparticles form stable colloidal dispersions in water (Fig. 5b, inset).
In addition to water, organic solvents can be also used for the preparation of metal nanoparticles. Especially polyols acting as both reducing agent and solvent are popular. Ni nanoparticles of controlled size in the range 30–100 nm were synthesized in ethylene glycol with polyvinylpyrrolidone and dodecylamine as a protecting agent.82 Whereas the morphology of the Ni nanoparticles was mainly controlled by the amount of dodecylamine, particle size and size distribution were dependent on the concentration of the metal source and the dodecylamine-to-polyvinyl pyrrolidone ratio. Under optimized reaction conditions, Ni nanoparticles of 63 nm can be produced, forming chain-like structures (Fig. 5c). In a related approach, Gou et al. grew Ag nanowires in ethylene glycol and in the presence of polyvinylpyrrolidone.83 The authors proposed that microwave irradiation accelerates nanowire formation by polarizing the conducting electrons, leading to a local “hot spot” effect at the wire ends, which promotes nanowire growth.
Also Tsuji et al. and Patel et al. extensively explored the polyol process with polyvinylpyrrolidone in combination with microwave heating.84–87 Variation of precursor, precursor concentration, and salt additions gave access to Au, Ag, Pt, Au/Pd, Au/Ag, Au/Pt core shell nanoparticles with different sizes and shapes.
The dependency of the penetration depths of microwaves on their wavelength makes it possible to control the penetration depths through variation of the frequency. Jiang et al. studied the polyol synthesis of silver nanoparticles using a microwave reactor with variable frequency.88 The center frequency of the microwave was 6.425 GHz with a bandwidth of 1.15 GHz and a sweeping time of 0.1 s. The authors proposed that the variable frequency microwave produced a particularly homogeneous heating, which resulted in narrow particle size distributions, enhanced nucleation and thus increased yield, however without altering the particle growth mechanism. Also ionic liquids, known as excellent microwave-absorbing agents due to their high ionic conductivity and polarizability, have been used for the preparation of metal nanostructures. Ren et al. not only took advantage of the microwave absorbing properties of ionic liquids, but also of their template effect for the fabrication of gold nano- and microstructures.89
3.3 Metal oxides
Komarneni's group is, without any doubts, one of the pioneers of microwave-hydrothermal synthesis of inorganic powders. Already in 1992 they reported the synthesis of TiO2, ZrO2, Fe2O3, KNbO3 and BaTiO3 using a combination of microwave and hydrothermal techniques.90 In the following years they applied microwave-hydrothermal processing to phosphates91 and metals.92 In 1998, Li and Wei described the microwave-induced hydrolysis of different ferric salts leading to the formation of uniform α-Fe2O3 nanoparticles.93 They found that the pH played an outstanding role in determining particle size and shape, high ion concentration accelerated the nucleation process, and that the addition of phosphate ions produced ellipsoidal nanoparticles. Hu and Yu also performed a systematic study on the effect of the reaction conditions on the morphology of α-Fe2O3 nanoparticles.94 They demonstrated fine shape control by adjusting the initial Fe3+ to PO43− molar ratio, resulting (among other shapes such as sheets, rings and spheres) in ellipsoidal hematite nanoparticles with tunable aspect ratios in the range of 1.1 to 6.3 (Fig. 6a-c). Komarneni et al. used the microwave-hydrothermal approach for the synthesis of nanophase ferrites (NiFe2O4, ZnFe2O4, CoFe2O4 and MnFe2O4).95,96 They underlined that the short reaction times represented ideal conditions for scaling-up the production via continuous flow synthesis.95 | ||
| Fig. 6 TEM or SEM overview images of metal oxide nanoparticles prepared by microwave-assisted liquid phase routes. (a)–(c) α-Fe2O3 with different aspect ratios (reproduced from ref. 94 with permission of Wiley-VCH), (d) indium tin oxide nanocrystals (inset: ITO nanoparticles dispersed in diethylene glycol) (reproduced from ref. 126 with permission of Elsevier), (e)–(f) monodisperse and single-crystalline ZnO colloidal nanocrystal clusters (reproduced from ref. 131 with permission of Wiley-VCH), (g) NiFe2O4, (h) BaTiO3 nanoparticles, (i) TiO2 nanocrystals (reproduced from ref. 139 with permission of the American Chemical Society). | ||
The microwave-hydrothermal method gave access to a large variety of binary and ternary oxides such as ZnO,97 CuO,98 PdO,99 Nd2O3,100 CeO2,101,102 and gadolinium-doped CeO2,103 In2O3,104 Tl2O3,104 SnO2,105,106 HfO2,107 BiVO4,108 ZnAl2O4,109 tungstates,110,111 Bi2MoO6,112 KNbO3,113 BaTiO3,,114–117 CaTiO3,118 and BaZrO3.119 In addition to studying the influence of precursor composition, concentration and heating temperature, Nyutu et al. showed that the microwave frequency could also affect the particle size, phase purity and morphology of tetragonal BaTiO3.116 Other unusual effects observed under microwave heating include a gradual phase transition from tetragonal to monoclinic BiVO4 upon extending the irradiation time,108 or the promotion of the growth of Gd-doped CeO2 nanorods by oriented attachment.103
High boiling points and high dielectric loss factors make organic solvents like ethylene glycol or benzyl alcohol perfect alternatives to an aqueous system, adding all the advantages of nonhydrolytic/nonaqueous nanoparticle synthesis120 to microwave chemistry. The polyol method, for example, was applied to TiO2,121 CeO2,122 Co1−xZnxFe2O4,123 Mn/CoFe2O4,124 BaTiO3, BaZrO3 and PbTiO3.125 Furthermore, doping of metal oxides was also achieved in the polyol–microwave approach, e.g., indium tin oxide (Fig. 6d),126 Mn/Ni/Co/Cr-doped ZnO,127,128 and Al- and In-doped ZnO nanoparticles.129 In the polyol route, the solvent, often ethylene glycol or diethylene glycol, acts as complexing and capping agent, and thus limits the particle growth and prevents agglomeration.130 Giri et al. took advantage of these features for the synthesis of Co1−xZnxFe2O4 to tune the particle size.123 Hu et al. used the polyol route for the synthesis of monodisperse ZnO colloidal nanocrystal clusters with tunable sizes (Fig. 6e and f).131 The diameter of the clusters composed of small primary nanocrystals can be varied from 57 to 274 nm by simply varying the amount of zinc oxide precursor. In addition to polyols, benzyl alcohol was also used to prepare various oxides such as Fe3O4,33 MnO,33 Mn3O4,33 CoO,33 NiFe2O4 (Fig. 6g)132 and BaTiO3 (Fig. 6h).33 In this case, subtle tuning of the crystal size through the initial precursor concentration33 and the irradiation time is easily possible as well.33,133 The strong connection between organic and inorganic reaction pathways and the high sensitivity of organic reactions towards microwave irradiation represents the basis for size controlled synthesis of metal oxides in organic solvents.
Ionic liquids are particularly suitable solvents, because they have a high thermal stability, a low vapor pressure, can dissolve many organic, inorganic and organometallic compounds, and they can act as template to control particle shape and assembly behavior.134,135 Taking advantage of these combined effects, Jacob et al. developed a fast method to synthesize Fe2O3 and different metal fluoride nanoparticles,136 whereas Wang and Zhu produced ZnO needles or flowers.137 Ionic liquids were also used for the preparation of highly conductive indium tin oxide,138 TiO2 (Fig. 6i),139 ZnFe2O4,140 and CuO nanocrystals.141 A more detailed discussion about the use of ionic liquids in the microwave-assisted preparation of functional nanomaterials can be found somewhere else.142
3.4 Metal chalcogenides
The third big class of functional materials synthesized by microwave-assisted liquid-phase routes are metal chalcogenides. In contrast to the generally applied nonhydrolytic colloidal synthesis of metal chalcogenides,48 microwave routes are often performed in aqueous media. For example, water-dispersible ZnSe quantum dots with quantum yields of 17% were obtained by reacting ZnCl2 with NaHSe in the presence of 3-mercaptopropionic acid as stabilizer in water.143 The high luminescence was attributed to the sulfur ions released from the stabilizer and that form a ZnSe(S) alloy shell on the surface of the ZnSe nanocrystals. Enhanced photoluminescence was also observed for microwave-grown CdTe/CdS144 and CdSe/ZnS145 core–shell nanocrystals. Other water-based microwave routes were reported for self-assembled ZnS nanoballs,146 CdSe nanoparticles with narrow photoluminescence emission in aqueous dispersion,147 alloyed CdSe–CdS quantum dots,148 or CdTe nanocrystals.149 The group of Gedanken was particularly active in the field of metal chalcogenide preparation, including aqueous (e.g., II–VI binary chalcogenides such as CdSe, PbSe, and Cu2−xSe),150 as well as nonaqueous systems. Especially the adaptation of the polyol route gave access to a large variety of binary chalcogenides such as PbTe,151 PbSe,151 CdSe,152 SnS,153 SnS2,153 CdSe,152 In2S3,154 Bi2Se3,155 MoSe2,156 or ternary compounds like CuCr2Se4,157 Cu2SnSe4,158 CuInTe2,159 CuInSe2,159 and Cd1-xZnxSe.160 Another organic solvent suitable for the preparation of metal chalcogenides is formaldehyde. Liao et al. reported the preparation of a series of nanocrystalline Cu, Hg, Zn, Bi, and Pb sulfides with different shapes and particle sizes by reacting metal salts dissolved in formaldehyde with thioacetamide.161 Dimethylformamide in combination with alkylamines was used for the preparation of ZnS nanorods and nanowires, ZnSe nanowires, and CdSe nanorods (Fig. 7a).162 Murugan et al. studied the formation of CdS nanocrystals by reacting cadmium sulfate with thiourea in water and in different organic solvents such as ethanol, ethylene diamine and ethylene glycol.163 The type of solvent determines the morphology of the particles (spherical or rod-like), whereas the molar ratio of Cd to thiourea influenced the crystal structure (hexagonal or cubic). | ||
| Fig. 7 TEM or SEM images of (a) CdSe nanoparticles (reproduced from ref. 162 with permission of the American Chemical Society), (b) carbon coated LiFePO4 nanoparticles (reproduced from ref. 170 with permission of the American Chemical Society), (c) silica coated InP/ZnS nanocrystals (reproduced from ref. 182 with permission of Wiley-VCH). | ||
Similar to metal and metal oxide synthesis, in the case of chalcogenides ionic liquids have also been used as solvent. One example is the formation of metal sulfide M2S3 (M = Bi, Sb) nanorods in 1-butyl-3-methylimidazolium tetrafluoroborate.164
3.5 Metal phosphates
Metal phosphates have extensively been studied for applications as phosphors, catalysts and as cathode materials in lithium ion batteries, and in all cases microwave chemistry opened up new synthesis pathways. Several lanthanide orthophosphates LnPO4 (Ln = La, Ce, Nd, Sm, Eu, Gd and Tb) were prepared by microwave heating of an aqueous solution of Ln(III) nitrate and NH4H2PO4.165 Bühler and Feldmann obtained LaPO4:Ce,Tb nanocrystals in high yields.166 The authors proposed that the high quantum yields and the possibility of synthesis-independent surface conditioning makes this synthesis strategy particularly interesting for technical applications, including biomedical applications.In the field of catalytically active phosphates, Jhung et al. reported the synthesis of nanoporous nickel phosphates.167 The fast crystallization time of about 1 min under microwave irradiation made it possible to perform the synthesis in a continuous manner.
Lithium transition metal phosphates are currently in the focus of scientific and technological interest, because they represent promising alternative cathode materials in lithium ion batteries.168 Murugan et al. presented an elegant approach combining microwave chemistry with solvothermal processing.169 Highly crystalline olivine LiFePO4 nanorods were directly obtained within 5 min at 300 °C from lithium hydroxide, iron acetate, and phosphoric acid in tetraethyleneglycol. Coating of the nanorods with carbon (Fig. 7b) or combining them with carbon nanotubes led to nanocomposites with high discharge capacity and good cyclability.170,171 In addition to tetraethyleneglycol, benzyl alcohol is also suitable for the efficient preparation of LiFePO4, LiFe0.5Mn0.5PO4 and LiMnPO4 powders.172
3.6 Zeolites
The preparation of zeolites using microwave chemistry has a long tradition. An early example is the microwave-hydrothermal synthesis of zeolite Y and ZSM-5.173 However, the microwave synthesis of nanoporous materials, including zeolites and mesoporous molecular sieves goes far beyond the scope of this Feature Article. The interested reader is referred to two excellent reviews by Cundy174 and Tompsett et al.163.7 Others
The compositional variety as well as the structural complexity of the nanomaterials synthesized by microwave-assisted liquid-phase approaches is growing rapidly. Examples along these lines include the synthesis of Sn1.24Ti1.94O3.66(OH)1.50F1.42,175 KMgF3,176 carbon nanotube/MnO2 composites,177 or manganite nanotubes in alumina templates.178 Carbon nanofibers and nanotubes have also been grown under microwave irradiation.179 Structurally more complex materials such as organic–inorganic hybrids consisting of poly(3,4-ethylenedioxythiophene/RuO2·xH2O for supercapacitor applications,180 biocompatible CdTe/CdS/ZnS core–shell quantum dots,181 or silica-coated InP/ZnS nanocrystals (Fig. 7c) as converter material in white LEDs182 have also been reported.4. Expected benefits of microwaves for nanomaterials' synthesis
4.1 Introduction
The focus of this section lies on the presentation of the advantages arising from the use of dielectric heating and their consequences for the preparation of inorganic nanoparticles. We have chosen selected reports from the literature, which give insight into the current understanding of the microwave–matter interaction. We will discuss examples that show increased reaction rates for nanoparticle formation under microwave irradiation. Due to the lack of well-established theories and mechanistic studies for nanoparticle formation, the impressive results obtained so far in this field will be interpreted based on the knowledge gained from organic synthesis, and on the dissolution and interface chemistry of heterogeneous systems. Finally, we will draw first conclusions regarding the production of more uniform nanoparticles and the tunability of nanoparticle morphology by applying microwave heating.4.2 Rate enhancement—fast synthesis
Microwave energy can favourably change the kinetics, the selectivity and the yield of a chemical reaction. The fundamental difficulties facing all the comparative studies between microwave and conventional heating, as well as between various microwave studies themselves, is the accurate determination of local temperatures and the inconsistencies between the different experiments in terms of field homogeneity, power delivery and reactor geometry.37 A very active area of research, where microwave energy has been demonstrated to beneficially influence the reaction kinetics and selectivity, is the synthesis of nanoporous materials.16 The first patent for the microwave-assisted synthesis of various zeolites was issued to Mobil in 1988.183 Although several subsequent studies all proved shorter reaction times in comparison to the classical hydrothermal technique, the results from different laboratories were not consistent.184 When Conner et al. studied the synthesis of silicalite employing two different microwave oven geometries, they found that understanding of the electromagnetic field distribution within the microwave cavity is essential and highly influences the morphology and the yield of the obtained silicate crystals.184 A follow-up study by the same group confirmed that the enhancement of the reaction rates was dependent on many complex factors, including vessel size, volume of the precursor reacted, microwave power delivery, and microwave cavity design.185 The rates of nucleation and crystallization, for example, were more rapid in a multimode oven than in a single mode oven with a more uniform field. Obviously, a nonuniform microwave field distribution appears to be a specific factor in the rate enhancement of some zeolite syntheses. Another important factor is the type of starting material. The ability of microwaves to influence the dissolution rate of various precursors (due to different dielectric properties) strongly affects the nucleation and crystallization rates.185 A similar observation was made during the microwave-assisted preparation of zinc oxide nanoparticles.133 It was found that microwave irradiation greatly enhances the dissolution rate of zinc acetate, which was used as solid precursor. However, in contrast to the aqueous zeolite synthesis, the preparation of the ZnO nanoparticles was performed in benzyl alcohol as a purely organic medium. It is important to know that in nonaqueous routes to metal oxide nanoparticles organic reaction pathways play a dominant role.28,186 The organic species (i.e., the solvent and the organic constituents of the precursor) undergo chemical reactions that are responsible for supplying the “monomers” for nucleation and growth of the inorganic nanoparticles. The high sensitivity of organic reactions towards microwave irradiation makes it obvious to combine nonaqueous liquid-phase synthesis with microwave chemistry, thus potentially providing a way to control the kinetics of “monomer”, and thus, nanoparticle formation. Monitoring the formation of benzyl acetate as organic product from the esterification reaction between acetate and benzyl alcohol and ZnO crystal size and yield with reaction time made it possible to correlate the kinetics of the organic side reaction with the growth kinetics of the ZnO nanoparticles.133 The esterification reaction, which represents the chemical basis for producing the monomers for ZnO formation, was found to be first order, whereas the growth of the ZnO nanoparticles followed the Lifshitz–Slyozov–Wagner model for coarsening. The rate constants kesterification for different temperatures can be determined directly from the slopes of the straight lines in Fig. 8a following the Arrhenius equation. Fig. 8b shows a plot of the natural logarithm of the rate constants kesterificationvs. the inverse temperature. The activation energy Eesterificationa calculated from the slope amounts to about 37 kJ mol−1 with a pre-exponential factor A of 9.5.133 The crystal size evolution shows a linear dependency of the radius cubed on the reaction time (Fig. 8c), as proposed by the Lifshitz–Slyozov–Wagner model for crystal coarsening. The rate constants kgrowth, extracted from the slopes of these straight lines, also follow the Arrhenius equation (Fig. 8d) and give an activation energy Egrowtha of 60 kJ mol−1 and a pre-exponential factor A of 21.3.133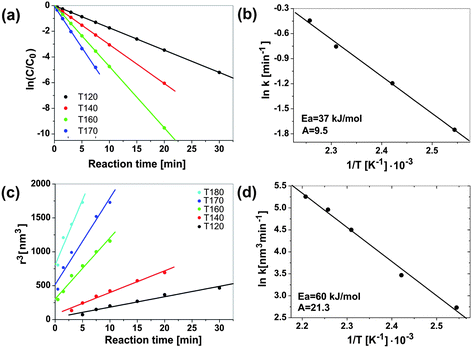 | ||
| Fig. 8 Benzyl acetate formation: (a) plot of ln(C/C0) vs. time for benzyl acetate formation for different times (fitted to a first order rate law); and (b) corresponding Arrhenius plot ln kesterificationvs. 1/T. ZnO crystal growth: (c) crystal radius r3vs. reaction time (according to Lifshitz–Slyozov–Wagner); and (d) corresponding Arrhenius plot ln kgrowthvs. 1/T. Figure partly reproduced from ref. 133 with permission of the American Chemical Society. | ||
Discussion of these kinetic and thermodynamic data in relation to other results presented in the literature is difficult due to the lack of comparable investigations. Nevertheless, it is interesting to see that the activation energy of 37 kJ mol−1 for benzyl acetate formation in the zinc acetate–benzyl alcohol system corresponds to the values of 30–45 kJ mol−1, which were reported for zeolite-catalyzed esterification reactions of benzyl alcohol with acetic acid.187 More conclusions can be drawn from a comparison of the syntheses of ZnO in the microwave reactor with conventional heating in an oil bath (Fig. 9). According to Fig. 9a, microwave irradiation clearly affects the organic esterification reaction, i.e., the zinc acetate concentration decreases much faster during microwave heating. For microwave heating kesterification is 0.124 min−1—an order of magnitude larger than that for conventional heating (Fig. 9c).133 In addition to the esterification reaction, also the crystal growth is considerably enhanced by microwave heating (Fig. 9b). The corresponding rate constants kgrowth are 15.36 nm3 min−1 for microwave and 3.9 nm3 min−1 for conventional heating (Fig. 9d), which means that crystal growth under microwave irradiation is about four times faster.133 Although in this particular case all the data support the acceleration of both the esterification reaction and the crystal growth, nanoparticle formation in solution includes additional steps, such as dissolution of precursor; formation of intermediate polynuclear species, which then nucleate and grow; formation of amorphous intermediary particles, which then nucleate; or reorganization and agglomeration of primary particles. Each of these steps can be influenced by microwave irradiation,16 and selective heating in a homogeneous (monomer–solvent) or heterogeneous (nuclei–particles–solvent) system might lead to unique synthesis conditions. In the case of ZnO formation, the enhanced esterification reaction, which also results from the faster dissolution of the precursor molecules, should have great consequences for the nucleation. As a conclusion, one can say that microwave irradiation greatly accelerates ZnO formation by (a) facilitating the dissolution of zinc acetate as precursor in benzyl alcohol, (b) by increasing the rate constants for the esterification reaction, resulting in faster production of monomer and consequently in an earlier nucleation event, and (c) by increasing the rate constants for crystal growth. The fact that different species absorb microwave energy to varying extents means that they are at different “effective temperatures” relative to each other. Consequently, all the species in a reaction solution, which contribute to the formation of the oxide, differ in their microwave absorption behavior, and therefore all the molecular processes, including hydrolysis and condensation, will be affected by microwave irradiation.16 On the other hand, the study of ZnO formation in benzyl alcohol does not provide any information about the influence of microwave irradiation on the nucleation itself. But taking into account the fact that various (molecular and/or solid) species with potentially different microwave absorbing properties are involved in nucleation, an influence of microwaves can be expected. As a matter of fact, work by Serrano et al.,188 and more recently by Jhung et al. on the microwave-mediated synthesis of microporous materials has shown that microwave irradiation accelerates both the nucleation and growth, however with a more pronounced effect on the nucleation stage.189 In some cases, the growth of inorganic nanoparticles does not just proceed along the attachment of ions, atoms or molecules onto the surface of a growing nuclei, but involves particle-mediated processes such as oriented attachment or mesocrystal formation.190–192 It seems that these “non-classical” crystallization pathways are also influenced by microwave irradiation. Leite et al. demonstrated that the use of microwave heating drastically decreased the treatment time to obtain gadolinium-doped ceria nanorods through oriented attachment of the spherical building blocks.103 This observation was explained on the basis that oriented attachment was a statistical process and that microwave heating increased the effective collision rate. If it turns out that microwaves can in general be used to influence and control the oriented attachment process, then this technique would open exciting opportunities for the assembly of nanoparticles.
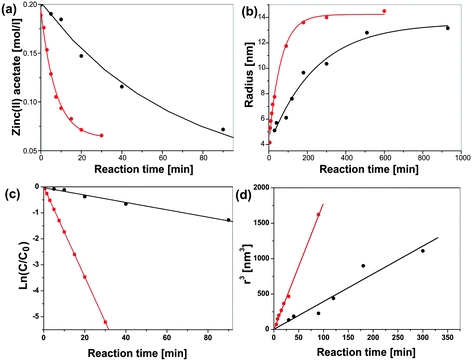 | ||
| Fig. 9 Comparison of (a) zinc acetate consumption (fitted with a pseudo first order kinetic model) and (b) evolution of the ZnO crystal size (fitted with a double exponential model) with reaction time under microwave irradiation and under conventional heating in an oil bath at 120 °C. Figure reproduced from ref. 133 with permission of the American Chemical Society. | ||
The formation of inorganic particles in liquid phase by nucleation and growth represents a heterogeneous process, i.e., the solid–liquid interface, and thus the sorption properties of the solid, should play a dominant role.16 In several publications Conner et al. addressed the question of the influence of microwaves on the sorption properties of oxides.193–195 The oxides studied, silica and silicalite zeolite, are nearly transparent to microwaves, which means that surface-adsorbed molecules will be heated selectively, resulting in higher temperatures at the surface, where sorption occurs, compared to the solid particle. Depending on the capability for absorbing microwave energy, different absorbates result in different local temperatures. It was found that the effective surface temperature increased when using adsorbates with higher permittivity.193 Taking into account how important the organic–inorganic interface is during nanoparticle formation,196 such studies cannot be overestimated. Although conclusive explanations why microwaves are able to provide such an immense enhancement of reaction rates during crystallization and growth of inorganic nanoparticles are still scarce, the concept that microwaves can change the “effective temperatures” of individual species or, in other words, that microwaves can selectively vary the relative intermediate energies of species in a reaction sequence, thus providing a more favorable reaction coordinate, is able to explain the increased reaction rates and selectivities. For a detailed discussion of this model the interested reader is referred to a recent paper by Conner et al., which summarizes the findings on the influence of microwaves on the chemistry at interfaces.195
4.3 Selective heating
The ability to absorb microwave energy can vary by orders of magnitude among different compounds and materials, enabling the selective heating of the species with the highest absorbing properties. This peculiar feature has been applied in catalysis to selectively heat the supported catalytic components, whereas the support itself often remained microwave transparent.197 Similar to microwave-assisted organic synthesis, there has also been an intense debate in the field of heterogeneous catalysis about the question, why microwave irradiation is able to increase the reaction rates and the product selectivity. Also here, theoretical and experimental studies showed that specific athermal effects are implausible and enhancements of reaction rates and product selectivity under microwave conditions must be attributed to thermal effects which result from differences between the local temperature at the reaction sites and the overall average temperature.197,198 As a matter of fact, it was shown that remarkable temperature gradients or hot-spots exist within the catalyst beds.199 In addition to catalysis, selective heating is involved in many other applications, in which one material has to be selectively heated, while the adjacent component must stay cool due to temperature intolerance, e.g., microwave curing of adhesives or processing of surface coatings on sheets, fibers, or fabrics.200 Of course, this effect bears interesting opportunities for nanoparticle research. For example, it was shown that microwaves can be used for the efficient purification of single-walled carbon nanotubes.201 The residual metal catalysts are selectively heated, leading to both the oxidation and rupturing of the carbon passivation layer over the metal particles, so that a mild subsequent acid treatment is enough to remove them from the sample. Similarly, selective heating of iron nanoparticles by microwave irradiation enables the patterned growth of carbon nanofibers and nanotubes.202 The short reaction times are impressive, because only a few minutes of irradiation were enough to get the carbon nanostructures with lengths of several micrometres. Selective heating should also be a powerful tool to activate the surface of a substrate, whether it is a flat substrate or a curved particle, and to deposit another material on top of it. As a matter of fact, several papers reported the microwave-assisted solution deposition of inorganic nanoparticles such as CdS,203 indium tin oxide,204 ZnO,205 TiO2,206 BaWO4,111 Eu:YVO4,207 or various ferrites132 on conducting glass substrates or silicon. Also core–shell nanoparticles have been prepared in the microwave by coating preformed nanoparticles with another material, e.g., CdTe/CdS,144 CdSe/ZnS,145 Ni/Fe3O4,132 or Au@Ag.208 However, whether these examples involve selective heating or not, and whether microwaves are particularly suitable to deposit a material on another one remains to be answered in future studies.Many nonpolar solvents are very popular in organic synthesis as well as in nanoparticle preparation, but due to their low absorbing properties they are basically excluded from the microwave technique. To overcome this problem without switching to a polar solvent, a small quantity of a strongly microwave absorbing solvent or salt can be added to an otherwise low absorbing medium. Ionic liquids are particularly suitable as co-solvents for increasing the absorbance level. In comparison to heterogeneous systems discussed above, leading to local differences in temperature, the situation in homogeneous mixtures of solvents is insofar different that the “hot” molecules immediately dissipate their energy to the surrounding solvent molecules.
Nonpolar solvents without polar additives can be applied in cases, in which the inorganic precursor or the forming nanoparticles are strongly microwave absorbing species. A few years ago Gerbec et al. published a detailed study on the microwave-assisted synthesis of semiconducting nanoparticles such as InGaP, InP and CdSe.209 The authors investigated the influence of the type of reactants, the applied power, the reaction time and temperature on the quality of the microwave-generated nanomaterials. They proposed that nanoparticle formation and growth is initiated by selective microwave heating of polar constituents in the reaction, resulting in elevated local temperatures. The selective heating of precursors, intermediates, or nanoparticles relative to the solvent should make it possible to achieve higher temperatures in the microwave than by conventional heating. The use of nonpolar solvents together with polar precursors and surfactants is very common in the synthesis of semiconducting nanoparticles,48,210 and therefore the combination of such routes with microwave chemistry will reveal additional synergies. The first results in this direction were recently presented by Washington and Strouse for the microwave synthesis of CdS and CdSe quantum dots.211,212 Both approaches made use of the high microwave absorbing properties of the trioctyl phosphine-containing precursors, enabling triggering of nucleation and control over crystal growth. These examples give a first impression of the fascinating possibilities that microwaves might have in selectively activating different precursors in a reaction mixture.
Selected heating effects similar to heterogeneous media (i.e., producing thermal gradients) come into play, when bi- or multiphasic liquid/liquid systems are irradiated. Considering how many synthesis methods for inorganic nanoparticles rely on such approaches,52,213 also here many more surprising results can be expected in the future.214,215
4.4 Size and shape control
Since the pioneering work of LaMer216 it is known that separation of nucleation and growth is required to obtain uniform colloidal particles with a narrow size distribution. This concept has also been adopted in nanoparticle synthesis. To prepare highly uniform nanocrystals it is necessary to induce a single nucleation event and to prevent any additional nucleation during the subsequent growth process.60 It is obvious that both the temperature of the reaction batch as well as the way of heating strongly influence nucleation and growth, and thus the crystal size and shape of the final product. Inhomogeneities in the growth process can be amplified by thermal gradients in the reaction mixture, thus producing poor nucleation processes and consequently broad size distributions. In this regard, dielectric volumetric heating, able to heat a reaction mixture very homogeneously and very rapidly, should provide ideal conditions for the preparation of nanoparticles. This assumption was confirmed in the case of II–VI and III–V quantum dot synthesis.209 The use of microwaves not only eliminated any thermal gradients, but also made superfluous the need for high-temperature injection for size focusing. Excellent control over nucleation and growth, which followed a La Mer scheme, was also reported for the microwave-assisted synthesis of NaYF4 nanocrystals.217 The authors found that the differently doped nanocrystals were of high quality regarding all relevant properties as compared with the corresponding nanoparticles prepared with other methods. Some of the samples showed a more than 30-fold increase in luminescence intensity.217The simplest way to control the crystal size by microwaves is through the irradiation time. The observation that prolonged irradiation time also increases the particle size seems to be rather general in microwave-directed nanoparticle syntheses. In the case of CoO and ZnO nanoparticle synthesis in benzyl alcohol, the crystal size continuously increases with irradiation time.33,133 XRD analysis (Fig. 10a) of the CoO samples obtained after different reaction times in the range of 30 s to 20 min revealed that already after 30 s the main reflections of CoO are present. After one minute, the CoO sample is phase-pure and longer heat treatment just results in crystallite growth from about 5 to 8 nm, which can easily be monitored by the narrowing of the 200 reflection. Furthermore, at the same temperature the yield increases with reaction time from about 60% to nearly 80%. A similar behaviour was found for ZnO nanoparticles synthesized from zinc acetate and benzyl alcohol. Fig. 10b nicely illustrates the relation between the yield of formed ZnO powder (black curve), crystal size (green curve) and zinc acetate concentration (red curve) at 120 °C for different irradiation times. It takes about five minutes of irradiation time to obtain phase-pure ZnO nanoparticles with crystal sizes of 8–9 nm (Fig. 10b, green curve). Between 5 and 35 min of irradiation time, the crystal size grows continuously to 15–16 nm, while zinc acetate is consumed due to the esterification reaction occurring parallel to ZnO formation. Its concentration drops from the initial concentration to about 35–40% within 18 min, from when on it remains constant (Fig. 10b, red curve). The yield of ZnO increases with time, reaching a maximum value of about 80% after about 18 min and then also remains constant. After about 18 min the system reaches a dynamic equilibrium between the dissolved monomer molecules and the solid metal oxide, whereas the crystal size still increases with irradiation time, indicating that Ostwald ripening becomes the dominant growth mode for longer reaction times.
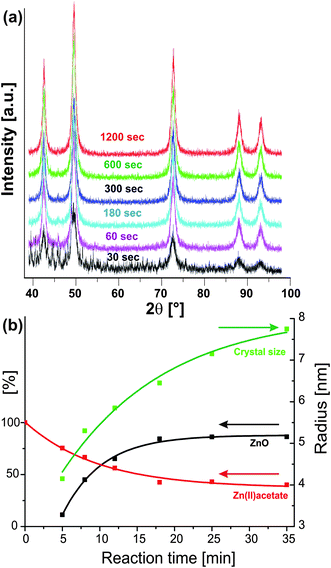 | ||
| Fig. 10 (a) X-Ray diffraction patterns of CoO nanoparticles obtained from the reaction of cobalt acetate with benzyl alcohol after different reaction times of 30 s to 20 min (reproduced from ref. 33 with permission of the Royal Society of Chemistry). (b) Plot of the time-dependent evolution of crystal size (green curve), yield of ZnO powder (black curve), and zinc acetate concentration (red curve) obtained in the zinc acetate–benzyl alcohol system at 120 °C (reproduced from ref. 133 with permission of the American Chemical Society). | ||
Another example of size control, although on a higher hierarchical level, was reported for monodisperse ZnO colloidal nanocrystal clusters.131 The reaction of zinc acetate with the very microwave-sensitive solvent diethylene glycol leads to the formation of spherical ZnO nanocrystals, which spontaneously assemble into spherical superlattices (cf.Fig. 6e and f). The size of these nanoparticle clusters can be varied from 57 to 274 nm by adjusting the precursor concentration. Interestingly, these raspberry-like agglomerates show a single-crystalline scattering behavior, proving that the nanoparticulate building blocks are crystallographically highly aligned with respect to each other. The authors proposed that the microwaves induce “hot surfaces” on the ZnO nuclei, speeding up the nanocrystal growth and the assembly into the clusters.131
Most of the examples in literature about the size and shape control of nanoparticles using microwaves rely on changing “classical” synthesis parameters like precursor concentration or precursor-to-additive ratio rather than microwave-specific features. Hu and Yu for example reported the continuous aspect-ratio tuning of hematite nanocrystals.94 Forced hydrolysis of FeCl3 under microwave conditions and in the presence of NH4H2PO4 yielded monodispersed elongated spindles (Fig. 6a-c). The aspect ratio depended on the ammonium phosphate concentration. Analogously, Kundu et al. achieved gold nanoparticles with different shapes through variation of the reactant concentrations (Fig. 5a),77 and Polshettiwar et al. reported the self-assembly of metal oxides into various 3D nanostructures (Fig. 11) by changing the precursor concentration and the reaction temperature.218
 | ||
| Fig. 11 SEM images of various metal oxide nanostructures (reproduced from ref. 218 with permission of the American Chemical Society). | ||
Nyutu et al. recently showed that the microwave frequency and the bandwidth sweep time can, as a matter of fact, have an effect on the phase purity, particle size, morphology, surface area and porosity of BaTiO3 nanoparticles.116 Nevertheless, it is obvious that the size- and shape-controlled synthesis of inorganic nanoparticles using microwave-related features is still in its infancy and the full potential has not yet been tapped at all. We are still far away from reaching the sophistication of morphology control as for example provided by the widely popular non-hydrolytic colloidal routes.48 But the combination of microwave chemistry with such state-of-the-art synthesis approaches should provide an immensely versatile tool for the fabrication of compositionally, morphologically and also structurally complex nanomaterials.
4.5 Energy and green aspects of dielectric heating
A rapidly growing part of nanoparticle synthesis technology is dedicated to the development of resource- and environmentally friendlier processes. Of particular importance is a reduction in the amounts of solvents and hazardous substances required, and the more efficient use of energy.71,219,220 Synthetic chemists have neglected alternative heating sources for chemical reactions for a long time. Most of the time, they still rely on isomantles, oil, sand and water baths or heat guns. The intensive research efforts in the last few years have produced a rapidly growing number of daily life products based on nanotechnology. With shorter reaction times, yet producing higher yields, the microwave technique has the potential to greatly contribute to green chemistry.221,222 Nevertheless, too much optimism has to be avoided, because microwave chemistry is usually applied to small amounts, neglecting the problems that might arise during scale-up from mmol to kmol.21 Another energy-related issue is the fact that often the microwave power applied is unnecessarily large (i.e., more energy is introduced to the system than thermodynamically required), lowering the efficiency factor to 0.05–0.01. It is not surprising that a reaction volume of 2.5 ml can rapidly be heated when irradiated with 250 W microwave power. A comparable power is used in conventional heating mantles for much larger volumes. Furthermore, the conversion of electric energy into microwave energy has an efficiency of only 0.5 to 0.65.21 Although microwave-assisted synthesis procedures are generally claimed to be very energy efficient, this topic has barely been scientifically explored. As a matter of fact, two recent studies have shown that microwave heating is not only often less energy-efficient than conventional processing,223 but even sometimes a rather inefficient process.224 Microwave heating has to provide substantial advantages in comparison to conventional heating to be competitive. The rate enhancement, the yield, or the selectivity must significantly be improved, or the properties of the final product have to be unique.5. Conclusions and outlook
Without any doubt, the use of microwave chemistry opens up unique opportunities for nanomaterials' synthesis that cannot be provided by any other synthesis technique. Although potential benefits of microwave irradiation as a heating tool have mainly been established in the context of organic synthesis, it can be expected that similar effects and advantages will also greatly contribute to the development of improved synthesis protocols for nanomaterials. Currently, rate enhancement as a consequence of the efficient and fast heating process of a reaction mixture by microwaves represents the most important benefit, significantly shortening the reaction times. Although such time-saving approaches are highly desired, we believe that the greatest potential of microwave-assisted syntheses lies in the possibility to selectively heat compounds with different microwave-absorbing properties. Control over local temperatures in a homogeneous (to the naked eye) or heterogeneous system should make it possible to selectively deposit one material on top of another one (core–shell particles, thin film deposition on substrates), to grow nanoparticles within a non-absorbing material (in situ formation of nanocomposites or nanoparticle formation within emulsion droplets), or to increase the reactivity of polar species, thus improving the selectivity of chemical reactions. Most of these issues have hardly been addressed yet. Therefore it can be expected that in the next few years unprecedented, compositionally complex multi-component materials will be synthesized by using the unique features of microwave chemistry. On the other hand, fundamental principles have not yet been established and microwave-assisted routes are, as most of the other synthesis strategies for inorganic nanomaterials, mainly based on trial-and-error experiments. Questions about the influence of microwaves on nucleation and growth of inorganic nanomaterials remain to be answered. The in situ study of nanoparticle formation under microwave irradiation will help to solve some of these open problems. Methods for the measurement of local temperatures with high resolution have to be developed to understand the effect of selective heating, and to take advantage of it in a rational way. Energy efficiency of microwave approaches and their scale-up to industrial quantities have to be addressed. In conclusion, one can say that there are still a lot of controversial and challenging topics around in the field of microwave chemistry, but at the same time, we can expect innovative ideas and input from this fascinating area of research, thus setting the basis for new concepts in the broad field of Nanoscience and Nanotechnology.Acknowledgements
Financial support by ETH Zürich and the Swiss National Science Foundation (Project No. 200021_124632) is gratefully acknowledged. We thank Martin Kubli for proof-reading of the manuscript.References
- B. L. Hayes, Microwave Synthesis: Chemistry at the Speed of Light, CEM Publishing, Matthews, 2002 Search PubMed.
- R. Gedye, F. Smith, K. Westaway, H. Ali, L. Baldisera, L. Laberge and J. Rousell, Tetrahedron Lett., 1986, 27, 279–282 CrossRef CAS.
- R. J. Giguere, T. L. Bray, S. M. Duncan and G. Majetich, Tetrahedron Lett., 1986, 27, 4945–4948 CrossRef CAS.
- A. K. Bose, M. S. Manhas, B. K. Banik and E. W. Robb, Res. Chem. Intermed., 1994, 20, 1–11 CrossRef CAS.
- C. R. Strauss and R. W. Trainor, Aust. J. Chem., 1995, 48, 1665–1692 CrossRef CAS.
- L. Perreux and A. Loupy, Tetrahedron, 2001, 57, 9199–9223 CrossRef CAS.
- C. O. Kappe, Angew. Chem., Int. Ed., 2004, 43, 6250–6284 CrossRef CAS.
- A. de la Hoz, A. Diaz-Ortiz and A. Moreno, Chem. Soc. Rev., 2005, 34, 164–178 RSC.
- C. O. Kappe, Chimia, 2006, 60, 308–312 CrossRef CAS.
- C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 71–193 Search PubMed.
- C. O. Kappe, D. Dallinger and S. S. Murphree, Practical Microwave Synthesis for Organic Chemists, Wiley-VCH: Weinheim, 2009 Search PubMed.
- K. J. Rao, B. Vaidhyanathan, M. Ganguli and P. A. Ramakrishnan, Chem. Mater., 1999, 11, 882–895 CrossRef CAS.
- M. Rajamathi and R. Seshadri, Curr. Opin. Solid State Mater. Sci., 2002, 6, 337–345 CrossRef CAS.
- S. Komarneni, Curr. Sci., 2003, 85, 1730–1734 CAS.
- S. Z. Shi and J.-Y. Hwang, J. Mineral. Mater. Charact. Eng., 2003, 2, 101–110 Search PubMed.
- G. A. Tompsett, W. C. Conner and K. S. Yngvesson, ChemPhysChem, 2006, 7, 296–319 CrossRef CAS.
- C. Leonelli and W. Lojkowski, Chem. Today, 2007, 25, 34–38 Search PubMed.
- V. Polshettiwar, M. N. Nadagouda and R. S. Varma, Aust. J. Chem., 2009, 62, 16–26 CrossRef CAS.
- S. Balaji, D. Mutharasu, N. S. Subramanian and K. Ramanathan, Ionics, 2009, 15, 765–777 CrossRef CAS.
- C. Gabriel, S. Gabriel, E. H. Grant, B. S. J. Halstead and D. M. P. Mingos, Chem. Soc. Rev., 1998, 27, 213–223 RSC.
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC.
- D. A. C. Stuerga and P. Gaillard, J. Microwave Power Electromagn. Energy, 1996, 31, 87–100.
- D. A. C. Stuerga and P. Gaillard, J. Microwave Power Electromagn. Energy, 1996, 31, 101–113.
- D. M. P. Mingos and D. R. Baghurst, Chem. Soc. Rev., 1991, 20, 1–47 RSC.
- D. Jezequel, J. Guenot, N. Jouini and F. Fievet, J. Mater. Res., 1995, 10, 77–83 CrossRef CAS.
- C. Feldmann and H. O. Jungk, Angew. Chem., Int. Ed., 2001, 40, 359–362 CrossRef CAS.
- C. Feldmann, Adv. Funct. Mater., 2003, 13, 101–107 CrossRef CAS.
- M. Niederberger and G. Garnweitner, Chem.–Eur. J., 2006, 12, 7282–7302 CrossRef CAS.
- N. Pinna and M. Niederberger, Angew. Chem., Int. Ed., 2008, 47, 5292–5304 CrossRef CAS.
- I. Djerdj, D. Arcon, Z. Jaglicic and M. Niederberger, J. Solid State Chem., 2008, 181, 1571–1581 CrossRef CAS.
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582–596 CrossRef CAS.
- I. Bilecka and M. Niederberger, Electrochim. Acta, 2010 DOI:10.1016/j.electacta.2009.1012.1066.
- I. Bilecka, I. Djerdj and M. Niederberger, Chem. Commun., 2008, 886–888 RSC.
- S. A. Galema, Chem. Soc. Rev., 1997, 26, 233–238 RSC.
- N. Kuhnert, Angew. Chem., Int. Ed., 2002, 41, 1863–1866 CrossRef CAS.
- D. Obermayer, B. Gutmann and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 8321–8324 CrossRef CAS.
- B. Ondruschka and W. Bonrath, Chimia, 2006, 60, 326–329 CrossRef CAS.
- G. A. Tompsett, B. Panzarella, W. C. Conner, K. S. Yngvesson, F. Lu, S. L. Suib, K. W. Jones and S. Bennett, Rev. Sci. Instrum., 2006, 77, 124101 CrossRef.
- G. R. Robb, A. Harrison and A. G. Whittaker, PhysChemComm, 2002, 5, 135–137 RSC.
- B. Panzarella, G. Tompsett, W. C. Conner and K. Jones, ChemPhysChem, 2007, 8, 357–369 CrossRef CAS.
- A. G. Whittaker, A. Harrison, G. S. Oakley, I. D. Youngson, R. K. Heenan and S. M. King, Rev. Sci. Instrum., 2001, 72, 173–176 CrossRef CAS.
- A. Harrison, R. Ibberson, G. Robb, G. Whittaker, C. Wilson and D. Youngson, Faraday Discuss., 2002, 122, 363–379 Search PubMed.
- N. E. Leadbeater and J. R. Schmink, Nat. Protoc., 2008, 3, 1–7 Search PubMed.
- J. R. Schmink, J. L. Holcomb and N. E. Leadbeaterr, Chem.–Eur. J., 2008, 14, 9943–9950 CrossRef CAS.
- J. R. Schmink, J. L. Holcomb and N. E. Leadbeater, Org. Lett., 2009, 11, 365–368 CrossRef CAS.
- G. R. Patzke, F. Krumeich and R. Nesper, Angew. Chem., Int. Ed., 2002, 41, 2446–2461 CrossRef.
- C. Burda, X. Chen, R. Narayanan and M. A. El-Sayed, Chem. Rev., 2005, 105, 1025–1102 CrossRef CAS.
- Y. W. Jun, J. S. Choi and J. Cheon, Angew. Chem., Int. Ed., 2006, 45, 3414–3439 CrossRef CAS.
- A. Gurav, T. Kodas, T. Pluym and Y. Xiong, Aerosol Sci. Technol., 1993, 19, 411–452 CAS.
- F. E. Kruis, H. Fissan and A. Peled, J. Aerosol Sci., 1998, 29, 511–535 CrossRef CAS.
- R. Strobel and S. E. Pratsinis, J. Mater. Chem., 2007, 17, 4743–4756 RSC.
- B. L. Cushing, V. L. Kolesnichenko and C. J. O'Connor, Chem. Rev., 2004, 104, 3893–3946 CrossRef CAS.
- X. Wang and Y. Li, Chem. Commun., 2007, 2901–2910 RSC.
- C. N. R. Rao, S. R. C. Vivekchand, K. Biswasa and A. Govindaraja, Dalton Trans., 2007, 3728–3749 RSC.
- S. G. Kwon and T. Hyeon, Acc. Chem. Res., 2008, 41, 1696–1709 CrossRef CAS.
- W. T. Yao and S. H. Yu, Adv. Funct. Mater., 2008, 18, 3357–3366 CrossRef.
- D. S. Wang, T. Xie and Y. Li, Nano Res., 2009, 2, 30–46 Search PubMed.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- L. L. Hench and J. K. West, Chem. Rev., 1990, 90, 33–72 CrossRef CAS.
- J. Park, J. Joo, S. G. Kwon, Y. Jang and T. Hyeon, Angew. Chem., Int. Ed., 2007, 46, 4630–4660 CrossRef CAS.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS.
- K. Byrappa and T. Adschiri, Prog. Cryst. Growth Charact. Mater., 2007, 53, 117–166 CrossRef CAS.
- K. S. Suslick and G. J. Price, Annu. Rev. Mater. Sci., 1999, 29, 295–326 CrossRef CAS.
- A. Gedanken, Ultrason. Sonochem., 2004, 11, 47–55 CrossRef CAS.
- A. Huczko, Appl. Phys. A: Mater. Sci. Process., 2000, 70, 365–376 CrossRef CAS.
- L. A. Estroff and A. D. Hamilton, Chem. Mater., 2001, 13, 3227–3235 CrossRef CAS.
- C. Sanchez, H. Arribart and M. M. Giraud Guille, Nat. Mater., 2005, 4, 277–288 CrossRef CAS.
- Y. Oaki and H. Imai, J. Mater. Chem., 2007, 17, 316–321 RSC.
- R. L. Brutchey and D. E. Morse, Angew. Chem., Int. Ed., 2006, 45, 6564–6566 CrossRef CAS.
- B. Bhushan, Philos. Trans. R. Soc. London, Ser. A, 2009, 367, 1445–1486 CrossRef CAS.
- Y. Mao, T. J. Park, F. Zhang, H. Zhou and S. S. Wong, Small, 2007, 3, 1122–1139 CrossRef CAS.
- K. Lu, Int. Mater. Rev., 2008, 53, 21–38 Search PubMed.
- S. Das, A. K. Mukhopadhyay, S. Datta and D. Basu, Bull. Mater. Sci., 2009, 32, 1–13 Search PubMed.
- A. G. Whittaker and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 2000, 1521–1526 RSC.
- A. G. Whittaker and D. M. P. Mingos, J. Chem. Soc., Dalton Trans., 2002, 3967–3970 RSC.
- Y. L. Luo, Mater. Lett., 2007, 61, 1873–1875 CrossRef CAS.
- S. Kundu, L. H. Peng and H. Liang, Inorg. Chem., 2008, 47, 6344–6352 CrossRef CAS.
- F. K. Liu, C. J. Ker, Y. C. Chang, F. H. Ko, T. C. Chu and B. T. Dai, Jpn. J. Appl. Phys., 2003, 42, 4152–4158 CrossRef CAS.
- V. K. Sharma, R. A. Yngard and Y. Lin, Adv. Colloid Interface Sci., 2009, 145, 83–96 CrossRef CAS.
- J. Chen, J. Wang, X. Zhang and Y. L. Jin, Mater. Chem. Phys., 2008, 108, 421–424 CrossRef CAS.
- B. Hu, S. B. Wang, K. Wang, M. Zhang and S. H. Yu, J. Phys. Chem. C, 2008, 112, 11169–11174 CrossRef CAS.
- D. S. Li and S. Komarneni, J. Am. Ceram. Soc., 2006, 89, 1510–1517 CrossRef CAS.
- L. F. Gou, M. Chipara and J. M. Zaleski, Chem. Mater., 2007, 19, 1755–1760 CrossRef CAS.
- M. Tsuji, M. Hashimoto, Y. Nishizawa, M. Kubokawa and T. Tsuji, Chem.–Eur. J., 2005, 11, 440–452 CrossRef CAS.
- M. Tsuji, N. Miyamae, M. Hashimoto, M. Nishio, S. Hikino, N. Ishigami and I. Tanaka, Colloids Surf., A, 2007, 302, 587–598 CrossRef CAS.
- M. Tsuji, K. Matsumoto, P. Jiang, R. Matsuo, X. L. Tang and K. S. N. Karnarudin, Colloids Surf., A, 2008, 316, 266–277 CrossRef CAS.
- K. Patel, S. Kapoor, D. P. Dave and T. Mukherjee, Res. Chem. Intermed., 2006, 32, 103–113 CrossRef CAS.
- H. J. Jiang, K. S. Moon, Z. Q. Zhang, S. Pothukuchi and C. P. Wong, J. Nanopart. Res., 2006, 8, 117–124 CrossRef CAS.
- L. Z. Ren, L. J. Meng, Q. H. Lu, Z. F. Fei and P. J. Dyson, J. Colloid Interface Sci., 2008, 323, 260–266 CrossRef CAS.
- S. Komarneni, R. Roy and Q. H. Li, Mater. Res. Bull., 1992, 27, 1393–1405 CrossRef CAS.
- S. Komarneni, Q. H. Li and R. Roy, J. Mater. Chem., 1994, 4, 1903–1906 RSC.
- S. Komarneni, R. Pidugu, Q. H. Li and R. Roy, J. Mater. Res., 1995, 10, 1687–1692 CAS.
- Q. Li and Y. Wei, Mater. Res. Bull., 1998, 33, 779–782 CrossRef CAS.
- X. L. Hu and J. C. Yu, Adv. Funct. Mater., 2008, 18, 880–887 CrossRef CAS.
- S. Komarneni, M. C. D'Arrigo, C. Leonelli, G. C. Pellacani and H. Katsuki, J. Am. Ceram. Soc., 1998, 81, 3041–3043 CAS.
- X. H. Liao, J. J. Zhu, W. Zhong and H. Y. Chen, Mater. Lett., 2001, 50, 341–346 CrossRef CAS.
- J. F. Huang, C. K. Xia, L. Y. Cao and X. R. Zeng, Mater. Sci. Eng., B, 2008, 150, 187–193 CrossRef CAS.
- Y. Zhao, J. J. Zhu, J. M. Hong, N. S. Bian and H. Y. Chen, Eur. J. Inorg. Chem., 2004, 4072–4080 CrossRef CAS.
- K. M. Wang, T. Huang, H. F. Liu, Y. X. Zhao, H. M. Liu and C. T. Sun, Colloids Surf., A, 2008, 325, 21–25 CrossRef CAS.
- M. Zawadzki, J. Alloys Compd., 2008, 451, 297–300 CrossRef CAS.
- Y. Tao, F. H. Gong, H. Wang, H. P. Wu and G. L. Tao, Mater. Chem. Phys., 2008, 112, 973–976 CrossRef CAS.
- H. M. Yang, C. H. Huang, A. D. Tang, X. C. Zhang and W. G. Yang, Mater. Res. Bull., 2005, 40, 1690–1695 CrossRef CAS.
- M. Godinho, C. Ribeiro, E. Longo and E. R. Leite, Cryst. Growth Des., 2008, 8, 384–386 CrossRef CAS.
- C. R. Patra and A. Gedanken, New J. Chem., 2004, 28, 1060–1065 RSC.
- J. Jouhannaud, J. Rossignol and D. Stuerga, J. Solid State Chem., 2008, 181, 1439–1444 CrossRef CAS.
- G. C. Xi, Y. T. He, Q. Zhang, H. Q. Xiao, X. Wang and C. Wang, J. Phys. Chem. C, 2008, 112, 11645–11649 CrossRef CAS.
- S. A. Eliziario, L. S. Cavalcante, J. C. Sczancoski, P. S. Pizani, J. A. Varela, J. W. M. Espinosa and E. Longo, Nanoscale Res. Lett., 2009, 4, 1371–1379 Search PubMed.
- H. M. Zhang, J. B. Liu, H. Wang, W. X. Zhang and H. Yan, J. Nanopart. Res., 2008, 10, 767–774 CrossRef CAS.
- M. Zawadzki, Solid State Sci., 2006, 8, 14–18 CrossRef CAS.
- A. Michailovski, R. Kiebach, W. Bensch, J. D. Grunwaldt, A. Baiker, S. Komarneni and G. R. Patzke, Chem. Mater., 2007, 19, 185–197 CrossRef CAS.
- R. Wang, C. Liu, J. Zeng, K. W. Li and H. Wang, J. Solid State Chem., 2009, 182, 677–684 CrossRef CAS.
- J. H. Bi, L. Wu, H. Li, Z. H. Li, X. X. Wang and X. Z. Fu, Acta Mater., 2007, 55, 4699–4705 CrossRef CAS.
- A. J. Paula, R. Parra, M. A. Zaghete and J. A. Varela, Mater. Lett., 2008, 62, 2581–2584 CrossRef CAS.
- B. L. Newalkar, S. Komarneni and H. Katsuki, Mater. Res. Bull., 2001, 36, 2347–2355 CrossRef.
- W. A. Sun, C. H. Li, J. Q. Li and W. Liu, Mater. Chem. Phys., 2006, 97, 481–487 CrossRef CAS.
- E. K. Nyutu, C. H. Chen, P. K. Dutta and S. L. Suib, J. Phys. Chem. C, 2008, 112, 9659–9667 CrossRef CAS.
- J. M. Lee, D. P. Amalnerkar, Y. K. Hwang, S. H. Jhung, J. S. Hwang and J. S. Chang, J. Nanosci. Nanotechnol., 2007, 7, 952–959 CrossRef CAS.
- M. L. Moreira, E. C. Paris, G. S. do Nascimento, V. M. Longo, J. R. Sambrano, V. R. Mastelaro, M. I. B. Bernardi, J. Andres, J. A. Varela and E. Longo, Acta Mater., 2009, 57, 5174–5185 CrossRef CAS.
- M. L. Moreira, J. Andres, J. A. Varela and E. Longo, Cryst. Growth Des., 2009, 9, 833–839 CrossRef CAS.
- M. Niederberger and N. Pinna, Metal Oxide Nanoparticles in Organic Solvents: Synthesis, Formation, Assembly and Application, Springer: London, 2009 Search PubMed.
- T. Yamamoto, Y. Wada, H. B. Yin, T. Sakata, H. Mori and S. Yanagida, Chem. Lett., 2002, 964–965 CrossRef CAS.
- M. Zawadzki, J. Alloys Compd., 2008, 454, 347–351 CrossRef CAS.
- J. Giri, T. Sriharsha and D. Bahadur, J. Mater. Chem., 2004, 14, 875–880 RSC.
- W.-W. Wang, Mater. Chem. Phys., 2008, 108, 227–231 CrossRef CAS.
- O. Palchik, J. J. Zhu and A. Gedanken, J. Mater. Chem., 2000, 10, 1251–1254 RSC.
- E. Hammarberg, A. Prodi-Schwab and C. Feldmann, Thin Solid Films, 2008, 516, 7437–7442 CrossRef CAS.
- J. Fidelus, R. R. Piticescu, R. M. Piticescu, W. Lojkowski and L. Giurgiu, Z. Naturforsch., B: Chem. Sci., 2008, 63, 725–729 CAS.
- W. Lojkowski, A. Gedanken, E. Grzanka, A. Opalinska, T. Strachowski, R. Pielaszek, A. Tomaszewska-Grzeda, S. Yatsunenko, M. Godlewski, H. Matysiak and K. J. Kurzydowski, J. Nanopart. Res., 2009, 11, 1991–2002 CrossRef CAS.
- E. Hammarberg, A. Prodi-Schwab and C. Feldmann, J. Colloid Interface Sci., 2009, 334, 29–36 CrossRef CAS.
- C. Feldmann, Solid State Sci., 2005, 7, 868–873 CrossRef CAS.
- X. L. Hu, J. M. Gong, L. Z. Zhang and J. C. Yu, Adv. Mater., 2008, 20, 4845–4850 CrossRef CAS.
- I. Bilecka, M. Kubli, E. Amstad and M. Niederberger, J. Sol–Gel Sci. Technol., 2010 DOI:10.1007/s10971-010-2165-1.
- I. Bilecka, P. Elser and M. Niederberger, ACS Nano, 2009, 3, 467 CrossRef CAS.
- M. Antonietti, D. Kuang, B. Smarsly and Y. Zhou, Angew. Chem., Int. Ed., 2004, 43, 4988–4992 CrossRef CAS.
- A. Taubert and Z. Li, Dalton Trans., 2007, 723–727 RSC.
- D. S. Jacob, L. Bitton, J. Grinblat, I. Felner, Y. Koltypin and A. Gedanken, Chem. Mater., 2006, 18, 3162–3168 CrossRef.
- W. W. Wang and Y. J. Zhu, Inorg. Chem. Commun., 2004, 7, 1003–1005 CrossRef CAS.
- G. Bühler, D. Thölmann and C. Feldmann, Adv. Mater., 2007, 19, 2224–2227 CrossRef.
- K. L. Ding, Z. J. Miao, Z. M. Liu, Z. F. Zhang, B. X. Han, G. M. An, S. D. Miao and Y. Xie, J. Am. Chem. Soc., 2007, 129, 6362–6363 CrossRef CAS.
- S. W. Cao, Y. J. Zhu, G. F. Cheng and Y. H. Huang, J. Hazard. Mater., 2009, 171, 431–435 CrossRef CAS.
- X. D. Xu, M. Zhang, J. Feng and M. L. Zhang, Mater. Lett., 2008, 62, 2787–2790 CrossRef CAS.
- G. Bühler, A. Zharkouskaya and C. Feldmann, Solid State Sci., 2008, 10, 461–465 CrossRef.
- H. F. Qian, X. Qiu, L. Li and J. C. Ren, J. Phys. Chem. B, 2006, 110, 9034–9040 CrossRef CAS.
- Y. He, H. T. Lu, L. M. Sai, W. Y. Lai, Q. L. Fan, L. H. Wang and W. Huang, J. Phys. Chem. B, 2006, 110, 13370–13374 CrossRef.
- J. Ziegler, A. Merkulov, M. Grabolle, U. Resch-Genger and T. Nann, Langmuir, 2007, 23, 7751–7759 CrossRef CAS.
- Y. Zhao, J. M. Hong and J. J. Zhu, J. Cryst. Growth, 2004, 270, 438–445 CrossRef CAS.
- M. Q. Chu, X. Y. Shen and G. J. Liu, Nanotechnology, 2006, 17, 444–449 CrossRef CAS.
- H. F. Qian, L. Li and J. C. Ren, Mater. Res. Bull., 2005, 40, 1726–1736 CrossRef CAS.
- L. Li, H. F. Qian and J. Ren, Chem. Commun., 2005, 528–530 RSC.
- J. J. Zhu, O. Palchik, S. G. Chen and A. Gedanken, J. Phys. Chem. B, 2000, 104, 7344–7347 CrossRef CAS.
- R. Kerner, O. Palchik and A. Gedanken, Chem. Mater., 2001, 13, 1413–1419 CrossRef CAS.
- O. Palchik, R. Kerner, A. Gedanken, A. M. Weiss, M. A. Slifkin and V. Palchik, J. Mater. Chem., 2001, 11, 874–878 RSC.
- C. R. Patra, A. Odani, V. G. Pol, D. Aurbach and A. Gedanken, J. Solid State Electrochem., 2007, 11, 186–194 CAS.
- C. R. Patra, S. Patra, A. Gabashvili, Y. Mastai, Y. Koltypin, A. Gedanken, V. Palchik and M. A. Slifkin, J. Nanosci. Nanotechnol., 2006, 6, 845–851 CrossRef CAS.
- R. Harpeness and A. Gedanken, New J. Chem., 2003, 27, 1191–1193 RSC.
- R. Harpeness, A. Gedanken, A. M. Weiss and M. A. Slifkin, J. Mater. Chem., 2003, 13, 2603–2606 RSC.
- D. Kim, A. Gedanken, Y. S. Tver'yanovich, D. W. Lee and B. K. Kim, Mater. Lett., 2006, 60, 2807–2809 CrossRef CAS.
- H. Grisaru, V. G. Pol, A. Gedanken and I. Nowik, Eur. J. Inorg. Chem., 2004, 1859–1864 CrossRef CAS.
- H. Grisaru, O. Palchik, A. Gedanken, V. Palchik, M. A. Slifkin and A. M. Weiss, Inorg. Chem., 2003, 42, 7148–7155 CrossRef CAS.
- H. Grisaru, O. Palchik, A. Gedanken, V. Palchik, M. A. Slifkin, A. N. Weiss and Y. R. Hacohen, Inorg. Chem., 2001, 40, 4814–4815 CrossRef CAS.
- X. H. Liao, J. J. Zhu and H. Y. Chen, Mater. Sci. Eng., B, 2001, 85, 85–89 CrossRef.
- A. B. Panda, G. Glaspell and M. S. El-Shall, J. Am. Chem. Soc., 2006, 128, 2790–2791 CrossRef CAS.
- A. V. Murugan, R. S. Sonawane, B. B. Kale, S. K. Apte and A. V. Kulkarni, Mater. Chem. Phys., 2001, 71, 98–102 CrossRef.
- Y. Jiang and Y. J. Zhu, J. Phys. Chem. B, 2005, 109, 4361–4364 CrossRef CAS.
- C. R. Patra, G. Alexandra, S. Patra, D. S. Jacob, A. Gedanken, A. Landau and Y. Gofer, New J. Chem., 2005, 29, 733–739 RSC.
- G. Bühler and C. Feldmann, Angew. Chem., Int. Ed., 2006, 45, 4864–4867 CrossRef.
- S. H. Jhung, J. W. Yoon, J. S. Hwang, A. K. Cheetham and J. S. Chang, Chem. Mater., 2005, 17, 4455–4460 CrossRef CAS.
- J. W. Fergus, J. Power Sources, 2010, 195, 939–954 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Electrochem. Commun., 2008, 10, 903–906 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, J. Phys. Chem. C, 2008, 112, 14665–14671 CrossRef.
- T. Muraliganth, A. V. Murugan and A. Manthiram, J. Mater. Chem., 2008, 18, 5661–5668 RSC.
- I. Bilecka, A. Hintennach, I. Djerdj, P. Novak and M. Niederberger, J. Mater. Chem., 2009, 19, 5125–5128 RSC.
- A. Arafat, J. C. Jansen, A. R. Ebaid and H. Vanbekkum, Zeolites, 1993, 13, 162–165 CAS.
- C. S. Cundy, Collect. Czech. Chem. Commun., 1998, 63, 1699–1723 CrossRef CAS.
- Y. H. Xie, S. Yin, H. Yamane, T. Hashimoto, H. Machida and T. Sato, Chem. Mater., 2008, 20, 4931–4935 CrossRef CAS.
- Z. W. Quan, P. P. Yang, C. X. Li, J. Yang, D. M. Yang, Y. Jin, H. Z. Lian, H. Y. Li and J. Lin, J. Phys. Chem. C, 2009, 113, 4018–4025 CrossRef CAS.
- J. Yan, Z. J. Fan, T. Wei, J. Cheng, B. Shao, K. Wang, L. P. Song and M. L. Zhang, J. Power Sources, 2009, 194, 1202–1207 CrossRef CAS.
- C. T. Sousa, A. M. L. Lopes, M. P. Proenca, D. C. Leitao, J. G. Correia and J. P. Araujo, J. Nanosci. Nanotechnol., 2009, 9, 6084–6088 CrossRef CAS.
- T. Druzhinina, S. Hoeppener and U. S. Schubert, Adv. Funct. Mater., 2009, 19, 2819–2825 CrossRef CAS.
- L. Chen, C. Z. Yuan, B. Gao, S. Y. Chen and X. G. Zhang, J. Solid State Electrochem., 2009, 13, 1925–1933 CrossRef CAS.
- Y. He, H. T. Lu, L. M. Sai, Y. Y. Su, M. Hu, C. H. Fan, W. Huang and L. H. Wang, Adv. Mater., 2008, 20, 3416–3421 CrossRef CAS.
- J. Ziegler, S. Xu, E. Kucur, F. Meister, M. Batentschuk, F. Gindele and T. Nann, Adv. Mater., 2008, 20, 4068–4073 CrossRef CAS.
- P. D. Chu and F. G. Vartuli, 1988, US Patent 4,778,666.
- W. C. Conner, G. Tompsett, K. H. Lee and K. S. Yngvesson, J. Phys. Chem. B, 2004, 108, 13913–13920 CrossRef CAS.
- B. Panzarella, G. A. Tompsett, K. S. Yngvesson and W. C. Conner, J. Phys. Chem. B, 2007, 111, 12657–12667 CrossRef CAS.
- G. Garnweitner and M. Niederberger, J. Mater. Chem., 2008, 18, 1171–1182 RSC.
- S. R. Kirumakki, N. Nagaraju and S. Narayanan, Appl. Catal., A, 2004, 273, 1–9 CrossRef CAS.
- D. P. Serrano, M. A. Uguina, R. Sanz, E. Castillo, A. Rodriguez and P. Sanchez, Microporous Mesoporous Mater., 2004, 69, 197–208 CrossRef CAS.
- S. H. Jhung, T. H. Jin, Y. K. Hwang and J. S. Chang, Chem.–Eur. J., 2007, 13, 4410–4417 CrossRef CAS.
- H. Cölfen and M. Antonietti, Angew. Chem., Int. Ed., 2005, 44, 5576–5591 CrossRef.
- M. Niederberger and H. Cölfen, Phys. Chem. Chem. Phys., 2006, 8, 3271–3287 RSC.
- Q. Zhang, S. J. Liu and S. H. Yu, J. Mater. Chem., 2009, 19, 191–207 RSC.
- S. J. Vallee and W. C. Conner, J. Phys. Chem. B, 2006, 110, 15459–15470 CrossRef CAS.
- S. J. Vallee and W. C. Conner, J. Phys. Chem. C, 2008, 112, 15483–15489 CrossRef CAS.
- W. C. Conner and G. A. Tompsett, J. Phys. Chem. B, 2008, 112, 2110–2118 CrossRef CAS.
- Y. Yin and A. P. Alivisatos, Nature, 2005, 437, 664–670 CrossRef CAS.
- X. L. Zhang and D. O. Hayward, Inorg. Chim. Acta, 2006, 359, 3421–3433 CrossRef.
- F. Chemat, D. C. Esveld, M. Poux and J. L. Di-Martino, J. Microwave Power Electromagn. Energy, 1998, 33, 88–94.
- X. L. Zhang, D. O. Hayward and D. M. P. Mingos, Chem. Commun., 1999, 975–976 RSC.
- C. H. Stern, J. Microwave Power Electromagn. Energy, 1998, 33, 207–215.
- A. R. Harutyunyan, B. K. Pradhan, J. P. Chang, G. G. Chen and P. C. Eklund, J. Phys. Chem. B, 2002, 106, 8671–8675 CrossRef CAS.
- T. Druzhinina, W. Weltjens, S. Hoeppener and U. S. Schubert, Adv. Funct. Mater., 2009, 19, 1287–1292 CrossRef CAS.
- Y. Liu, T. Tan, B. Wang, R. Zhai, X. Song, E. Li, H. Wang and H. Yan, J. Colloid Interface Sci., 2008, 320, 540–547 CrossRef CAS.
- M. Okuya, N. Ito and K. Shiozaki, Thin Solid Films, 2007, 515, 8656–8659 CrossRef CAS.
- A. M. Peiro, C. Domingo, J. Peral, X. Domenech, E. Vigil, M. A. Hernandez-Fenollosa, M. Mollar, B. Mari and J. A. Ayllon, Thin Solid Films, 2005, 483, 79–83 CrossRef CAS.
- E. Vigil, L. Saadoun, J. A. Ayllon, X. Domenech, I. Zumeta and R. Rodriguez-Clemente, Thin Solid Films, 2000, 365, 12–18 CrossRef CAS.
- H. Y. Xu, H. Wang, T. N. Jin and H. Yan, Nanotechnology, 2005, 16, 65–69 CrossRef CAS.
- M. Tsuji, N. Miyamae, S. Lim, K. Kimura, X. Zhang, S. Hikino and M. Nishio, Cryst. Growth Des., 2006, 6, 1801–1807 CrossRef CAS.
- J. A. Gerbec, D. Magana, A. Washington and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 15791–15800 CrossRef CAS.
- C. de Mello Donega, P. Liljeroth and D. Vanmaekelbergh, Small, 2005, 1, 1152–1162 CrossRef CAS.
- A. L. Washington and G. F. Strouse, Chem. Mater., 2009, 21, 2770–2776 CrossRef CAS.
- A. L. Washington and G. F. Strouse, Chem. Mater., 2009, 21, 3586–3592 CrossRef CAS.
- X. Wang, Q. Peng and Y. D. Li, Acc. Chem. Res., 2007, 40, 635–643 CrossRef CAS.
- E. Caponetti, D. C. Martino, M. Leone, L. Pedone, M. L. Saladino and V. Vetri, J. Colloid Interface Sci., 2006, 304, 413–418 CrossRef CAS.
- J. W. Doolittle and P. K. Dutta, Langmuir, 2006, 22, 4825–4831 CrossRef CAS.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847–4854 CrossRef CAS.
- H. Q. Wang and T. Nann, ACS Nano, 2009, 3, 3804–3808 CrossRef CAS.
- V. Polshettiwar, B. Baruwati and R. S. Varma, ACS Nano, 2009, 3, 728–736 CrossRef CAS.
- J. E. Hutchison, ACS Nano, 2008, 2, 395–402 CrossRef CAS.
- C. J. Murphy, J. Mater. Chem., 2008, 18, 2173–2176 RSC.
- B. A. Roberts and C. R. Strauss, Acc. Chem. Res., 2005, 38, 653–661 CrossRef CAS.
- C. R. Strauss and R. S. Varma, Top. Curr. Chem., 2006, 266, 199–231 CAS.
- T. Razzaq and C. O. Kappe, ChemSusChem, 2008, 1, 123–132 CrossRef CAS.
- R. Hoogenboom, T. F. A. Wilms, T. Erdmenger and U. S. Schubert, Aust. J. Chem., 2009, 62, 236–243 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |