Unusual corrosion process of gold nanoplates and the mechanism study†
Yingwen
Cheng
a,
Cuicui
Qiu
a,
Houyi
Ma
*ab,
Xiaokai
Zhang
c and
Xiaohu
Gu
a
aKey Laboratory for Colloid and Interface Chemistry of State Education Ministry, Shandong University, Jinan, 250100, China. E-mail: hyma@sdu.edu.cn; Fax: + 86-531-88564464; Tel: + 86-531-88364959
bBeijing National Laboratory for Molecular Science, Beijing, 100190, China
cAnalysis and Testing Center, College of Physics and Electronics, Shandong Normal University, Jinan, 250014, China
First published on 17th February 2010
Abstract
Herein we describe an unusual localized corrosion that initiates on large {111} surfaces and ends with the complete fragmentation of the gold nanoplates, which is expected to develop into a specific method for processing large metal nanomaterials into smaller ones with desired shapes.
Due to their fascinating physical and chemical properties,1 gold nanomaterials have received increasing interests in many research fields.2 Recently, a variety of synthetic methods have been developed to prepare gold nanomaterials with different shapes and morphology.3 However, the exploration of simple and practical methods for further tailoring the shapes, structures and morphology of the as-prepared gold nanostructures is still a challenging research topic in current nanoprocessing technology. In this paper we will focus on developing a novel nanoprocessing method based on the localized corrosion of gold nanostructures, by means of which we are able to make holes in nanoplates and even process them into irregular nanoplates and small nanoparticles.
Gold is one of the noble metals because of its chemical inertness. The corrosion of gold can take place only in the presence of both strong oxidants and complexing ions. Some recent studies have demonstrated that gold nanostructures can be corroded and even dissolved considerably in solutions containing cyanide or halide ions when using O2 or H2O2 as oxidizing agents.4,5 This type of interesting corrosion opens a new path for the nano-processing of gold nanostructures and has led to some unexpected results. For example, the dissolution (i.e. free corrosion) of the Au core from the polystyrene- and silica-coated Au nanorods in solutions containing KCN generated hollow nanotubes.6 The selective shortening of gold nanorods was achieved by mild oxidation with O2 in HCl solutions at elevated temperature.7
So far there has been only limited success in the aspect of selective chemical etching of gold nanostructures owing to the intrinsic difficulties involved. In particular, almost all the previous studies focused on the oxidation of gold nanorods4,6–9 or small nanospheres.10 Herein we present for the first time the spatially-directed oxidation of gold nanoplates by Au3+ ions in the presence of sodium dodecylbenzenesulfonate (SDBS), which took place selectively on submicron- or micron-scale {111} basal surfaces and finally broke nanoplates into small particles, with perforated nanoplates as intermediates. This new finding is of fundamental importance and will be helpful for having a better insight into the corrosion mechanism of micro/nanometer-scale gold materials.
Gold nanoplates are a class of particularly interesting single-crystalline structures, bounded by two large {111} basal surfaces. They are thermodynamically stable nanostructures as compared with nanorods and nanocubes, because of the existence of stable {111} planes.11 Although Au nanoplates were observed to melt and collapse first at (110) facets upon heating,12 the present study clearly indicates that the selective dissolution or localized corrosion was initiated at large (111) surfaces. Fig. 1A shows a typical SEM image of such etched gold nanoplates, which were prepared by 2-day slow reduction of AuCl4− ions at room temperature in 50 mL of aqueous solution containing 20 vol% ethylene glycol (EG), 0.5 mM HAuCl4 and 1 mM SDBS. The presence of SDBS is essential for formation of hexagonal and triangular nanoplates with large edge length of ∼1 μm during the reduction of gold ions. Interestingly, nearly all of the obtained nanoplates were etched to a certain degree, which was exemplified by many holes (inset in Fig. 1A) on their basal planes and indentations at their edges. Of all the chemicals used in this study, EG was commonly used as the reductant in polyol processes and HAuCl4 was applied as the gold precursor in the preparation of Au nanomaterials.12,13 It is therefore inferred that SDBS molecules play a key role in the unusual localized corrosion of gold nanoplates and the preferential oxidation of the gold atoms on the {111} surfaces.
 | ||
| Fig. 1 A set of representative SEM and TEM images of various irregular gold nanoplates. Inset in (A) is an enlarged image of a typical corroded nanoplate and the scale bar is 200 nm. | ||
The previous study reveals that the quantitative binding of AuCl4− anions to CTAB micelles is responsible for the spatially-directed oxidation of gold nanorods,9 but this mechanism does not work for our system. The critical micelle concentration (cmc) of SDBS at room temperature is around 3 mM,14 which is above the concentration at which the etched nanoplates were produced. Furthermore, repeated experiments confirmed that the SDBS concentration cannot determine whether the final nanoplates are etched or not. Fig. S1 (see the ESI†) shows a set of TEM images for the gold nanoplates obtained in 1 mM, 0.8 mM and 0.5 mM SDBS solutions, respectively. The perforated plates were observed in each case. However, when SDBS was above the cmc, the main products were small gold nanoparticles rather than nanoplates (Fig. S2†).
Fig. 1B and 1C also show the TEM images of several representative nanoplates generated in 1 mM SDBS solution. The white spots on the nanoplates indicate the etch-holes caused by the localized corrosion. By comparing the thickness (Fig. 1B) and sizes (Fig. 1C) of these gold nanoplates, we found that the larger or thicker gold nanoplates, the more readily they suffered from localized corrosion. The influence of thickness and basal sizes on the localized corrosion of nanoplates is probably attributable to the following reasons: (i) the thick plates have more stacking faults than thin ones; (ii) more SDBS molecules adsorb on the large basal surfaces, which increases the surface concentration of SDBS on the (111) planes.
After repeated experiments, we confirmed that the localized corrosion of gold nanoplates originated from the selective dissolution of surface atoms on the large (111) surfaces. An important characteristic of such corrosion is that an individual flat plate was perforated in the central area at first, followed by expansion of the perforated hole up to complete fragmentation of the whole plate. The TEM image shown in Fig. 1D presents several representative etched nanoplates. By comparing their morphology and shapes it is inferred that the localized corrosion of gold nanoplates underwent the stages of initiation, development, partial etching and complete fragmentation. The formation of a visible hole at the center indicated the beginning of the localized corrosion. For nanoplate a, the localized corrosion led to a pit at its center, which almost completely penetrated the whole plate. Plate b displayed a larger and brighter spot at the center. Comparison of the size and morphology of the two holes implies that the gold atoms around the precursor pit dissolved randomly. This non-preferential dissolution cannot result in a regular hole with a specific shape. Plate c is a commonly observed transition shape before serious etching of gold nanoplate. The large brightness contrast between the left side and right side reveals that the thickness of the left part was decreasing due to the layer-by-layer dissolution of gold atoms from the (111) surface. The TEM images of corroded gold nanoplates shown in Fig. S3† provide further evidence for the layer-by-layer corrosion fashion. Meanwhile, the corrosion region in plate c expanded from the central region to the edge. With continuous dissolution of the etched part, another transition state-like plate d appeared, whose left part almost completely disappeared and the rest started to be attenuated.
In order to study the mechanism for the selective dissolution of gold nanoplates, we investigated the interaction between AuCl4− complex anions and dodecylbenzenesulfonate (DBS) anions by using cyclic voltammetry (CV) and ultraviolet (UV)-visible spectroscopy. Fig. 2A shows the time evolution of the UV-visible adsorption spectra for the mixed solution of 1 mM SDBS and 0.5 mM HAuCl4. The characteristic absorption band of Au(III) ions,15 centered around 300 nm, was observed to decrease with increasing storage time. After 80 h, a slight protuberance around 500 nm emerged in the UV-visible spectrum; meanwhile, the solution turned from its original light yellow color to a light pink color. These changes reveal the formation of Au+ and gold nanoparticles.16Fig. 2B shows the cyclic voltammograms (CVs) measured in the same solution. A reduction peak was observed at ∼0.38 V in the first cycle, obviously lower than the value of 0.75 V in the absence of SDBS (black line in Fig. 2D), but the peak potential shifted positively to 0.71 V from the second cycle, accompanied by an obvious increase of oxidation current in the potential region between 0.71 V and 1.2 V. All these experimental results indicate that another stable Au species, Au(I), was gradually generated from the original Au(III) ions.
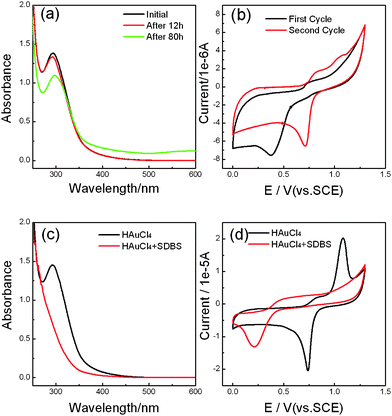 | ||
| Fig. 2 (A) Time evolution of the UV-visible absorption spectra for the aqueous solution of 0.5 mM HAuCl4 and 1 mM SDBS. The reaction time is marked in the plot. (B) The first potential scan (black line) and the second potential scan (red line) of the cyclic voltammograms (CVs) for the same solution as used in (A), scan rate: 50 mV s−1. (C). UV-visible absorption spectra of 0.5 mM HAuCl4 in the absence (red line) and presence (black line) of 5 mM SDBS. (D) CVs for 0.5 mM HAuCl4 in the absence (black line) and presence (red line) of 5 mM SDBS. Scan rate: 50 mV s−1. | ||
According to the crystal field theory,17 the complexing ability of SO32− ions is superior to that of Cl− ions. Thus, dodecylbenzenesulfonate ions (R–SO3−) would replace chloride ions in AuCl4− complex anions, forming new complex ions via the following reaction
| AuCl4− + 4 (R–SO3−) ↔ Au(R-SO3)4− + 4 Cl− | (1) |
| Au(R–SO3)4− + 2 e− ↔ Au(R–SO3)2− + 2 R–SO3− | (2) |
This theory is supported by the decrease of the concentration of Au(III) ions and the formation of gold nanoparticles (Fig. S4†). Due to the strong coordination ability of DBS ions, it is more difficult for free gold ions to dissociate from Au(R–SO3)4− (or Au(R–SO3)2−) ions than from AuCl4− (or AuCl2−) ions, which was reflected well in the negative shift in the reduction potential of gold ions.
The rate of the ligand exchange reaction in AuCl4− complex ions is strongly dependent on SDBS concentration. In the case of 5 mM SDBS, the characteristic absorption peak for Au(III) ions almost completely disappeared in a short time, as can be seen in Fig. 2C. Considering this reason and the appearance of an absorption peak for gold nanoparticles in the same solution after 1 month (see Fig. S4†), the presence of SDBS might be able to transform Au(III) ions to Au(I) ions. The CVs shown in Fig. 2D indicate that the reduction potential of gold ions shifted negatively to 0.21 V in the aqueous solution containing 5 mM SDBS formed by mixing 0.5 mM HAuCl4 with SDBS solution. The negative shift of 0.53 V is close to that of the redox potential of Au+/Au0 in the presence of CTAB micelles (0.6 V). The latter case was observed from the oxidation of gold nanoparticles by Au(III)–CTAB complexes.9
On the basis of the above UV-visible and CV results, we can draw the following conclusions: (i) Au3+ ions can be induced to transform into Au+ ions by SDBS since they are highly unstable; (ii) the complex compounds formed through the reaction of HAuCl4 with SDBS are difficult to reduce, perhaps because of the strong interaction between DBS ions and gold ions; and (iii) the extent of the transformation of Au3+ to Au+ strongly depends on the concentration of SDBS.
Seeing that the Au(R–SO3)2− complex is very stable, the following reaction,
| AuCl4− + 2Au0 + 2Cl− ↔ 3AuCl2− | (3) |
Finally, such a mechanism was confirmed by studying the corrosion behavior of gold nanoplates synthesized by means of a slow reduction method (see Fig. 3). Interestingly, when as-prepared gold nanoplates were exposed to 5 mM SDBS solution for 1 month, no obvious changes in shape and morphology were observed (Fig. 3B). Clearly, SDBS molecules themselves are unable to induce the dissolution of gold atoms. In strong contrast to this situation, the gold nanoplates suffered from severe corrosion in the presence of both SDBS and HAuCl4, as demonstrated by the observation that the large nanoplates become small nanoparticles and irregular nanoplates with some “tails” at the corners, through a process that is similar with the one described above (Fig. 3C–F). Therefore we can conclude that the collaboration of Au3+ ions and SDBS drive the dissolution of gold atoms on nanoplates.
 | ||
| Fig. 3 TEM images of the uncorroded gold nanoplate samples (A), those after being exposed to 5 mM SDBS only (B) and those exposed to the mixed solution of 5 mM SDBS and 0.5 mM HAuCl4 (C, D, E and F) for 1 month. Inset in (D) shows the SAED pattern taken from the region marked with a circle in Fig. 3D, indicating the crystalline nature of the corroded plates still held. | ||
In conclusion, we presented an interesting corrosion process of gold nanoplates that initiated on their basal {111} surfaces, which leads to complete fragmentation of the plates. This counter-Ostwald ripening process provides a way to process large particles into small ones. The combined action of DBS and AuCl4− anions plays a key role in the novel localized corrosion. The formation of stable Au(R–SO3)2− complex ions is probably the driving force for oxidation of the gold atoms on the plates. The present results are helpful for giving a deeper insight into the oxidation mechanisms of nanostructured noble metals. The localized corrosion of gold nanoplates can be expected to develop into a specific method to process large metal nanomaterials into small ones with desired shapes.
Acknowledgements
This work was supported by Beijing National Labotatory for Molecular Science, the National Basic Research Program of China (2009CB930103), and the National Science Foundation of China (20673067).Notes and references
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef CAS.
- N. L. Rosi and C. A. Mirkin, Chem. Rev., 2005, 105, 1547 CrossRef CAS.
- J. Shan and H. Tenhu, Chem. Commun., 2007, 44, 4580 Search PubMed.
- N. R. Jana, L. Gearheart, S. O. Obare and C. J. Murphy, Langmuir, 2002, 18, 922 CrossRef CAS.
- X. Kou, W. Ni, C.-K. Tsung, K. Chan, H.-Q. Lin, G. D. Stucky and J. Wang, Small, 2007, 3, 2103 CrossRef CAS.
- S. O. Obare, N. R. Jana and C. J. Murphy, Nano Lett., 2001, 1, 601 CrossRef CAS.
- C.-K. Tsung, X. Kou, Q. Shi, J. Zhang, M. H. Yeung, J. Wang and G. D. Stucky, J. Am. Chem. Soc., 2006, 128, 5352 CrossRef CAS.
- T. S. Sreeprasad, A. K. Samal and T. Pradeep, Langmuir, 2007, 23, 9463 CrossRef CAS.
- J. Rodríguez-Fernández, J. Pérez-Juste, P. Mulvaney and L. M. Liz-Marzán, J. Phys. Chem. B, 2005, 109, 14257 CrossRef CAS.
- (a) S. Singh and B. L. V. Prasad, J. Phys. Chem. C, 2007, 111, 14348 CrossRef CAS; (b) H. Huang, C. He, Y. Zeng, X. Xia and X. Yu, Colloids Surf., A, 2008, 317, 56 CrossRef CAS.
- X. Liu, N. Wu, B. H. Wunsch, R. J. Barsotti Jr. and F. Stellacci, Small, 2006, 2, 1046 CrossRef CAS.
- C. Kan, X. Zhu and G. Wang, J. Phys. Chem. B, 2006, 110, 4651 CrossRef CAS.
- C. Li, W. Cai, B. Cao, F. Sun, Y. Li, C. Kan and L. Zhang, Adv. Funct. Mater., 2006, 16, 83 CrossRef CAS.
- S. K. Hait, P. R. Majhi, A. Blume and S. P. Moulik, J. Phys. Chem. B, 2003, 107, 3650 CrossRef CAS.
- A. K. Gangopadhayay and A. Chakravorty, J. Chem. Phys., 1961, 35, 2206.
- S. Eustis, H.-Y. Hsu and M. A. El-sayed, J. Phys. Chem. B, 2005, 109, 4811 CrossRef CAS.
- T. Hua, J. Yang, J. Chen and S. Liu, Principal of General Chemistry, 2nd ed.; Peking University Press, Beijing, 1993, chapter 14 Search PubMed.
- C. Li, K. L. Shuford, Q.-H. Park, W. Cai, Y. Li, E. J. Lee and S. O. Cho, Angew. Chem., Int. Ed., 2007, 46, 3264 CrossRef CAS.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990 CrossRef CAS.
Footnote |
| † Electronic Supplementary Information (ESI) available: Additional experimental details and TEM images. See DOI: 10.1039/b9nr00229d/ |
| This journal is © The Royal Society of Chemistry 2010 |