Theoretical studies on structures, 13C NMR chemical shifts, aromaticity, and chemical reactivity of finite-length open-ended armchair single-walled carbon nanotubes†
Lei Vincent
Liu
a,
Wei Quan
Tian
b,
Ya Kun
Chen
a,
Yu Adam
Zhang
a and
Yan Alexander
Wang
*a
aDepartment of Chemistry, University of British Columbia, Vancouver, BC V6T 1Z1, Canada. E-mail: yawang@chem.ubc.ca
bState Key Laboratory of Theoretical and Computational Chemistry, Institute of Theoretical Chemistry, Jilin University, Changchun, Jilin 130023, China
First published on 22nd October 2009
Abstract
The geometries, chemical shifts, aromaticity, and reactivity of finite-length open-ended armchair single-walled carbon nanotubes (SWCNTs) have been studied within density functional theory. The widely used model of capping hydrogen atoms at the open ends of a SWCNT changes the chemical activity of the SWCNT and destabilizes the frontier molecular orbitals. The edge π-orbital of the open ends enhances both π- and σ-aromaticity of the first belt of hexagons of carbon atoms at the open ends. The effect of the open ends on the structure and chemical reactivity of the SWCNT reaches only the first several layers of the hexagons of carbon atoms. Additions of carbene and dichlorocarbene to the nanotube reveal that the open ends have higher reactivities than the inner regions.
1. Introduction
Carbon nanotubes (CNTs) have been extensively studied during the last 18 years, since Iijima's discovery of multi-walled CNTs in 19911 and single-walled carbon nanotubes (SWCNTs) in 1993.2 Various wide-ranging intriguing potential applications based on the unique properties of CNTs have been proposed, including hydrogen storage,3 molecular sieves,4drug delivery,5 chemical sensors,6 and molecular electronics.7 For the first two applications, short open-ended nanotubes are required to overcome the limitation of diffusion.Many methods have been developed to cut CNTs, such as controlled oxidation by piranha solutions,8fluorination,9 ultrasound power,10ball milling,11 and STM and AFM techniques.12,13 Once CNTs have been cut open, their open ends possess extra interesting properties and offer additional potential applications. It has been discreetly predicted that chemical reactions may take place more readily on the flexible open ends of CNTs, because those carbon atoms at the open-end edges can change their hybridization more easily than the carbon atoms on the rigid sp2 sidewall.14 This property induces regioselectivity to the functionalization of CNTs. In practice, however, the insolubility of as-prepared CNTs imposes severe limitations on the applications of CNTs, and functionalization of CNTs offers a solution to overcome such a problem.15,16 Experimentalists and theoreticians have extensively studied the functionalization of CNTs by carbene and its analogues17–23 and have found that such functionalization is able to engineer the electronic structure of SWCNTs.18,23
Compared with closed CNTs, open-ended CNTs have higher conductivity and much improved field-emission properties.24,25 However, theoretical studies on the geometries, electronic structures, and reactivities of open-ended SWCNTs are sparse.25–30 In this work, we thus hope to provide additional detailed studies of open-ended SWCNTs.
Recently, 13C NMR has been applied to study the electronic structures and dynamics of SWCNTs and to characterize functionalized SWCNTs.31–34 The possibility of using 13C NMR to separate metallic and semiconducting SWCNTs has also been proposed.35,36 Theoretically, 13C NMR chemical shifts have already been predicted for hydrogen-capped and carbon-capped closed SWCNTs.35,37,38 For comparison, we will also provide NMR information for open-ended SWCNTs.
In the following, we will present our theoretical studies on the geometrical and electronic structures, 13C NMR chemical shifts, aromaticity, and chemical reactivity of open-ended SWCNTs. Chemical reactions of SWCNTs with carbene and dichlorocarbene are used to discuss the reactivity of open-ended SWCNTs.
2. Model selection and computational methods
Two models are commonly used for theoretical studies of finite-size open-ended SWCNTs. The more widely used model saturates one open end with hydrogen atoms and leaves the other open end untouched for subsequent studies,25,26,39–41 whereas the other less used model has both ends unsaturated.27,28 We carried out comparative studies on these two models based on two open-ended (5,5) armchair SWCNTs consisting of 70 carbon atoms: C70 (in D5h symmetry) without capping atoms (Fig. 1a) and C70H10 (in C5v symmetry) with capping hydrogen atoms at one open end (Fig. 1b).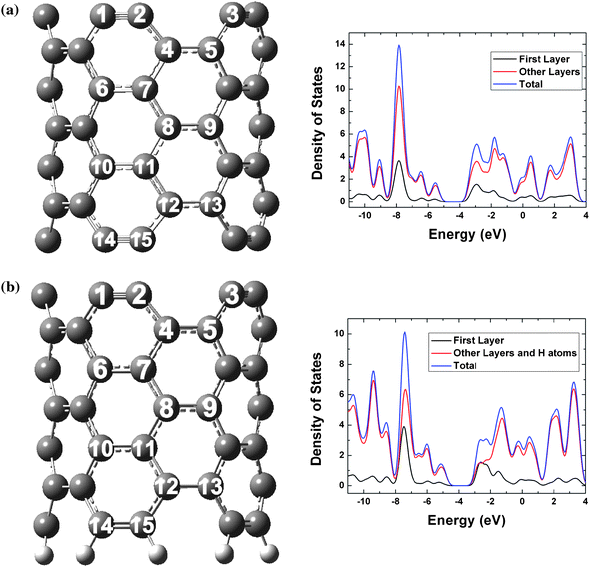 | ||
| Fig. 1 Structures and density of states of open-ended (5,5) SWCNTs: (a) C70 without any capping atoms and (b) C70H10 with one end capped with hydrogen atoms. Carbon atoms are grey; hydrogen atoms are white. The first layer only includes the ten outmost carbon atoms. The Fermi levels are −4.37 and −3.88 eV for C70 and C70H10, respectively. | ||
Geometries were optimized by using the hybrid density functional B3LYP42,43 with the 6-31G(d) basis set, which has been shown to accurately reproduce experimental structures of finite-length armchair SWCNTs before.14 Partial charges and bonding properties were analyzed by using the NBO 3.1 module44 implemented in the Gaussian 03 package.45 Data of bond lengths, bond orders, and NBO partial charges are collected in Table 1. For the first three layers of carbon atoms of C70 and C70H10 at the unsaturated open end, bond lengths, bond orders, and partial charges are quite similar. Hence, we can conclude that the influence of the capping hydrogen atoms at one open end on the overall structure of the SWCNT does not significantly go beyond the first three layers from the capped end. The capping hydrogen atoms hardly affect the structure of the other open end if the model of the open-ended SWCNT is long enough (i.e., more than 5 layers of carbon atoms or bigger than C50 for our model system).
| Bond | Bond lengths | Bond orders | Atom | Partial charges | |||
|---|---|---|---|---|---|---|---|
| C70 | C70H10 | C70 | C70H10 | C70 | C70H10 | ||
| C1–C2 | 1.240 | 1.241 | 2.462 | 2.462 | C2 | +0.060 | +0.054 |
| C2–C4 | 1.404 | 1.405 | 1.253 | 1.253 | C4 | −0.066 | −0.068 |
| C4–C5 | 1.432 | 1.433 | 1.200 | 1.200 | C7 | +0.007 | +0.007 |
| C4–C7 | 1.442 | 1.442 | 1.251 | 1.253 | C8 | −0.003 | −0.003 |
| C6–C7 | 1.444 | 1.446 | 1.255 | 1.252 | C11 | +0.007 | −0.003 |
| C7–C8 | 1.436 | 1.435 | 1.192 | 1.194 | C12 | −0.066 | −0.022 |
| C8–C9 | 1.417 | 1.413 | 1.317 | 1.322 | C15 | +0.060 | −0.206 |
| C8–C11 | 1.436 | 1.435 | 1.192 | 1.186 | — | — | — |
| C10–C11 | 1.444 | 1.437 | 1.255 | 1.229 | — | — | — |
| C11–C12 | 1.442 | 1.416 | 1.251 | 1.298 | — | — | — |
| C12–C13 | 1.432 | 1.450 | 1.200 | 1.199 | — | — | — |
| C12–C15 | 1.404 | 1.431 | 1.253 | 1.223 | — | — | — |
| C14–C15 | 1.240 | 1.372 | 2.462 | 1.612 | — | — | — |
We also analyzed the electronic structures of C70 and C70H10 based on local density of states (LDOS) and frontier molecular orbitals (FMOs). The LDOS plots are shown in Fig. 1. The first layer of the ten outmost carbon atoms of C70 contributes very little to the total density of states (DOS) near the region where the highest occupied molecular orbital (HOMO) lies and contributes a relatively bigger amount to the total DOS near the region where the lowest unoccupied molecular orbital (LUMO) resides. For C70H10, the first layer also contributes a small amount to the total DOS near the HOMO region, but the LUMO has a much larger contribution from the open end without capping atoms. This suggests that the hydrogen capping atoms artificially introduce localized electronic states in the LUMO region for the first layer of carbon atoms. Without rationally taking this artifact into account in theoretical modeling, one might be misled to overestimate the chemical reactivity and field emissions of the open ends.25
A few important FMOs of C70 and C70H10 are exhibited in Fig. 2. The HOMO and the LUMO of C70 are very similar to those of C70H10, while some other corresponding FMOs are quite different. The orbital energies of the HOMO and the LUMO of C70 are both 0.49 eV lower than those of C70H10. In other words, C70 and C70H10 have the same HOMO–LUMO gap, 2.15 eV. It is nonetheless clear that the hydrogen capping atoms decrease the ionization potential of the open-ended SWCNT, which might artificially affect the field-emission threshold and intensity as a result.
 | ||
| Fig. 2 Frontier molecular orbitals of open-ended (5,5) SWCNTs: (a) C70 without capping atoms and (b) C70H10 with one end capped by hydrogen atoms. HOMO–n (p) is the nth orbital below the HOMO with orbital energy p eV. LUMO+m (q) is the mth orbital above the LUMO with orbital energy q eV. | ||
As also shown in Fig. 2, there is no localized electronic state on the first layer of carbon atoms near the HOMO regions for either C70 or C70H10; this fact is reflected in the LDOS analysis. On the other hand, localized (virtual) electronic states do exist on the first layers of C70 and C70H10. The LUMO+1 to LUMO+4 of C70 (Fig. 2a) clearly have localized electronic states, which are mainly the p-electrons along the tube axis at the open ends. The LUMO+1 and LUMO+2 of C70H10 (Fig. 2b) represent strongly localized electronic states, also composed of the p-electrons along the tube axis, on the first layer carbon atoms of the unsaturated open end. However, the symmetry of such edge π-orbitals is broken by the capping of hydrogen atoms in C70H10 and the orbitals are mainly concentrated at the open end without capping atoms.
Although an open-ended (5,5) SWCNT with and without capping atoms at one open end, do have almost identical geometries at the unsaturated open end, their electronic structures are very different. Fig. 2 evidently shows that the capping hydrogen atoms not only break the symmetry of some virtual orbitals but also modify many occupied orbitals. It is clear that artificially capping one end with hydrogen atoms significantly modifies the electronic structure of the nanotube to such an extent that this model has many unrealistic features. Therefore, we will only employ open-ended (5,5) SWCNTs without capping atoms as models to study the properties of open-ended armchair SWCNTs hereafter.
B3LYP/6-31G(d) was employed to optimize a series of finite-length open-ended (5,5) SWCNTs, C10n (n = 4–18). 13C NMR chemical shifts were then computed using GIAO-HF/6-31G(d).14,46Tetramethylsilane was used as the reference compound for NMR. The predicted 13C NMR chemical shift of open-ended (5,5) SWCNT C60 is 148.16 ppm, reasonably close to the experimental result of 142.68 ppm,47 given the large system size and the relatively small basis set used. Such a good agreement gave us confidence to use this level of theory to predict the 13C NMR chemical shifts of similar SWCNTs. Nucleus-independent chemical shift (NICS) calculations were also performed at the same level of theory to evaluate the local aromaticity of open-ended (5,5) SWCNTs.48 To appreciate the chemical reactivity of finite-length open-ended armchair SWCNTs, we studied the chemical reaction of carbene and dichlorocarbene with C70, using the same level of theory but without symmetry constraints.
3. Results and discussions
Formation energies for all of the (5,5) SWCNTs studied are plotted in Fig. 3, which shows a monotonic increase of the formation energies from C40 to C180. The formation energy for C180 is −6.97 eV, which is almost identical to the formation energy of C60 fullerene, −6.98 eV. This indicates that the open ends destabilize the open-ended SWCNTs, thus possibly enhancing their chemical reactivity. | ||
| Fig. 3 Formation energies per carbon atom for open-ended (5,5) SWCNTs C40–C180. The formation energy results from subtracting the energy of a free carbon atom from the average energy of the nanotube per carbon atom. | ||
Bond lengths of the optimized structures are listed in Table 2. The hexagons at the open ends closely resemble o-benzyne (1,2-didehydrobenzene). The bond lengths of CC bond a (indexed in Fig. 4a and Fig. S1 of the ESI† ) are around 1.24 Å, which are similar to the corresponding bond length of o-benzyne, 1.25 Å, at the same level of theory and agree very well with the experimental CC bond distances of o-benzyne, 1.24 ± 0.2 Å.49 Within the C30n, C30n+10, and C30n+20 series, the bond lengths of the same bonds are very similar.
| a | b | c | d | e | f | g | h | i | |
|---|---|---|---|---|---|---|---|---|---|
| C40 | 1.240 | 1.406 | 1.445 | 1.465 | — | — | — | — | — |
| C50 | 1.234 | 1.416 | 1.412 | 1.453 | 1.463 | — | — | — | — |
| C60 | 1.246 | 1.392 | 1.444 | 1.443 | 1.442 | 1.443 | — | — | — |
| C70 | 1.240 | 1.404 | 1.432 | 1.442 | 1.444 | 1.436 | 1.417 | — | — |
| C80 | 1.238 | 1.412 | 1.419 | 1.447 | 1.446 | 1.429 | 1.416 | 1.440 | — |
| C90 | 1.245 | 1.396 | 1.443 | 1.443 | 1.443 | 1.438 | 1.428 | 1.420 | 1.443 |
 | ||
| Fig. 4 13C NMR chemical shifts (CS) and nucleus-independent chemical shifts (NICS) of finite-length open-ended (5,5) SWCNTs. (See also Fig. S1 of the ESI† ). | ||
Vibrational frequencies of the stretching modes of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C triple bonds on the open ends of C70 are 2004 (A2″), 2032 (E1″), 2032 (E1″), 2034 (A1′), 2042 (E1′), 2042 (E1′), 2058 (E2′), 2058 (E2′), 2059 (E2″), and 2059 cm−1 (E2″). In comparison, the vibrational frequency of the stretching mode of the C≡C triple bonds of o-benzyne is 2026 cm−1 (A1) at the same level of theory, and the experimental value is 1846 cm−1 (observed in a neon matrix50).
C triple bonds on the open ends of C70 are 2004 (A2″), 2032 (E1″), 2032 (E1″), 2034 (A1′), 2042 (E1′), 2042 (E1′), 2058 (E2′), 2058 (E2′), 2059 (E2″), and 2059 cm−1 (E2″). In comparison, the vibrational frequency of the stretching mode of the C≡C triple bonds of o-benzyne is 2026 cm−1 (A1) at the same level of theory, and the experimental value is 1846 cm−1 (observed in a neon matrix50).
13C NMR chemical shifts and NICS of the finite-length open-ended (5,5) SWCNTs are shown in Fig. 4 (and Fig. S1 of the ESI). Monitoring the 13C NMR chemical shifts at the open ends unveils an unique feature for open-ended (5,5) SWCNTs: the big chemical shifts of the carbon atoms at the open ends, around 200 ppm, are very close to the chemical shifts (201.74 ppm) of the CC triple bond of o-benzyne at the same level of theory, whereas other chemical shifts are well separated, at least 55 ppm away.
Chemical shifts of the carbon atoms on the sidewall mainly range from 116 to 139 ppm, except that C50 has a chemical shift at 144.26 ppm and the C30n series have lower chemical shifts around 100 ppm for the second, fifth, and eighth layers of carbon atoms. Within the C30n, C30n+10, and C30n+20 series, the same numbered carbon atoms have similar 13C NMR chemical shifts. As the nanotube elongates, the 13C NMR chemical shifts of the middle layers approach 120 ppm, and the wave pattern in the chemical shifts for C30n, C30n+10, and C30n+20 becomes more and more evident (Fig. 4b). This wave pattern also manifests itself in the chemical shifts along the tube elongation direction if the first-layer carbon atoms from the open end are excluded from consideration (see Fig. S1 of the ESI).
The occupied orbitals have some localized electron density at the open end (see Fig. 2 and Fig. S2 of the ESI). Such localization at the open end can form strong electron currents in the first-layer hexagons of carbon atoms and thus may result in high aromaticity.48 The NICS values have been taken as criteria for aromaticity in many systems.51–55 The NICS at the hexagonal centers, NICS(0), represents the σ-aromaticity of a ring, whereas the NICS at the place 1 Å above the rings, NICS(1), represents the π-aromaticity of a ring.51 The predicted NICS data for the open-ended SWCNTs are shown in Fig. 4 (and Fig. S1 of the ESI). Overall, both NICS numbers, NICS(0) and NICS(1), of the hexagons of carbon atoms become more positive as the ring moves further away from the open end. Thus, the NICS, or the aromaticity, of the hexagon of carbon atoms probably converges to a value in a long SWCNT, according to the evolution pattern of the NICS values from C40 to C180. The NICS values of hexagons A, B, C, and D in Fig. 4 further reveal that the NICS values of the hexagons of carbon atoms at the same position from the open end (of different tubes) also become more positive as the tube gets longer. The wave pattern in the NICS values for C30n, C30n+10, and C30n+20 is also evident as the tube elongates. Except for hexagon A (Fig. 4c), the π-aromaticity is usually stronger than the σ-aromaticity in all other rings, which is probably due to the fact that the orbital hybridization of the carbon atoms at the open end is roughly between sp (triple bond) and sp2 (double bond), thus enhancing both the σ-electron overlap and the π-electron conjugation. However, the enhancement of the σ-electron overlap is strongly influenced by the more confined overall space for electron conjugation.
Similar to the widely observed oscillation pattern of the orbital energies of the HOMO and the LUMO and the HOMO–LUMO gap for finite-length SWCNTs,14,30,56,57 the same behavior is confirmed for the open-ended SWCNTs C40–C180 in Fig. 5. Moreover, the HOMO and the LUMO of the open-ended SWCNTs C40–C180 clearly show interesting oscillation properties in the shapes of molecular orbitals (see Fig. S2 of the ESI).14,30,56 For C30n and C30n+10 tubes, the HOMO has π-bonding along the tube axis, whereas the LUMO has π-bonding perpendicular to the tube axis. However, for C30n+20 tubes, the HOMO has π-bonding perpendicular to the tube axis, whereas the LUMO has π-bonding along the tube axis. This bonding pattern of the FMOs dictates different reaction behaviors (e.g., toward cycloaddition) for the tubes of different lengths. On the other hand, if the models for open-ended SWCNTs are too short, such as C40 and C50 (see Fig. S2 of the ESI), one might obtain a less conclusive understanding because the p-orbitals along the tube axis of the edge carbon atoms contribute greatly to the LUMOs. Certainly, much greater efforts are required to reveal the correct bonding and reaction patterns by working with much longer SWCNTs, such that the edge effects can be effectively minimized.
 | ||
| Fig. 5 (a) The HOMO and LUMO orbital energies and (b) HOMO–LUMO gaps of open-ended (5,5) SWCNTs C40–C180. | ||
To comprehend the reactivity of open-ended SWCNTs, we studied the adsorptions of carbene and dichlorocarbene at eight different bonding sites on C70 (Fig. 1a): C1–C2, C2–C3, C2–C4, C4–C5, C4–C7, C6–C7, C7–C8, and C8–C9. Table 3 gathers adsorption energies, bond lengths, and bond orders of the aforementioned eight adsorption occurrences. The adsorption energy Ead is defined as Ead = Etotal − (Etube + Emol), where Etotal is the energy of C70 with the adsorbed molecule, Etube is the total energy of the clean C70 without adsorbate, and Emol is the energy of a triplet carbene or a singlet dichlorocarbene. From the data displayed in Table 3, adsorption on C1–C2 is obviously the most exothermic, −131.0 kcal/ mol−1 for carbene and −95.5 kcal mol−1 for dichlorocarbene, because of the relaxation of the high-tension strain within the C1–C2 bond. Upon adsorption, the C1–C2 bond elongates from 1.24 to ca.1.35 Å and the bond order decreases significantly from 2.46 to as little as 1.40. In other words, the C1–C2 bond transforms effectively from a triple bond to a double bond upon adsorption. Adsorption on C2–C3 is the second most exothermic: the system releases 113.2 and 77.5 kcal mol−1 for carbene and dichlorocarbene adsorptions, respectively. After adsorption, C2–C3 shortens from 3.10 to ca. 2.38 Å. According to the data of bond lengths and bond orders, the adducts formed from carbene and dichlorocarbene additions on sites C4–C7 and C7–C8 are triangular structures, while at other sites on the sidewall they have open structures. Evidently, the correlation of the structure of adducts with finite-length SWCNTs depends on the positions of the adsorption sites.
| Bond | Adsorption energies | Bond lengths | Bond orders | |||||
|---|---|---|---|---|---|---|---|---|
| CH2 | CCl2 | none | CH2 | CCl2 | none | CH2 | CCl2 | |
| C1–C2 | −131.0 | −95.5 | 1.240 | 1.344 | 1.351 | 2.462 | 1.455 | 1.402 |
| C2–C3 | −113.2 | −77.5 | 3.095 | 2.376 | 2.385 | 0.008 | 0.011 | 0.009 |
| C2–C4 | −104.5 | −66.0 | 1.404 | 2.329 | 2.327 | 1.253 | 0.110 | 0.054 |
| C4–C5 | −108.8 | −68.1 | 1.432 | 2.385 | 2.412 | 1.200 | 0.038 | 0.037 |
| C4–C7 | −74.0 | −32.9 | 1.442 | 1.706 | 1.695 | 1.251 | 0.705 | 0.726 |
| C6–C7 | −108.5 | −51.9 | 1.444 | 2.309 | 2.322 | 1.255 | 0.080 | 0.080 |
| C7–C8 | −53.9 | −13.4 | 1.436 | 1.580 | 1.576 | 1.192 | 0.841 | 0.851 |
| C8–C9 | −89.1 | −32.8 | 1.417 | 2.210 | 2.205 | 1.317 | 0.132 | 0.153 |
The above adsorption studies confirm that the open ends of armchair SWCNTs closely resemble o-benzyne and have higher reactivity than the sidewall. The use of o-benzyne in organic synthesis has been extensively investigated by Pellissier and Santelli,58 who identified three types of reactions on o-benzyne: pericyclic reactions, nucleophilic additions, and transition-metal-catalyzed reactions. Such existing o-benzyne chemistry can certainly be utilized to functionalize the tip of open-ended armchair SWCNTs with many different functional groups for various applications. Recently, the first buckybowl aryne corannulyne has been synthesized and functionalized,59 and it is thus worthwhile to attempt similar experimental trials on open-ended SWCNTs.
4. Conclusions
In summary, within density functional theory, the geometries, chemical shifts, aromaticity, and reactivity of finite-length open-ended armchair SWCNTs have been studied. Capping hydrogen atoms at the open end of a SWCNT modify the electronic structure and enhance the chemical activity of the SWCNT. The edge π-orbital of the open end increases both the π- and σ-aromaticity of the first belt of hexagons of carbon atoms. However, the effect of the open end on the structure and chemical reactivity of a SWCNT only reaches its immediate neighboring region (three layers of carbon atoms along the tube elongation axis). Additions of carbene and dichlorocarbene on the tube reveal that the open end has much higher reactivity than the inner regions, which should be utilized to functionalize SWCNTs with novel properties and applications.Acknowledgements
The financial support from the Natural Sciences and Engineering Research Council of Canada is gratefully acknowledged. WestGrid provided us the necessary computational resources. L.V.L. acknowledges support from a Gladys Estella Laird Fellowship and a Charles A. McDowell Fellowship from the Department of Chemistry at the University of British Columbia. W.Q.T. acknowledges funding from Jilin University and from the Chinese Natural Science Foundation under Grant No. 20473031.References
- S. Iijima, Nature, 1991, 354, 56 CrossRef CAS.
- S. Iijima and T. Ichihashi, Nature, 1993, 363, 603 CrossRef CAS.
- C. Liu, Y. Y. Fan, M. Liu, H. T. Cong, H. M. Cheng and M. S. Dresselhaus, Science, 1999, 286, 1127 CrossRef CAS.
- Q. Wang, S. R. Challo, D. S. Sholl and J. K. Johnson, Phys. Rev. Lett., 1999, 82, 956 CrossRef CAS.
- A. Bianco, K. Kostarelos and M. Prato, Curr. Opin. Chem. Biol., 2005, 9, 674 CrossRef CAS.
- J. Kong, N. R. Franklin, C. W. Zhou, M. G. Chapline, S. Peng, K. J. Cho and H. J. Dai, Science, 2000, 287, 622 CrossRef CAS.
- P. Avouris, Acc. Chem. Res., 2002, 35, 1026 CrossRef CAS.
- K. J. Ziegler, Z. Gu, H. Peng, E. L. Flor, R. H. Hauge and R. E. Smalley, J. Am. Chem. Soc., 2005, 127, 1541 CrossRef CAS.
- Z. Gu, H. Peng, R. Hauge, R. E. Smalley and J. L. Margrave, Nano Lett., 2002, 2, 1009 CrossRef CAS.
- K. L. Lu, R. M. Lago, Y. K. Chen, M. L. H. Green, P. J. F. Harris and S. C. Tsang, Carbon, 1996, 34, 814 CrossRef CAS.
- N. Pierard, A. Fonseca, Z. Konya, I. Willems, G. Van Tendeloo and J. B. Nagy, Chem. Phys. Lett., 2001, 335, 1 CrossRef CAS.
- L. C. Venema, J. W. G. Wildoer, H. L. T. Twinsta, C. Dekker, A. G. Rinzler and R. E. Smalley, Appl. Phys. Lett., 1997, 71, 2629 CrossRef CAS.
- J. Y. Park, Y. Yaish, M. Brink, S. Rosenblatt and P. L. McEuen, Appl. Phys. Lett., 2002, 80, 4446 CrossRef CAS.
- Y. Matsuo, K. Tahara and E. Nakamura, Org. Lett., 2003, 5, 3181 CrossRef CAS.
- A. Hirsch, Angew. Chem., Int. Ed., 2002, 41, 1853 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS.
- J. Chen, M. A. Hamon, H. Hu, Y. Chen, A. M. Rao, P. C. Eklund and R. C. Haddon, Science, 1998, 282, 95 CrossRef CAS.
- H. Hu, B. Zhao, M. A. Hamon, K. Kamaras, M. E. Itkis and R. C. Haddon, J. Am. Chem. Soc., 2003, 125, 14893 CrossRef CAS.
- K. Kamaras, M. E. Itkis, H. Hu, B. Zhao and R. C. Haddon, Science, 2003, 301, 1501 CrossRef CAS.
- X. Lu, F. Tian and Q. Zhang, J. Phys. Chem., 2003, 107, 8388 Search PubMed.
- Y. Y. Chu and M. D. Su, Chem. Phys. Lett., 2004, 394, 231 CrossRef CAS.
- H. F. Bettinger, Org. Lett., 2004, 6, 731 CrossRef CAS.
- J. Zhao, Z. Chen, Z. Zhou, H. Park, P. Schleyer and J. Lu, ChemPhysChem, 2005, 6, 598 CrossRef CAS.
- L. B. Zhu, Y. Y. Sun, D. W. Hess and C.-P. Wong, Nano Lett., 2006, 6, 243 CrossRef CAS.
- G. Zhou, W. Duan and B. Gu, Phys. Rev. Lett., 2001, 87, 095504 CrossRef CAS.
- S. Hou, Z. Shen, X. Zhao and Z. Xue, Chem. Phys. Lett., 2003, 373, 308 CrossRef CAS.
- Y.-J. Xu and J.-Q. Li, Chem. Phys. Lett., 2005, 412, 439 CrossRef CAS.
- X. Y. Zhu, S. M. Lee, Y. H. Lee and T. Frauenheim, Phys. Rev. Lett., 2000, 85, 2757 CrossRef CAS.
- Y. Kumeda, Y. Fukuhiro, T. Takesugu and T. Hirano, Chem. Phys. Lett., 2001, 333, 29 CrossRef CAS.
- (a) J. Li, Y. Zhang and M. Zhang, Chem. Phys. Lett., 2002, 364, 328 CrossRef CAS; (b) J. Li, Y. Zhang and M. Zhang, Chem. Phys. Lett., 2002, 364, 338 CrossRef CAS.
- X.-P. Tang, A. Kleinhammes, H. Shimoda, L. Fleming, K. Y. Bennoune, S. Sinha, C. Bower, O. Zhou and Y. Wu, Science, 2000, 288, 492 CrossRef CAS.
- C. Goze Bac, S. Latil, L. Vaccarini, P. Bernier, P. Gaveau, S. Tahir, V. Micholet, R. Aznar, A. Rubio, K. Meternier and F. Beguin, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 100302 CrossRef.
- H. Peng, L. B. Alemany, J. L. Margrave and V. N. Khabashesku, J. Am. Chem. Soc., 2003, 125, 15174 CrossRef CAS.
- L. S. Cahill, Z. Yao, A. Adronov, J. Penner, K. R. Moonoosawmy, P. Kruse and G. R. Goward, J. Phys. Chem. B, 2004, 108, 11412 CrossRef CAS.
- S. Latil, L. Henrard, C. Goze Bac, P. Bernier and A. Rubio, Phys. Rev. Lett., 2001, 86, 3160 CrossRef CAS.
- A. Kitaygorodskiy, W. Wang, S.-Y. Xie, Y. Lin, K. A. Shiral Fernando, X. Wang, L. Qu, B. Chen and Y.-P. Sun, J. Am. Chem. Soc., 2005, 127, 7517 CrossRef CAS.
- E. Zurek and J. Autschbach, J. Am. Chem. Soc., 2004, 126, 13079 CrossRef CAS.
- N. A. Besley, J. J. Titman and M. D. Wright, J. Am. Chem. Soc., 2005, 127, 17948 CrossRef CAS.
- C. Kim, K. Seo, B. Kim, N. Park, Y. S. Choi, K. A. Park and Y. H. Lee, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 115403 CrossRef.
- Y. G. Hwang and Y. H. Lee, J. Kore. Phys. Soc., 2003, 42, 267 Search PubMed.
- G. Mpourmpakis, G. E. Froudakis, A. N. Andriotis and M. Menon, Appl. Phys. Lett., 2005, 87, 193105 CrossRef.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 37, 2467 CrossRef CAS.
- NBO Version 3.1, E. D. Glendening; A. E. Carpenter; F. Weinhold ( 1995) Search PubMed; A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 Search PubMed.
- Gaussian 03, Revision B.05, M. J. Frischet al., Gaussian, Inc., Pittsburgh PA, 2003 Search PubMed.
- J. R. Cheeseman, G. W. Trucks, T. A. Keith and M. J. Frisch, J. Chem. Phys., 1996, 104, 5497 CrossRef CAS.
- R. Taylor, J. P. Hare, A. K. Abdul-Sada and H. W. Kroto, J. Chem. Soc., Chem. Commun., 1990, 1423 RSC.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, J. Am. Chem. Soc., 1996, 118, 6317 CrossRef CAS.
- A. M. Orendt, J. C. Facelli, J. G. Radziszewski, W. J. Horton, D. M. Grant and J. Michl, J. Am. Chem. Soc., 1996, 118, 846 CrossRef CAS.
- J. G. Radziszewski, B. A. Hess, Jr. and R. Zahradnik, J. Am. Chem. Soc., 1992, 114, 52 CrossRef.
- H. Fallah-Bagher-Shaidaei, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Org. Lett., 2006, 8, 863 CrossRef.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS.
- Z.-Z. Liu, W. Q. Tian, J.-K. Feng, G. Zhang, W.-Q. Li, Y.-H. Cui and C.-C. Sun, Eur. J. Inorg. Chem., 2006, 2808 CrossRef CAS.
- D.-L. Chen, W. Q. Tian, J.-K. Feng and C.-C. Sun, J. Chem. Phys., 2007, 126, 074313 CrossRef.
- D.-L. Chen, W. Q. Tian and C.-C. Sun, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 75, 013201 CrossRef.
- A. Rochefort, D. R. Salahub and P. Avouris, J. Phys. Chem. B, 1999, 103, 641 CrossRef CAS.
- J. Cioslowski, N. Rao and D. Moncrieff, J. Am. Chem. Soc., 2002, 124, 8485 CrossRef CAS.
- H. Pellissier and M. Santelli, Tetrahedron, 2003, 59, 701 CrossRef CAS.
- A. Sygula, R. Sygula and P. W. Rabideau, Org. Lett., 2005, 7, 4999 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 13C NMR chemical shifts (CS), nucleus-independent chemical shifts (NICS), and HOMO–LUMO contour maps of open-ended SWCNTs C40–C180. See DOI: 10.1039/b9nr00159j |
| This journal is © The Royal Society of Chemistry 2010 |