Nanoscale assembly of high-temperature oxidation-resistant nanocomposites
Xiao
Peng
State Key Laboratory for Corrosion and Protection, Institute of Metal Research, Chinese Academy of Sciences, 62 Wencui Rd., Shenyang 110016, China
First published on 8th October 2009
Abstract
Structural considerations for designing a high-temperature oxidation-resistant metallic material are proposed, based on the dependence of the material structure on a promotion of the development of a protective scale of chromia or alumina. The material should have numerous sites on its surface for nucleating the protective oxides at the onset of oxidation and abundant grain boundaries in deeper areas for simultaneously supplying sufficient flux of the protective-oxide-forming elements toward the surface for a rapid linkage of the oxide nuclei through their lateral growth. Based on these considerations, we fabricated, using an electrochemical deposition method, novel nanocomposites which have a nanocrystalline metal matrix containing Cr and/or Al nanoparticles dispersed at the nano length scale. The validity of the design considerations is verified by comparing the high-temperature oxidation of a typical Ni–Cr nanocomposite system with two types of conventional Ni–Cr materials having similar or higher Cr content but different structure: one is a composite having a nanocrystalline Ni matrix containing Cr microparticles dispersed at the microscale and the other are micron-grained Ni–Cr alloys with the Cr distribution at the atomic length scale.
1. Introduction
Most metallic materials in service at high temperatures unavoidably grow a scale of oxides. Among the thermally grown oxides (TGOs), only those with a high compactness and thermodynamic stability as well as a slow growth rate are oxidation-resistant. Such typical TGOs are chromia and alumina. Unfortunately, most conventionally coarse-grained (CG) commercial alloys (grain size > 1 µm) cannot guarantee the formation of chromia or alumina scales, because of some limitations of the content of Cr and/or Al (e.g., high content would increase the alloy's brittleness and decrease its high-temperature strength or creep resistance). To improve the oxidation resistance of alloys, adding high Cr and/or Al coatings has been a preferred choice in most cases. In the late 1960s Giggins et al.1 and afterward Merz2 reported that grain refinement promoted the formation of TGO chromia on Cr-containing alloys, and an enhanced Cr diffusion to the oxidation front through the increased grain boundaries (GBs) was considered to be responsible. In the last decade, the effect of alloy grain refinement on oxidation has been extended into the nano regime.3–8 Nanocrystallization, without any increase in Cr and/or Al content, has been regarded as another approach to increase the oxidation resistance of alloys. Magnetron sputtering has so far been a popular technique to fabricate oxidation-resistant nanocrystalline (NC) alloys,3,4,6,7 which are single solid solutions in a non-equilibrium state9 where the protective oxide-forming components such as Cr and/orAl exists as solute atoms. Although the fundamental effect of alloy nanocrystallization on oxidation remains unclear, the result that grain refinement promotes the selective oxidation of alloys provides a basis for designing an oxidation-resistant material by means of nanostructuring it, not through conventionally increasing its content of Cr and/or Al. In this contribution, we present structural considerations for nanoscale assembly of novel oxidation-resistant metal-based nanocomposites using an electrochemical deposition method.2. Structural considerations
The oxidation of alloys is generally divided into two stages: an initial and transient stage in which a scale of the most thermodynamically-stable oxide in a given condition establishes and a steady-state stage in which the oxide scale grows in the direction normal to the surface. If a Cr- and/or Al-containing alloy is to become oxidation-resistant, it is vital to form a protective scale of chromia or alumina during the initial and transient stage. Based on a model for the scaling process of a pure metal,10 the selective oxidation of Cr or Al to form the corresponding protective oxide scale during the initial and transient stage can be divided into four sequential steps, as schematically illustrated in Fig. 1 and addressed as follows: (1) adsorption of oxygen gas to the metal surface, (2) nucleation of numerous individual protective oxides, (3) lateral growth of the protective oxide nuclei to form a continuous layer, (4) thickening of the oxide layer (entering the steady-state oxidation stage). For the oxidation of most Fe-, Ni- or Co-based alloys, the limited content of Cr and/or Al, together with the CG structure, normally causes the alloys to be covered by a non-protective scale of the base-metal oxides because of an easy linkage of the nuclei of the oxides, although they nucleate simultaneously with chromia and/or alumina at the onset of oxidation. Accordingly, structural considerations to make steps (2) and (3) operative are essential for designing oxidation-resistant materials. | ||
| Fig. 1 Schematic process for the selective formation of a protective oxide scale during the initial and transient oxidation stage. Olive area: a protective oxide; orange area: the base-metal oxide. | ||
With this in mind, we first consider seeding particles for growing a protective oxide scale into a metal matrix. When oxidation starts, the protective oxide will nucleate on the particles adjacent to the surface, as schematically demonstrated in Fig. 2. Assuming that the particles are spherical in shape and their spatial distribution in a unit cell follows a simple cubic structure, the dependence of the interparticle spacing (s) on the particle size (r) can be calculated using the following equation
 | (1) |
 | (2) |
 | ||
| Fig. 2 Schematic diagram showing protective oxides nucleating on the surface particles and the spacing between the oxide nuclei depends on the particle size at a given particle content. | ||
That is, the interparticle spacing decreases with the particle size, as illustrated in Fig. 3. For example, s is reduced from 1 µm to 50 nm if the particle size becomes 20 times smaller, and further to 10 nm if it becomes 100 times smaller. Such a close interparticle spacing would permit an easier linkage of the oxide nuclei (islands) initially formed on the particles through their lateral growth.
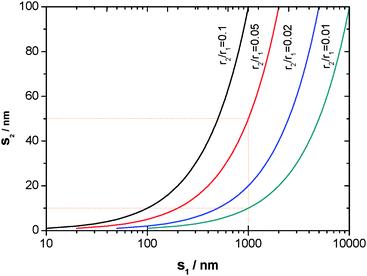 | ||
| Fig. 3 Dependence of the spacing (s2) between r2-sized particles on that (s1) between r1-sized reference particles. | ||
The second consideration is to promote the lateral growth of the protective oxide nuclei (islands) presented in Fig. 2, by enhancing the diffusion of this oxide-forming element to the surface through refining the grains of the metal matrix of the above composites. Here, an effective diffusivity (Deff) is used, which is a summation of lattice (Dl) and GB diffusivity (DGB) as given below11
| Deff = (1 − f)Dl + fDGB | (3) |
 | (4) |
If regarding D*eff as the effective diffusivity of the protective oxide-forming element in the 10 µm-sized alloy, the dependence of the Deff/D*eff ratio on d is presented in Fig. 4 (where δ ≈ 0.5 nm12). Clearly, the Deff/D*eff ratio is dramatically increased when the grain size is decreased down to the nano regime. For example, if d is reduced from the reference 10 µm to 100 nm, Deff becomes one order of magnitude larger than D*eff in the case of Dl/DGB = 103, and two orders of magnitude larger when Dl/DGB is equal to 105. Therefore, the initial nuclei (islands) of the protective oxide will quickly link together if the metal matrix dispersing the particles is nanograined.
 | ||
| Fig. 4 Variation of the ratio of effective diffusivity (Deff) in a d-grained metal to the effective diffusivity (D*eff) in a 10 µm grained reference metal with a decrease of d. | ||
In summary, the deliberately designed materials will characteristically exhibit the nanosize effects of the seeding particles and the matrix grains on the formation of the protective scale during oxidation. Based on these structural considerations, nanocomposites having a nanocrystalline metal matrix with a nano length scale distribution of nanoparticles of Cr and/or Al would be oxidation resistant at high temperatures, because the nanoparticles adjacent to the surface will act as “diffusionless nucleating sites” for chromia or alumina at the onset of oxidation, and at the same time the nanoparticles in deeper areas will act as “reservoirs” for supplying Cr or/Al to the surface through the abundant GBs in the nanocrystalline metal matrix for the rapid linkage of the formed chromia or alumina nuclei (islands) to establish a continuous layer.
After the formation of the protective oxide scale, the flux of the oxide-forming elemental atoms from the composite interior to the scale–composite interface would be kinetically reduced to a very low value equal to that from the interface to the scale surface required for the steady-state growth of the oxide scale. In this case, decrease of Deff (eqn (4)) with the coarsening of the nanograins of the metal matrix of the nanocomposites, because they are not thermally stable in the structure13,14 during oxidation, may not significantly affect the stable oxide growth.
3. Fabrication and characterization
Electrodeposition is a sophisticated technique with the merits of simple processing and ease of fabrication, cost-efficient, high productivity and good compositional control. In electrodeposition the metal species are dissolved cations or complexes, and are reduced on the metallic cathode through the reaction: Mz+ + ze− → M. The as-deposited metals, according to their chemical, physical and mechanical properties, have been extensively used as different surface decorative or functional (magnetic, catalytic, optical, etc.) coatings, as well as structural coatings against friction and liquid corrosion, as reviewed elsewhere.15 To further increase the properties (in particular the mechanical, tribological and wet corrosion properties) of these electrodeposits, suitable solid particles are normally added by composite electrodeposition with a metal matrix from a particle-loaded electrolytic bath. Composite electrodeposition was first reported by Grazen in the 1960s.16 Since then, this technique has been widely used for developing various metal-based composites, most of which contain dispersed second-phase particles of oxide or non-oxide ceramics.15 However, works on an application of composite deposition to develop oxidation-resistant chromia or alumina formers are still lacking.Electrodeposition is a typical method to fabricate nanocrystalline metals, and electrodeposited nanocrystalline Ni has been reported elsewhere.17,18 On this basis, nanoparticles of Cr or/Al may be seeded into a nanocrystalline Ni matrix using the composite deposition method to fabricate the oxidation-resistant nanocomposites proposed. Fig. 5 shows the set-up for assembly of electrodeposited nanocomposites (ENCs). The insets on the upper left and right are typical TEM images of the Cr and Al nanoparticles, respectively. Cr nanoparticles were prepared using a direct-current plasma process. They were gathered from the condensed downstream gas of Cr from the chamber where pure Cr was used as an anode and evaporated in an Ar atmosphere at a voltage of 30 V and the current ranged from ∼200 A. The as-prepared Cr nanoparticles were mainly spherical, but some appeared to be cubic. The Al nanoparticles were prepared in an Ar atmosphere at a voltage of ∼32 kV using a wire electrical explosion method. They were spherical in shape. The Ni-based ENCs were electrodeposited from a conventional sulfate or citrate bath with nanoparticle loading. Two Ni plates were used as anodes and another Ni plate after being ground to 800 grit SiC paper for depositing the ENCs were used as cathodes.
 | ||
| Fig. 5 Electrodeposition set-up for the nanoscale assembly of nanoparticles into a nanocrystalline metal matrix. | ||
During electrodeposition, the bath was agitated by a reciprocating perforated plastic plate driven by a motor. The agitation-concomitant hydrodynamic forces caused the nanoparticles to suspend in the electrolyte and to convectively transport toward the metallic cathode. The nanoparticles, before they were embedded into the Ni matrix on the cathode, generally experienced several consecutive steps which can be summarized as follows:15,19,20 1) forming ionic clouds on the surface, 2) transferring toward the cathode through three successive layers consisting of a convention layer (typical width <1 mm), a concentration boundary layer (namely, a diffusion layer generally hundreds of microns in width) and an electrical double layer (normally with width in nano regime), 3) being adsorbed on the cathode surface, and finally 4) being entrapped within the Ni matrix. The deposition content of nanoparticles was profoundly affected by the bath-loaded particle content and also slightly by the processing parameters (current density, pH, temperature and hydrodynamics). Moreover, adding some surfactants and additives can greatly increase the co-deposition content.21
Using the above method, various Ni-based ENCs such as Ni–Cr,22,23 Ni–Al24 and Ni–Cr–Al25 could be developed. The fine structures of the ENCs were characterized using TEM. The nanoparticles were, in general, individually dispersed in the Ni matrix, but some agglomerates were also seen. A typical fine structure of the as-deposited ENCs can be seen in Fig. 6. The white spherical areas were the co-deposited particles (Al particles in this case) in various size ranges. No cracks or pores were visible between the particles and Ni. The average grain size of the Ni matrix was in the range 30–60 nm. The rather uniform rings of the corresponding selected-area electron diffraction (SAED) pattern in the inset indicate random crystallographic orientations of the nanograins of the Ni matrix. The grain size of the Ni matrix could be further refined by adding suitable additives such as saccharin or by using pulse power.15,26
 | ||
| Fig. 6 Bright-field TEM image of an as-prepared Ni–Al nanocomposite with the inset showing the corresponding SAED pattern. | ||
4. High-temperature oxidation
4.1 Nanosize effect of particles
It is proposed from the first structural consideration that the nanocomposites seeding the nanosized particles of Cr and/or Al tend to be chromia- or alumina-formers (Fig. 2). To confirm this, we took electrodeposited Ni–Cr composites which have a nanocrystalline Ni matrix but different sizes of Cr particles. Fig. 7 shows the comparison of the oxidation kinetics at 900 °C in air of the Ni–Cr composites containing different sizes and numbers of dispersed Cr particles. There are several observations. First, the composite containing on average ∼2.4 µm-sized Cr of 12.4 wt% (14.8 ΔV%) is oxidized faster than the electrodeposited pure Ni. Second, the ENC containing ∼39 nm-sized Cr particles is oxidized as fast as the Ni–12.4Cr composite when the nanoparticle content is 9.0 wt% (10.9 ΔV%), and its oxidation rate is greatly reduced when the content becomes 10.9 wt% (13.1ΔV%). Third, the ENC seeded with smaller Cr nanoparticles (mean size: 21 nm) has the slowest scaling rate, although it has only 7.5 wt% Cr (9.1 ΔV %).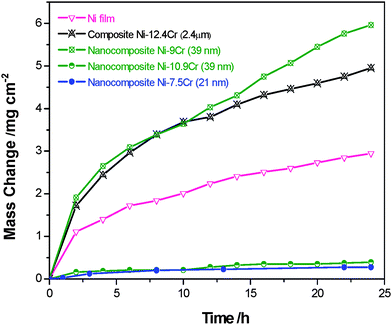 | ||
| Fig. 7 Oxidation kinetics in air at 900 °C of the Ni-based composites assembled using Cr particles of different sizes and content. | ||
The scales on these electrodeposited composites after oxidation in air at 900 °C were characterized using X-ray diffraction (XRD) and scanning electron microscopy/energy dispersive X-ray analysis (SEM/EDXA). For the cross-sectional investigation, surface oxide scales were protected in advance by electroless Ni plating. Typical surface and cross-sectional features of the scales are shown in Fig. 8. The pure Ni deposits, during 24 h oxidation, grow a NiO scale with a large-grained and porous structure (Fig. 8a). A NiO scale of similar structure also occurs on the Ni–12.4Cr composite oxidized for 24 h (Fig. 8b); however, the thicker scale suggests that the NiO grows more quickly on this composite than on the pure Ni. The ENC Ni–9.0Cr for 20 h oxidation grows a NiO scale with the above surface and cross-sectional morphological characteristics. In contrast, the ENC Ni–10.9Cr forms a finer-grained, thinner Cr2O3 scale. Fig. 8c shows the SEM top and cross-sectional views of the scale on the ENC Ni–7.5Cr (particles ∼21 nm size) oxidized at 900 °C in air but for 80 h. The scale is only Cr2O3, which is obviously dense and very fine-grained.
 | ||
| Fig. 8 Typical surface and cross-sectional morphologies of the scales on (a) pure Ni, (b) ENC Ni–12.4Cr and (c) ENC Ni–7.5 Cr. | ||
The Ni–12.4Cr (particles ∼2.4 µm size) cannot form a chromia scale. The main reason lies in that the interparticle spacing is in the micrometre size (2.5 µm calculated using eqn (2)). Such a micron length scale spacing makes linkage of the chromia islands locally formed on the surface particles impossible before the oxide is entirely “engulfed” by the faster growing NiO scale. The incorporation of chromia would increase the defects in the growing NiO scale in these conditions (see the cross-sectioned scale in Fig. 8b) and thus the oxidation rate of the composite. However, for Ni–9.0Cr (particles ∼39 nm size), the calculated interparticle spacing is in the nano range (53 nm), slightly larger than the interparticle spacing (46 nm) for the Ni–10.9Cr containing the same sized particles, a NiO scale still grows. This means that the nano length scale distribution of Cr particles is a necessary but not sufficient condition for the formation of an external chromia scale. There exists a critical interparticle spacing which permits the composites to be a chromia former. The critical value should be in the range 46–53 nm for the ENCs containing 39 nm-sized particles. On this basis, it is understandable that Ni–7.5Cr with nanoparticles distributed on an ∼33 nm length scale forms an exclusive external chromia scale during oxidation, although it is lower in Cr content than the above Ni–9.0Cr and Ni–12.4Cr composites.
Hence, the oxidation resistance of the Ni–Cr composite system strongly depends on the size of the Cr particles. Assembly of finer particles tends to reduce the critical Cr content for the formation of an exclusive external Cr2O3 scale.
4.2 Nanosize effect of metal-matrix grains
To clarify the second structural consideration that nanocrystallization of the metal-matrix of the nanocomposites proposed promotes the formation of a protective scale of chromia or alumina, the oxidation at 900 °C in air of the ENC Ni–7.5 Cr (particles ∼21 nm size) was compared with CG alloys Ni–10Cr and Ni–20Cr (mean grain size: ∼30 µm) which were prepared using arc melting as reported previously.23 The oxidation curves are presented in Fig. 9. Clearly, among the three samples, the ENC has the lowest oxidation rate. The scale formed on the Ni–10Cr alloy during 24 h is typically featured as non-protective NiO, as seen in Fig. 10a. Fig. 10b presents the surface and cross-section of the Ni–20Cr alloy oxidized for 24 h. The finer-grained surface oxides are a mixture of NiO and spinel NiCr2O4 based on combined analyses of XRD and EDXA data. Below the thin mixed oxides, a Cr2O3 layer developed, as seen in the scale cross-section.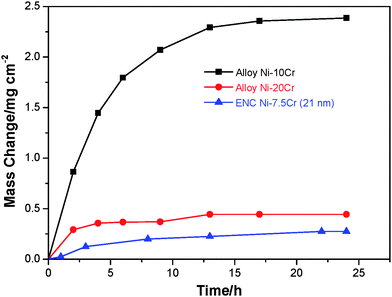 | ||
| Fig. 9 Oxidation kinetics of ENC Ni–7.5 Cr and two arc-melted Ni–Cr alloys in air at 900 °C. | ||
 | ||
| Fig. 10 Typical surface and cross-sectional morphologies of the scales on (a) Ni–10Cr and (b) Ni–20Cr alloys. (Note: The dark layers in (a) and (b) are the gaps between the scales and the top electroless Ni plating). | ||
The oxidation behaviors of the two CG alloys may be explained by comparison to the model proposed for the selective oxidation of the ENCs. Cr2O3 and NiO nucleate together at the onset of oxidation, but the Cr2O3 nuclei cannot link together before they are “engulfed” by the faster growing NiO. Consequently, a NiO layer preferentially develops. The continuous growth of the layer progressively reduces the partial pressure of O2 (PO2) at the NiO–alloy interface. When the PO2 decreases to a value limit, fresh Cr2O3 nucleates and then grows laterally to form a continuous layer. This should be the case for the Ni–20Cr alloy but not for the Ni–10Cr, because the much higher Cr concentration in the former can supply the amount of Cr to the interface needed for the Cr2O3 layer formation. Afterward, the top NiO ceased to grow. It accounts for the result that the surface NiO is finer-grained on the Ni–20Cr alloy (Fig. 10b) than on the Ni–10Cr alloy (Fig. 10c).
The ENC Ni–7.5Cr (Fig. 8c), although it has a lower content and distribution uniformity of Cr in comparison to the two arc-melted alloys, exhibits a greatly-increased ability for thermally developing an exclusive external scale of chromia. This follows the second structural consideration above. The thin Cr2O3 scale is retained during 80 h exposure of the ENC, suggesting that its structural evolution with oxidation does not affect the steady-state growth of this oxide. This is consistent with the assumption that the thermal stability of the ENCs would not significantly influence the stable growth of the protective oxide scale once it can form during the initial and transient oxidation stage. In fact, our recent study shows that the electrodeposited nanocrystalline Ni grains, although coarsened, are still submicron-sized.27 The ultrafine-grained structure is able to sustain the transport of the protective-oxide-forming element at a relatively fast rate, at which the amount of the elemental atoms arriving at the interface can reach the value required for constant thickening of the protective oxide scale. Thus, one can expect that if the Ni–Cr ENCs have appropriate thickness and Cr content, they would maintain good long-term stability and durability at high temperatures, through a slow, continuous and exclusive growth of an external scale of chromia. The case will be applicable to the oxidation of other chromia- or alumina-forming ENC systems.24,25,28
5. Conclusions
This work will constitute a frame of reference for fabricating oxidation-resistant materials through deliberately nanostructuring, not conventionally increasing the Cr and/or Al content. The nanocomposites which integrate a certain amount of Cr and/or Al nanoparticles with a nanocrystalline metal matrix are oxidation-resistant during oxidation at high temperatures, because of the combined nanosize effects of the particles and metal-matrix grains on the promoted formation of a protective scale of chromia or alumina. Such typical nanocomposites can be assembled using an electrochemical deposition method. The chromia- or alumina-forming nanocomposites may also be applied to metals with a poor oxidation resistance as substitutes for protective coatings manufactured using other techniques such as physical or chemical vapor deposition.Acknowledgements
The author is grateful to support from “One Hundred Talents Plan” sponsored by the Chinese Academy of Sciences and from the National Natural Science Foundation of China (project Grant Nos. 50674082 and 50571108).References
- C. S. Giggins and F. S. Pettit, J. Electrochem. Soc., 1971, 118, 1782 CrossRef CAS.
- M. D. Merz, Metall. Trans., 1979, 10A, 71 Search PubMed.
- X. Peng, F. Wang, in Oxidation resistant nanocrystalline coatings, ed. W. Gao and Z. Li, Woodhead Pub. Ltd, Cambridge, England, 2008, ch. 15, pp. 456–475 Search PubMed.
- W. Gao, Z. Y. Liu and Z. W. Li, Adv. Mater., 2001, 13, 1001 CrossRef CAS.
- S. Mrowec, High Temp. Mater. Proc., 2003, 22, 1 Search PubMed.
- G. F. Chen and H. Y. Lou, Mater. Lett., 2000, 45, 286 CrossRef CAS.
- Z. Liu, W. Gao, L. Karl and F. Wang, Acta Mater., 1998, 46, 1691 CrossRef CAS.
- L. Ajdelsztajn, J. A. Picas, G. E. Kim, F. L. Bastian, J. Schoenung and V. Provenzano, Mater. Sci. Eng., A, 2002, 338, 33 CrossRef.
- S. M. Shin, M. A. Ray, J. M. Rigsbee and J. E. Greene, Appl. Phys. Lett., 1983, 43, 249 CrossRef CAS.
- P. Kofstad, in High Temperature Corrosion, Elsevier Applied Science, London and New York, 1998 Search PubMed.
- E. W. Hart, Acta Metall., 1957, 5, 597 CrossRef CAS.
- B. Fultz and H. N. Frase, Hyperfine Interact., 2000, 130, 81 CrossRef CAS.
- U. Klement, U. Erb, A. M. El-Sherik and K. T. Aust, Mater. Sci. Eng., A, 1995, 203, 177 CrossRef.
- F. Czerwinski, A. Zielinska-Lipiec and J. A. Szpunar, Acta Mater., 1999, 47, 2553 CrossRef CAS.
- C. T. J. Low, R. G. A. Wills and F. C. Walsh, Surf. Coat. Technol., 2006, 201, 371 CrossRef CAS.
- A. E. Grazen, US Pat., 3061525, 1962.
- A. M. El-Sherik, U. Erb, G. Palumbo and K. T. Aust, Scr. Metall. Mater., 1992, 27, 1185 CrossRef CAS.
- H. Gleiter, Acta Mater., 2000, 48, 1 CrossRef CAS.
- J. R. Roos, J. P. Celis, J. Fransaer and C. Buelens, J. Met., 1990, 42, 60 Search PubMed.
- N. Guglielmi, J. Electrochem. Soc., 1972, 119, 1009 CrossRef CAS.
- C. Zhang. MSc thesis, Institute of Metal Research, Chinese Academy of Sciences, 2005.
- Y. Zhang, X. Peng and F. Wang, Mater. Lett., 2004, 58, 1134 CrossRef CAS.
- C. Zhang, X. Peng, J. Zhao and F. Wang, J. Electrochem. Soc., 2005, 152, B321 CrossRef CAS.
- Y. Zhou, X. Peng and F. Wang, Scr. Mater., 2004, 50, 1429 CrossRef CAS.
- X. Yang, X. Peng, C. Xu and F. Wang, J. Electrochem. Soc., 2009, 156, C167 CrossRef CAS.
- A. M. El-sherik and U. Erb, J. Mater. Sci., 1995, 30, 5743 CrossRef CAS.
- L. Zheng, X. Peng, F. Wang, in preparation.
- Z. Huang, X. Peng, C. Xu and F. Wang, J. Mater. Res., 2007, 22, 3166 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |