New therapeutic potential for psychoactive natural products
Katherine M.
Prevatt-Smith
and
Thomas E.
Prisinzano
*
Department of Medicinal Chemistry, The University of Kansas, Lawrence, KS 66045-7582, USA. E-mail: prisinza@ku.edu; Fax: +1-785-864-5326; Tel: +1-785-864-3267
First published on 26th October 2009
Abstract
Much of our knowledge in neuroscience was discovered through the study of mind-altering natural products. However, although much has been learned about human physiology and basic biological processes, the underlying causes of CNS disorders and other disease states are still elusive. Based on its main past successes, the continued study of mind-altering compounds promises to yield novel agents that may be developed into medications and to identify new targets for the treatment of diseases. This Highlight describes the history of investigations into several classes of mind-altering natural products and relates recent and potential therapeutic uses for these agents.
 Katherine M. Prevatt-Smith | Katherine M. Prevatt-Smith received a B. S. (2006) in Chemistry from Emory University in Atlanta, GA. During her undergraduate studies, she spent a summer studying dopamine medicinal chemistry in the laboratory of Dr. Amy H. Newman at the National Institute on Drug Abuse in Baltimore, MD. After earning her degree, she moved to the University of Kansas to pursue her Ph.D. under the supervision of Professor Thomas E. Prisinzano. Her research focuses on the synthesis and structure–activity relationships of natural products with psychoactive activity. |
 Thomas E. Prisinzano | Thomas E. Prisinzano received a B.S. (1995) in Chemistry from the University of Delaware. He earned his Ph.D. studying serotonin medicinal chemistry with Richard A. Glennon in the School of Pharmacy at Virginia Commonwealth University. He was an Intramural Research Training Award Fellow at the National Institutes of Health with Kenner C. Rice, where he studied the chemistry and pharmacology of drugs subject to abuse and potential treatment agents. He is now an Associate Professor of Medicinal Chemistry at the University of Kansas. His research focuses on the identification, structure elucidation, and synthesis of natural products with central nervous system activity. |
Introduction
Naturally occurring mind-altering substances are among the oldest compounds used by mankind. For millennia the plants containing these compounds have been ingested for both spiritual and recreational purposes. Documented examples of mind-altering substance use include the soma of ancient India,1,2 the teonanácatl of the Toltecs in pre-Columbian Central America,3 the use of peyote as a sacrament of the Native American Church (NAC),4 and the chewing of khat in East Africa and the Middle East.5,6 The World Health Organization estimates that 10 million people worldwide chew khat (Catha edulis Forsk., Celastraceae), the active component of which is cathinone (1), an amphetamine like-alkaloid.5,6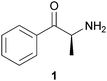
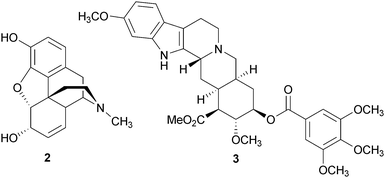
It was not until the nineteenth century that technology was adequate to isolate the principle agents responsible for the mind-altering effects of these plants. In 1803 Surtürner isolated morphine (2) from the opium poppy, Papaver somniferum L. (Papaveraceae).7,8 Although 2 was used as a drug throughout the 1800s, because of its complex nature, the correct structure of 2 was not proposed until 1925, by Gulland and Robinson.9–11 Interestingly, the structure was not finally confirmed until the total synthesis by Gates and Tschudi in 1953.12,13 Over a century of research into the chemistry and pharmacology of 2 and its derivatives has led to the discovery of the opioid receptors and the endogenous opioids, and significantly advanced our understanding of pain. The study of alkaloids from Rauvolfia serpentina Benth. (Apocynaceae), such as reserpine (3), which can evoke depression, has lead to the development of the monoamine neurotransmitter deficit theories that describe the underlying causes of depression.14 Studies of 3 have also advanced our understanding of the norepinephrine, serotonin, and dopamine neurotransmitter syntheses and release processes.
Lysergic acid diethylamide (LSD) is perhaps the most popularly known mind-altering substance. LSD (4) is related to a class of compounds called ergolines or ergot alkaloids, which are produced by a parasitic fungus that grows on cereal grains.15,16 One of the most powerful alkaloids produced by this fungus is lysergic acid amide (LSA) or ergine (5). Consumption of this compound via contaminated food can cause a number of symptoms including gangrene, hallucinations, and convulsions. Some have hypothesized that ergot poisoning may have contributed to the furor of the Salem witchcraft trials of 1692.15 Alkaloid 4 is a synthetic derivative of 5 and was made by Albert Hofmann in 1943.16 Alkaloid 4 and other compounds that produce effects on the mind have been called hallucinogens because they cause sensory distortions and elicit hallucinations. This term has been applied across a breadth of structurally different compounds that produce different mind-altering effects. The most accepted definition of a hallucinogenic substance is one described by Hollister.17 According to Hollister, hallucinogenic agents are those agents which when administered in a single effective dose (a) consistently produce noticeable changes in thought, mood, and perception without memory impairment, (b) produce neither narcosis nor excessive stimulation, (c) produce no severe autonomic side effects, and (d) are non-addictive. Thus, this definition has been extremely useful in narrowing the list of substances to which the term hallucinogen can be applied. However, the term hallucinogen has been used to describe the mind-altering effects of several classes of agents including: serotonergics, anticholinergics, cannabinoids, and opioid agonists.
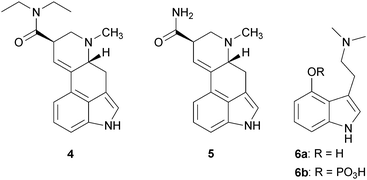
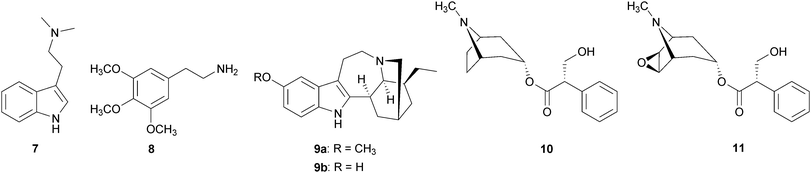
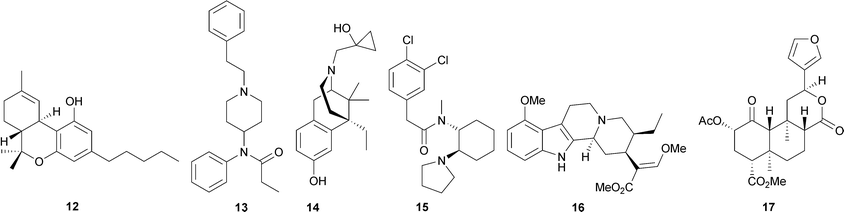
Serotonergics exert their effect as full or partial agonists at the serotonin 5-HT2A receptor. This class includes compounds such as 4, psilocybin (6b), N,N-dimethyltryptamine (DMT, 7), mescaline (8), and ibogaine (9a).18,19 Anticholinergics are antagonists at muscarinic cholinergic receptors. Examples of anticholinergics are the alkaloids L-hyoscyamine (10), atropine (racemic (−)-hyoscyamine), and scopolamine (11), isolated from members of the Solanaceae plant family.20 Cannabinoids include Δ9-tetrahydrocannabinol (Δ9-THC, 12) from the plant Cannabis sativa L. (Cannabaceae). Agonists at opioid receptors include synthetically derived small molecules such as fentanyl (13), bremazocine21 (14) and U50,48822 (15), and also the natural products 2, mitragynine (16), isolated from the Southeast Asian herb Mitragyna speciosa Korth. (Rubiaceae), and salvinorin A (17), isolated from the Mexican mint plant Salvia divinorum L. Epling & Jativa (Lamiaceae).
Serotonergics
Serotonergic hallucinogens, often called ‘classical hallucinogens’, are known to produce profound sensory distortions and hallucinations. Chemically, these compounds can be divided into three categories: phenylethylamines, tryptamines, and ergolines.18 Alkaloid 8, considered the prototypical example of a phenylethylamine, is the principal hallucinogenic component in the peyote cactus, Lophophora williamsii J. M. Coult. (Cactaceae).18 Alkaloid 6b and psilocin (6a), found in ‘magic mushrooms’ (Psilocybe cubensis Singer, Strophariaceae), and 7 found in Psychotria viridis Ruiz & Pav. (Rubiaceae) are naturally occurring tryptamines.20 Tryptamine 7 in combination with β-carbolines such as harmine (18) from Banisteriopsis caapi C. V. Morton (Malpighiaceae) are the active components of the brewed tea ayahuasca, which is used by the indigenous peoples of the Amazon for both spiritual and medicinal purposes.20,23 Tryptamine 7 is known to be hallucinogenic when smoked, inhaled, or injected, but is only orally active when the monoamine oxidase (MAO) enzymes in the intestines and liver are inhibited by co-ingestion of 18 and/or its related alkaloids.20,23 Finally, the category of ergolines is related to the category of tryptamines. Whereas tryptamines are structurally relatively simple and have considerable conformational flexibility, ergolines, exemplified by 4, are structurally rigid compounds.18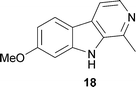
Serotonergic hallucinogens have historically been used for the study of brain function and mental illness. In 1952 Osmond and Smythies observed that many hallucinogenic compounds are structurally similar to endogenous neurotransmitters and hypothesized that the hallucinogenic experience could be a model for psychosis,24 hence the term ‘psychotomimetic’ has sometimes been used to describe these compounds. Therefore the study of hallucinogenic compounds such as 4 and 8 could lead to insights into the nature of schizophrenia and other mental disturbances. Indeed, there were many clinical studies of hallucinogens through the 1950s and 1960s, mostly focusing on 4 as a precipitant of a model psychosis. These efforts ceased by the 1970s due to concern about the unstructured use of such compounds in popular culture.25 Past clinical and pharmacological studies with serotonergic hallucinogens have recently been extensively reviewed by Nichols.18
Because some serotonergic hallucinogens have such a dramatic effect on the mind, it is not surprising that there is a recent resurgence of research interest in the 5-HT2A receptor for its importance in psychiatric disorders as well as in cognitive functions. Currently, there are two broad research areas where there is clinical interest in classical hallucinogens. The first is in the treatment of mental illness. This arises from the continuing belief that hallucinogenic compounds may be able to alleviate certain psychiatric conditions. In fact, there is growing evidence that 4, 6b and 8 may all be useful for treating the symptoms of obsessive-compulsive disorder.26–29 Research exploring this correlation is beginning to emerge.30,31 There is also research interest in antagonists at the 5-HT2A receptor, as compounds with this activity appear to be able to improve the cognitive deficits associated with some psychiatric disorders.32–35 Additionally, there is growing evidence suggesting that ayahuasca may be useful in treating depression and other mood disorders as well as alcohol or other substance abuse.23 Finally, agonists at the 5-HT2C receptor are currently being investigated as appetite suppressants and treatments for obesity.36–39
Ibogaine (9a) is a naturally occurring indole alkaloid (related to the category of tryptamines) that has long been known for its activities as a CNS stimulant and hallucinogen.40 Indole alkaloid 9a is isolated from the shrub Tabernanthe iboga Baill. (Apocynaceae), which is native to western Africa. This plant has been used by indigenous peoples to treat fatigue, hunger and thirst, and as a religious sacrament.41 Indole alkaloid 9a has been implicated as a possible drug abuse therapeutic due to anecdotal reports of its efficacy in eliminating withdrawal symptoms and long-term craving for cocaine and heroin.41 Its primary metabolite, noribogaine (9b), is also biologically active.42,43 Indole alkaloid 9a has a rather complex pharmacology, as it has reported affinities for several receptors, transporters, and ion channels.19,40 Among these are the adrenergic, dopaminergic, serotonergic, muscarinic, and opioidergic receptor systems.19,44,45 In self-administration studies in rats, a single 40 mg kg−1 injection of 9a significantly decreased cocaine intake.46,47 Cocaine-induced locomotor activity in rodents was also reduced by 9a.48,49 Furthermore, under open-label conditions of opioid detoxification in 33 human subjects, 9a eliminated signs of withdrawal and drug-seeking behavior in 25 subjects.50 This effect was sustained throughout the entire 72-hour period of post-treatment observation. There have been concerns about the clinical use of 9a, as high doses (100–300 mg kg−1) precipitate neurotoxicity.51 However, at doses effective for treatment of opiate and cocaine addiction (5–25 mg kg−1), no neurotoxicity has been observed.52 The above evidence suggests that 9a and its derivatives may be useful molecules for the development of medications to treat drug abuse.
The second area of interest is in the value of serotonergic hallucinogens as research tools for cognitive neuroscience. Research has shown that 5-HT2A receptors may play a role in the formation of working memory53 and in learning processes.54 As agonists at the 5-HT2A receptor, classical hallucinogens are quickly becoming important tools for studies of cognition.
Anticholinergics
The earliest and most well known anticholinergics were isolated from the plant family Solanaceae. This family includes jimsonweed or thorn-apple (Datura stramonium L.), henbane (Hyoscyamus niger L.), and deadly nightshade (Atropa belladonna L.).20 From these species come 10, 11 and atropine (racemic (−)-hyoscyamine),55 which have historically been used for their hallucinogenic effects, as they produce profound sensory distortion in low doses, as well as for the pain relief and anaesthesia they produce in higher doses.56 Atropine was first isolated from the roots of belladonna in 1831 by Mein, and then purified by Runge in 1833.57,58 In that same year, 10 was obtained from henbane by Geiger and Hesse.57,58 Recognition of the physiological action of these alkaloids dates back to the independent experiments of Langley and Erlich, from which the concept of a receptor emerged.55 These compounds act as antagonists at the muscarinic cholinergic receptors of the autonomic nervous system.59 In the CNS, muscarinic receptor roles include involvement in cognition, motor control, and cardiovascular and respiratory activity. In the peripheral nervous system (PNS), muscarinic receptor roles include heart rate control, smooth muscle contraction, and vasodilation.55,60 Given their anticholinergic activity, Solanaceae alkaloids are known to be poisonous in large doses.The muscarinic cholinergic receptors are a family of five receptors called M1–M5.60 Their identification and elucidation took over a century, and research in this area was hampered by the fact that in its inception, receptors were only a concept. Early attempts to characterize muscarinic receptors were based on structure–activity relationship (SAR) studies of cholinergic ligands such as atropine, leading to a proliferation of highly selective agonists and antagonists.59 The chemical advances were paralleled by biological, molecular pharmacological, and molecular biological advances that culminated in the ability to isolate, purify, sequence, and clone cholinergic receptors.61,62
An emerging area of research for muscarinic receptors is in allosteric modulators for the treatment of Alzheimer's disease and schizophrenia.63 Pharmacologically, it has been found that relatively non-selective muscarinic antagonists exacerbate cognitive deficits and psychotic behaviors in animal models, whereas agonists have an ameliorating effect.64 The muscarinic receptors thought to be involved are M1, M4, and M5.63,65 However, the discovery of ligands that are highly selective between the different muscarinic receptors has been hampered by the high sequence homology shared in their binding domains.63 Positive allosteric modulators (PAMs) are small molecules that bind to an allosteric site in the receptor and potentiate the activation of the receptor by its endogenous ligand. PAMs provide another, indirect, approach to developing therapies for CNS disorders. Recently, PAMs at M1,66 M4,64,67 and M565 have been reported.
Cannabinoids
Compounds from the cannabis plant have been ingested for centuries and are known to produce an analgesic effect in addition to hallucinogenic effects such as feelings of dissociation from reality. Cannabinoid molecules are divided into two categories: classical cannabinoids and nonclassical cannabinoids. Classical cannabinoids are tricyclic dibenzopyran derivatives that are either found in the cannabis plant or semi-synthetic analogs of these compounds.68 This group of molecules is exemplified by 12, the main hallucinogenic principle in the cannabis plant. Resorcinol 12 was isolated and its structure elucidated by Gaoni and Mechoulam in 1964.69,70 Nonclassical cannabinoids emerged from Pfizer SAR studies of the classical cannabinoids.68 These compounds lack the dihydropyran ring present in Δ9-THC, and CP47497 (19) represents a typical compound in this class.71 Further developments in the SAR of nonclassical cannabinoids led to CP55940 (20), the tritiated derivative of which ultimately lead to the discovery of the CB1 cannabinoid receptor in 1988.72,73 In 1993, a second cannabinoid receptor was found from a human cDNA library and designated CB2 based on its homology to the CB1 receptor and similar ligand binding profile.74 The cannabinoid receptors CB1 and CB2 are G-protein coupled receptors (GPCRs), and are thought to play roles in several physiological processes including metabolic regulation and pain.68,75 The discovery of these receptors quickly resulted in the identification of the endogenous cannabinoids (endocannabinoids) in the mid-1990s. The most well known endocannabinoids are anandamide76 and 2-arachidonyl glycerol (2-AG).77–79 Studies with both CB1 and CB2 receptors have found that anandamide is a low efficacy agonist, whereas 2-AG is a highly efficacious agonist.80,81 Resorcinol 12 acts as an agonist with efficacy similar to that of anandamide. It has been suggested that the hallucinogenic effects of 12 arise from the compound's ability to mimic the action of anandamide at cannabinoid receptors, while simultaneously antagonizing 2-AG at these same receptors. This hypothesis is supported by the observation that a single high dose of a CB1 receptor antagonist has only a limited ability to block the subjective effects of cannabis ingestion.82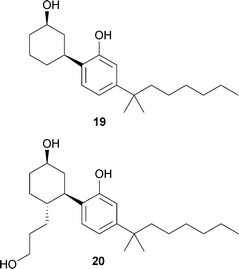
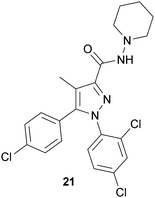
Synthetic 12 (dronabinol, Marinol®) is approved in the United States to treat nausea and vomiting brought on by cancer chemotherapy, as well as an appetite stimulant for cachexia in both cancer and AIDS. Recent advances in the understanding of the endocannabinoid system have broadened the therapeutic possibilities resulting from its manipulation. CB1 receptor antagonists have received the most attention of the potential drugs affecting the endocannabinoid system. Their primary indication is for obesity. The rationale behind this indication lies in the generally accepted notion that ingestion of cannabis enhances the appetite, resulting in increased consumption of rich foods. Therefore, CB1 receptor antagonists should function to reduce the appetite, thereby reducing caloric intake and body weight.75 The first reported CB1 receptor antagonist was rimonabant (21) (SR141716, Accomplia®).83 Pyrazole 21 has nanomolar affinity for CB1 receptors, and little affinity for CB2 receptors. One European clinical study with CB1 receptor antagonists found that a daily dose of 20 mg of 21 coupled with a reduced calorie diet led to an average sustained weight loss of 7 kg vs. an average sustained weight loss of 2 kg in the placebo group, treated with diet alone.84 Although 21 was not approved for use in the United States, the results of the clinical study suggest that CB1 antagonists may be useful drugs for treating obesity.
Another indication for CB1 receptor antagonists is in the treatment of drug abuse. Several studies in animals have observed that CB1 receptor antagonists such as rimonabant reduce the rewarding properties of opioid receptor agonists.85–87 In fact, these rewarding properties are absent in CB1 receptor knockout mice.88 However, opioid receptors do not seem to be involved in the hallucinogenic effects of CB1 receptor agonists, as opioid receptor antagonists do not block these effects.89 Also, the CB1 receptor seems to be involved in responses to both nicotine and alcohol; CB1 receptor antagonists are able to block nicotine-induced conditioned place preference (CPP)90 and to decrease alcohol consumption.91
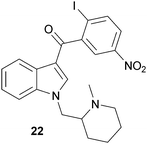
Extracts of the cannabis plant are known to produce an analgesic effect. A particularly attractive feature of selective CB2 receptor agonists as therapeutics is that they are devoid of any known hallucinogenic effects such as those associated with CB1 receptor agonists.75 Although the physiological role of the CB2 receptor has yet to be fully elucidated, several preclinical studies suggest that this receptor may be useful in chronic pain models. Specifically, CB2 receptor agonists such as AM1241 (22) have been shown to produce analgesia in neuropathic pain models as well as peripheral inflammatory models.92,93
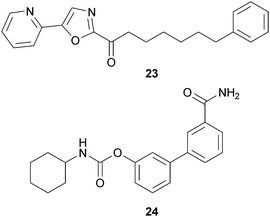
The newest research in the cannabinoid field is investigating the therapeutic potential of fatty acid amide hydrolase (FAAH) inhibitors. FAAH is the major degradative enzyme for anandamide94 and related fatty acid amides such as oleamide. Oleamide is a non-cannabinoid, endogenous fatty acid amide that, like anandamide, is metabolized by FAAH.95 Oleamide has been found to induce sleep in a manner that is indistinguishable from physiological sleep,96 and therefore it is possible that FAAH inhibitors could be used for the treatment of sleep disorders through increasing endogenous oleamide levels. Additionally, FAAH does not appear to metabolize 2-AG in vivo,97 so an inhibitor of FAAH would increase anandamide levels without affecting 2-AG levels. Preclinical studies in animals indicate that anandamide release is stimulated following nociception.98,99 Therefore, inhibitors of the degradation of this and related endocannabinoids might be therapeutically useful as analgesics. In fact, FAAH knockout mice demonstrate increased pain thresholds in several pain models.100 Importantly, FAAH inhibitors do not appear to produce the hallucinogenic effects observed in cannabinoid receptor agonists.99 Two families of FAAH inhibitors that produce analgesia in animal models have been developed, the α-ketoheterocycles (exemplified by OL-135 (23))99,101 and the carbamates (exemplified by URB597 (24)).102,103 These FAAH inhibitors have been found to be effective in both neuropathic104 and inflammatory105 pain models. The analgesia produced by these FAAH inhibitors is blocked by CB1 antagonists.99
Opioid receptor agonists
There are three types of opioid receptors: μ (MOP), named for its most well known agonist, morphine; κ (KOP), named for the agonist initially used to characterize it, ketocyclazocine (25); and δ (DOP), named for the mouse vas deferens tissue preparation.106 These receptors have all been characterized and cloned.107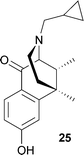
The opium alkaloids, such as 2 and its derivatives, are among the most potent analgesics currently used in the clinic. All of these compounds and many of their synthetic derivatives are selective MOP agonists. Administration of these compounds, while producing analgesia, is associated with several clinically significant side effects such as respiratory depression, constipation, and tolerance and dependence.7,8
Selective KOP agonists are also capable of producing clinically useful analgesia, but lack the respiratory depression, constipation, and addictive properties associated with MOP agonists.7,8 However, a side effect associated with activation of KOP receptors is dysphoria, a hallucinogenic-like effect that is the opposite of euphoria.108 Still, KOP agonists are targets for achieving pain relief without the negative side effects associated with MOP agonists. Although KOP agonists are known to produce dysphoric effects, there is still some hope that a clinically useful analgesic may be found. KOP selective agonists with only a peripheral action have been shown to be effective in relieving inflammatory pain.109–112 This analgesia occurs without the hallucinogenic-like side effects (dysphoria) associated with KOP agonists that have central activity.
Another important indication for KOP agonists is in the treatment of drug dependence. This is a chronic and relapsing condition that involves compulsive drug-seeking and drug-taking behavior.113 This behavior persists despite serious negative consequences. The chronic use of drugs of abuse leads to adaptive physiological changes in the brain that further lead to drug tolerance, physical dependence, craving, and eventually relapse.8,114 KOP receptors have been found to be important for opposing relapse and drug seeking behavior.115 In fact, in animal models, administration of KOP agonists has been found to decrease cocaine self-administration,116–119 inhibit cocaine-conditioned place preference and locomotor sensitization,120 and decrease cocaine-induced reinstatement of drug-seeking behavior.119,121 These results imply that KOP agonists could be used in the treatment of cocaine or other stimulant abuse.
Kratom (Mitragyna speciosa Korth., Rubiaceae) is an herb indigenous to Southeast Asia that is traditionally used to treat fever, diarrhea, fatigue, pain, and as a substitute for 2 in treating opioid addicts.122,123 The main component of kratom is the indole alkaloid mitragynine (16), which has been reported to have affinities for all three opioid receptors, though it appears to be relatively selective for MOP receptors.124,125 Additionally, other constituents of kratom and derivatives of 16, including mitragynine pseudoindoxyl (26) and 7-hydroxymitragynine (27), have also been found to have affinity for opioid receptors.125
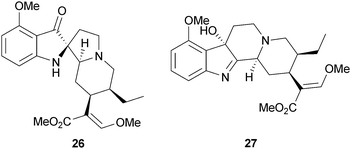
Research interest in 16 stems from its increasing use as a remedy for opioid withdrawal by individuals who self-treat chronic pain.126–128 Indole alkaloid 16 is known to produce antinociception in mice in the tail-pinch and hot-plate tests,124 in the tail-flick test,125 and in the PBQ writhing and formalin-induced pain response tests.129 Additionally, 16 has been observed to reduce locomotor activity in rats, an indication of a sedative effect.123 Furthermore, in a case report of a human subject, ingested kratom was reported to effectively attenuate opioid withdrawal.128 However, the exact mechanisms underlying the effect of 16 are currently unknown. It has been hypothesized that the MOP agonism of 16 might avert withdrawal symptoms, while KOP agonism might attenuate reinforcement and blunt cravings.128 The collective findings of the effects of 16 indicate that the molecule and its derivatives may be useful for the development of new analgesics and possibly for the treatment of opioid abuse. Analogs of mitragynine are currently being explored as potential analgesic alternatives to morphine.130–134
Salvinorin A (17), a neoclerodane diterpene, is the active hallucinogenic component in the Mexican mint plant Salvia divinorum L. Epling & Jativa (Lamiaceae).135–138 This plant has been used by the Mazatec Indians in Oaxaca, Mexico, as a hallucinogenic agent, and to relieve diarrhea, headache, and rheumatism.139–141 Smoking S. divinorum leaves is known to produce hallucinations lasting up to one hour.137,142 However, until recently, the target of the hallucinogenic effects was not clear, as 17 lacks activity at the targets of other known hallucinogens, specifically serotonin receptors, cholinergic receptors, and cannabinoid receptors.137,143 In 2002, 17 was identified as a potent and selective KOP agonist.143,144 This result is surprising considering that 17 lacks the basic nitrogen that has long been thought to be required for opioid activity. However, given the known hallucinogenic effects of other KOP agonists, this finding is not unprecedented.
Neoclerodane diterpene 17 produces a discriminative effect in both rats145 and non-human primates146 that is similar to other KOP agonists. It has also been shown to produce analgesia in mice that can be blocked by a KOP receptor antagonist.147,148 Additional work in rodents has shown that 17 is able to produce an aversive response in the conditioned place preference assay149 and block the locomotor-stimulant effects of cocaine.150 Further studies in non-human primates found that 17 produces facial relaxation and ptosis (drooping of the upper eyelid), acting as a high-efficacy KOP agonist in a translationally viable neuroendocrine biomarker assay.151,152 These collective findings suggest that 17 and its derivatives will be useful opioid ligands for the development of pharmacotherapies to treat drug abuse and other conditions involving opioid receptors.
Conclusions
Mind-altering natural products have played a vital role in the development of treatments for a number of diseases and have advanced our knowledge of basic biological processes in the CNS. The study of psychoactive natural products has led to the discovery of several receptor systems, as well as their endogenous ligands. The discovery of a variety of receptor-ligand systems has been a boon to neuroscience. Investigation of mind-altering natural products has also resulted in the development of useful biological probes that have furthered our understanding of the underlying causes of many CNS disorders. In addition, these biological probes have provided a deeper understanding of how a healthy system functions. Furthermore, structural modification of mind-altering natural products has led to the development of effective medications to treat a number of CNS disorders.At present, there is less drive to uncover new compounds with CNS activity and potential utility in neuroscience compared to the drive to uncover new compounds with anticancer potential. The etiology of some CNS disorders yet eludes us, and treatment – if available – is not always effective. Genomics and targeted SAR have largely replaced natural products screening in CNS drug discovery. Even with these advances in technology, CNS drugs entering clinical development have only approximately a 7% probability of reaching the marketplace, as compared to an industry average of 15%.153,154 Thus, it is imperative to find new chemical scaffolds for CNS drug discovery and development, as well as for potential biological probes that can be used to further enhance our understanding of neurobiology. As described above, mind-altering natural products continue to demonstrate potential for the treatment of CNS disorders and other disorders. Investigation of natural sources promises to yield novel structures and ever more selective agents that may be developed into medications. Furthermore, research into mind-altering natural products promises to further develop the field of neuroscience, deepening our understanding of human physiology and further expanding the chemical toolset available for study of the central nervous system.
References
- R. G. Wasson, J. Am. Orient. Soc., 1971, 91, 169–187 Search PubMed.
- D. H. H. Ingalls, J. Am. Orient. Soc., 1971, 91, 188–191 Search PubMed.
- J. L. Diaz, Annu. Rev. Pharmacol. Toxicol., 1977, 17, 647–675 CrossRef CAS.
- O. C. Stewart, Peyote Religion: A History, University of Oklahoma Press, Norman, OK, 1987 Search PubMed.
- A. Al-Motarreb, K. Baker and K. J. Broadley, Phytother. Res., 2002, 16, 403–413 CrossRef CAS.
- A. M. Feyissa and J. P. Kelly, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2008, 32, 1147–1166 CrossRef CAS.
- J. V. Aldrich and S. C. Vigil-Cruz, in Burger's Medicinal Chemistry and Drug Discovery, ed. D. A. Abraham, John Wiley, New York, 2003, vol. 6, pp. 329–441 Search PubMed.
- D. S. Fries, in Foye's Principles of Medicinal Chemistry, ed. D. A. Williams and T. L. Lemke, Lippincott Williams & Wilkins, Baltimore, MD, 5th edn., 2002, pp. 453–479 Search PubMed.
- J. M. Gulland and R. Robinson, Mem. Proc. Manchester Lit. Phil. Soc., 1925, 69, 79 Search PubMed.
- J. M. Gulland and R. Robinson, J. Chem. Soc., 1923, 123, 980–998 RSC.
- J. M. Gulland and R. Robinson, J. Chem. Soc., 1923, 123, 998–1011 RSC.
- M. Gates and G. Tschudi, J. Am. Chem. Soc., 1952, 74, 1109–1110 CrossRef.
- M. Gates and G. Tschudi, J. Am. Chem. Soc., 1956, 78, 1380–1393 CrossRef CAS.
- D. A. Slattery, A. L. Hudson and D. J. Nutt, Fundam. Clin. Pharmacol., 2004, 18, 1–21 CrossRef CAS.
- L. R. Caporael, Science, 1976, 192, 21–26 CrossRef CAS.
- A. Hofmann, J. Psychedelic Drugs, 1979, 11, 53–60 Search PubMed.
- L. E. Hollister, Chemical Psychoses, Charles C. Thomas, Springfield, IL, 1968 Search PubMed.
- D. E. Nichols, Pharmacol. Ther., 2004, 101, 131–181 CrossRef CAS.
- P. Sweetnam, J. Lancaster, A. Snowman, J. Collins, S. Perschke, C. Bauer and J. Ferkany, Psychopharmacology, 1995, 118, 369–376 CrossRef CAS.
- J. H. Halpern, Pharmacol. Ther., 2004, 102, 131–138 CrossRef CAS.
- D. Romer, H. Buscher, R. C. Hill, R. Maurer, T. J. Petcher, H. B. A. Welle, H. Bakel and A. M. Akkerman, Life Sci., 1980, 27, 971–978 CrossRef CAS.
- R. A. Lahti, P. F. Vonvoigtlander and C. Barsuhn, Life Sci., 1982, 31, 2257–2260 CrossRef CAS.
- D. J. McKenna, Pharmacol. Ther., 2004, 102, 111–129 CrossRef CAS.
- H. Osmond and J. Smythies, J. Ment. Sci., 1952, 98, 309–315 Search PubMed.
- C. S. Grob, G. R. Greer and M. Mangini, J. Psychoact. Drugs, 1998, 30, 315–319 Search PubMed.
- H. L. Leonard and J. L. Rapoport, Am. J. Psychiatry, 1987, 144, 1239–1240 Search PubMed.
- K. R. Hanes, J. Clin. Psychopharmacol., 1996, 16, 188–189 CrossRef CAS.
- F. A. Moreno and P. L. Delgado, Am. J. Psychiatry, 1997, 154, 1037–1038 Search PubMed.
- P. L. Delgado and F. A. Moreno, J. Psychoact. Drugs, 1998, 30, 359–366 Search PubMed.
- M. El Mansari and P. Blier, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2006, 30, 362–373 CrossRef CAS.
- S. Flaisher-Grinberg, O. Klavir and D. Joel, Int. J. Neuropsychopharmacol., 2008, 11, 811–825 Search PubMed.
- A. M. Barr, V. Lehmann-Masten, M. Paulus, R. R. Gainetdinov, M. G. Caron and M. A. Geyer, Neuropsychopharmacology, 2004, 29, 221–228 CrossRef CAS.
- D. J. Hellerstein, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2004, 28, 1347–1348 CrossRef CAS.
- A. V. Terry, Jr., J. J. Buccafusco and G. D. Bartoszyk, Psychopharmacology (Berlin, Ger.), 2005, 179, 725–732 Search PubMed.
- S. Akhondzadeh, M. Malek-Hosseini, A. Ghoreishi, M. Raznahan and S.-A. Rezazadeh, Prog. Neuropsychopharmacol. Biol. Psychiatry, 2008, 32, 1879–1883 CrossRef CAS.
- J. Dunlop, A. L. Sabb, H. Mazandarani, J. Zhang, S. Kalgaonker, E. Shukhina, S. Sukoff, R. L. Vogel, G. Stack, L. Schechter, B. L. Harrison and S. Rosenzweig-Lipson, J. Pharmacol. Exp. Ther., 2005, 313, 862–869 CAS.
- G. Yoon, H. J. Jeong, J. J. Kim and S. H. Cheon, Arch. Pharmacal Res., 2008, 31, 989–994 Search PubMed.
- W. J. Thomsen, A. J. Grottick, F. Menzaghi, H. Reyes-Saldana, S. Espitia, D. Yuskin, K. Whelan, M. Martin, M. Morgan, W. Chen, H. Al-Shamma, B. Smith, D. Chalmers and D. Behan, J. Pharmacol. Exp. Ther., 2008, 325, 577–587 CrossRef CAS.
- B. M. Smith, J. M. Smith, J. H. Tsai, J. A. Schultz, C. A. Gilson, S. A. Estrada, R. R. Chen, D. M. Park, E. B. Prieto, C. S. Gallardo, D. Sengupta, P. I. Dosa, J. A. Covel, A. Ren, R. R. Webb, N. R. A. Beeley, M. Martin, M. Morgan, S. Espitia, H. R. Saldana, C. Bjenning, K. T. Whelan, A. J. Grottick, F. Menzaghi and W. J. Thomsen, J. Med. Chem., 2008, 51, 305–313 CrossRef CAS.
- M. S. Levi and R. F. Borne, Curr. Med. Chem., 2002, 9, 1807–1818 CAS.
- D. C. Mash, C. A. Kovera, J. Pablo, R. F. Tyndale, F. D. Ervin, I. C. Williams, E. G. Singleton and M. Mayor, Ann. N. Y. Acad. Sci., 2000, 914, 394–401 CAS.
- M. H. Baumann, J. P. Pablo, S. F. Ali, R. B. Rothman and D. C. Mash, Ann. N. Y. Acad. Sci., 2000, 914, 354–368 CAS.
- M. H. Baumann, R. B. Rothman, J. P. Pablo and D. C. Mash, J. Pharmacol. Exp. Ther., 2001, 297, 531–539 CAS.
- S. D. Glick, I. M. Maisonneuve and S. M. Pearl, Brain Res., 1997, 749, 340–343 CrossRef CAS.
- S. D. Glick and I. M. Maisonneuve, Ann. N. Y. Acad. Sci., 1998, 844, 214–226 CrossRef CAS.
- S. L. T. Cappendijk and M. R. Dzoljic, Eur. J. Pharmacol., 1993, 241, 261–265 CrossRef CAS.
- S. D. Glick, M. E. Kuehne, J. Raucci, T. E. Wilson, D. Larson, R. W. Keller, Jr. and J. N. Carlson, Brain Res., 1994, 657, 14–22 CrossRef CAS.
- H. Sershen, A. Hashim, L. Harsing and A. Lajtha, Life Sci., 1992, 50, 1079–1086 CrossRef CAS.
- I. M. Maisonneuve, K. L. Rossman, R. W. Keller, Jr. and S. D. Glick, Brain Res., 1992, 575, 69–73 CrossRef CAS.
- K. R. Alper, H. S. Lotsof, G. M. Frenken, D. J. Luciano and J. Bastiaans, Am. J. Addictions (American Academy of Psychiatrists in Alcoholism and Addictions), 1999, 8, 234–242 Search PubMed.
- E. O'Hearn and M. E. Molliver, J. Neurosci., 1997, 17, 8828–8841 CAS.
- D. C. Mash, C. A. Kovera, B. E. Buck, M. D. Norenberg, P. Shapshak, W. L. Hearn and J. Sanchez-Ramos, Ann. N. Y. Acad. Sci., 1998, 844, 274–292 CrossRef CAS.
- G. V. Williams, S. G. Rao and P. S. Goldman-Rakic, J. Neurosci., 2002, 22, 2843–2854 CAS.
- J. A. Harvey, Learn Memory, 2003, 10, 355–362 Search PubMed.
- G. Grynkiewicz and M. Gadzikowska, Pharmacol. Rep., 2008, 60, 439–463 Search PubMed.
- R. S. Holzman, Anesthesiology, 1998, 89, 241–249 CrossRef CAS.
- G. Fodor, in The Alkaloids: Chemistry and Physiology, ed. R. H. F. Manske, Academic Press, New York, 1971, pp. 351–396 Search PubMed.
- W. Sneader, in Drug Discovery: A History, ed. W. Sneader, Wiley & Sons, Ltd., Chichester, 2005, pp. 115–150 Search PubMed.
- D. L. Lattin and E. K. Fifer, in Foye's Principles of Medicinal Chemistry, ed. D. A. Williams and T. L. Lemke, Lippincott Williams & Wilkins, Baltimore, MD, 5th edn., 2002, pp. 264–291 Search PubMed.
- M. P. Caulfield and N. J. M. Birdsall, Pharmacol. Rev., 1998, 50, 279–290 CAS.
- J. P. Changeux, A. Devillers-Thiery and P. Chemouilli, Science, 1984, 225, 1335–1345 CAS.
- M. Mishina, T. Kurosaki, T. Tobimatsu, Y. Morimoto, M. Noda, T. Yamamoto, M. Terao, J. Lindstrom, T. Takahashi and M. Kuno, Nature, 1984, 307, 604–608 CrossRef CAS.
- P. J. Conn, A. Christopoulos and C. W. Lindsley, Nat. Rev. Drug Discovery, 2009, 8, 41–54 CrossRef CAS.
- W. Y. Chan, D. L. McKinzie, S. Bose, S. N. Mitchell, J. M. Witkin, R. C. Thompson, A. Christopoulos, S. Lazareno, N. J. M. Birdsall, F. P. Bymaster and C. C. Felder, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10978–10983 CrossRef CAS.
- T. M. Bridges, J. E. Marlo, C. M. Niswender, C. K. Jones, S. B. Jadhav, P. R. Gentry, H. C. Plumley, C. D. Weaver, P. J. Conn and C. W. Lindsley, J. Med. Chem., 2009, 52, 3445–3448 CrossRef CAS.
- J. E. Marlo, C. M. Niswender, E. L. Days, T. M. Bridges, Y. Xiang, A. L. Rodriguez, J. K. Shirey, A. E. Brady, T. Nalywajko, Q. Luo, C. A. Austin, M. B. Williams, K. Kim, R. Williams, D. Orton, H. A. Brown, C. W. Lindsley, C. D. Weaver and P. J. Conn, Mol. Pharmacol., 2009, 75, 577–588 CrossRef CAS.
- A. E. Brady, C. K. Jones, T. M. Bridges, J. P. Kennedy, A. D. Thompson, J. U. Heiman, M. L. Breininger, P. R. Gentry, H. Yin, S. B. Jadhav, J. K. Shirey, P. J. Conn and C. W. Lindsley, J. Pharmacol. Exp. Ther., 2008, 327, 941–953 CrossRef CAS.
- A. C. Howlett, F. Barth, T. I. Bonner, G. Cabral, P. Casellas, W. A. Devane, C. C. Felder, M. Herkenham, K. Mackie, B. R. Martin, R. Mechoulam and R. G. Pertwee, Pharmacol. Rev., 2002, 54, 161–202 CrossRef CAS.
- Y. Gaoni and R. Mechoulam, J. Am. Chem. Soc., 1964, 86, 1646–1647 CrossRef CAS.
- R. Mechoulam and Y. Gaoni, Tetrahedron Lett., 1967, 8, 1109 CrossRef.
- L. S. Melvin, M. R. Johnson, C. A. Harbert, G. M. Milne and A. Weissman, J. Med. Chem., 1984, 27, 67–71 CrossRef CAS.
- W. A. Devane, F. A. Dysarz, M. R. Johnson, L. S. Melvin and A. C. Howlett, Mol. Pharmacol., 1988, 34, 605–613 CAS.
- L. A. Matsuda, S. J. Lolait, M. J. Brownstein, A. C. Young and T. I. Bonner, Nature, 1990, 346, 561–564 CAS.
- S. Munro, K. L. Thomas and M. Abushaar, Nature, 1993, 365, 61–65 CAS.
- K. Mackie, Annu. Rev. Pharmacol. Toxicol., 2006, 46, 101–122 CrossRef CAS.
- W. A. Devane, L. Hanus, A. Breuer, R. G. Pertwee, L. A. Stevenson, G. Griffin, D. Gibson, A. Mandelbaum, A. Etinger and R. Mechoulam, Science, 1992, 258, 1946–1949 CAS.
- R. Mechoulam, S. Ben-Shabat, L. Hanus, M. Ligumsky, N. E. Kaminski, A. R. Schatz, A. Gopher, S. Almog, B. R. Martin, D. R. Compton, R. G. Pertwee, G. Griffin, M. Bayewitch, J. Barg and Z. Vogel, Biochem. Pharmacol., 1995, 50, 83–90 CrossRef CAS.
- T. Sugiura, S. Kondo, A. Sukagawa, S. Nakane, A. Shinoda, K. Itoh, A. Yamashita and K. Waku, Biochem. Biophys. Res. Commun., 1995, 215, 89–97 CrossRef CAS.
- N. Stella, P. Schweitzer and D. Piomelli, Nature, 1997, 388, 773–778 CrossRef CAS.
- W. Gonsiorek, C. Lunn, X. D. Fan, S. Narula, D. Lundell and R. W. Hipkin, Mol. Pharmacol., 2000, 57, 1045–1050 CAS.
- K. Mackie, W. A. Devane and B. Hille, Mol. Pharmacol., 1993, 44, 498–503 CAS.
- M. A. Huestis, D. A. Gorelick, S. J. Heishman, K. L. Preston, R. A. Nelson, E. T. Moolchan and R. A. Frank, Arch. Gen. Psychiatry, 2001, 58, 322–328 CrossRef CAS.
- M. Rinaldi-Carmona, F. Barth, M. Héaulme, D. Shire, B. Calandra, C. Congy, S. Martinez, J. Maruani, G. Néliat, D. Caput, P. Ferrara, P. Soubrié, J. C. Brelière and G. Le Fur, FEBS Lett., 1994, 350, 240–244 CrossRef CAS.
- L. F. Van Gaal, A. M. Rissanen, A. J. Scheen, O. Ziegler and S. Rössner, Lancet, 2005, 365, 1389–1397 CrossRef CAS.
- F. Chaperon, P. Soubrié, A. J. Puech and M.-H. Thiébot, Psychopharmacology, 1998, 135, 324–332 CrossRef CAS.
- T. De Vries, J. Homberg, R. Binnekade, H. Raasø and A. Schoffelmeer, Psychopharmacology, 2003, 168, 164–169 CrossRef CAS.
- M. Solinas, L. V. Panlilio, K. Antoniou, L. A. Pappas and S. R. Goldberg, J. Pharmacol. Exp. Ther., 2003, 306, 93–102 CrossRef CAS.
- G. Cossu, C. Ledent, L. Fattore, A. Imperato, G. A. Böhme, M. Parmentier and W. Fratta, Behav. Brain Res., 2001, 118, 61–65 CrossRef CAS.
- S. R. Wachtel and H. de Wit, Drug Alcohol Depend., 2000, 59, 251–260 CrossRef CAS.
- C. Cohen, G. Perrault, C. Voltz, R. Steinberg and P. Soubrie, Behav. Pharmacol., 2002, 13, 451–463 CAS.
- J. E. Gallate, P. E. Mallet and I. S. McGregor, Psychopharmacology, 2004, 173, 210–216 CrossRef CAS.
- M. M. Ibrahim, H. Deng, A. Zvonok, D. A. Cockayne, J. Kwan, H. P. Mata, T. W. Vanderah, J. Lai, F. Porreca, A. Makriyannis and T. P. Malan, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10529–10533 CrossRef CAS.
- S. J. R. Elmes, M. D. Jhaveri, D. Smart, D. A. Kendall and V. Chapman, Eur. J. Neurosci., 2004, 20, 2311–2320 CrossRef.
- M. K. McKinney and B. F. Cravatt, Annu. Rev. Biochem., 2005, 74, 411–432 CrossRef CAS.
- D. L. Boger, H. Sato, E. L. Aaron, M. P. Hedrick, R. A. Fecik, H. Miyauchi, D. W. Gordon, B. J. Austin, M. P. Patricelli and B. F. Cravatt, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 5044–5049 CrossRef CAS.
- B. F. Cravatt, O. Prosperogarcia, G. Siuzdak, N. B. Gilula, S. J. Henriksen, D. L. Boger and R. A. Lerner, Science, 1995, 268, 1506–1509 CrossRef CAS.
- A. H. Lichtman, E. G. Hawkins, G. Griffin and B. F. Cravatt, J. Pharmacol. Exp. Ther., 2002, 302, 73–79 CrossRef CAS.
- J. M. Walker, S. M. Huang, N. M. Strangman, K. Tsou and M. C. Sanudo-Pena, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12198–12203 CrossRef CAS.
- A. H. Lichtman, D. Leung, C. C. Shelton, A. Saghatelian, C. Hardouin, D. L. Boger and B. F. Cravatt, J. Pharmacol. Exp. Ther., 2004, 311, 441–448 CrossRef.
- A. H. Lichtman, C. C. Shelton, T. Advani and B. F. Cravatt, Pain, 2004, 109, 319–327 CrossRef CAS.
- D. L. Boger, H. Miyauchi, W. Du, C. Hardouin, R. A. Fecik, H. Cheng, I. Hwang, M. P. Hedrick, D. Leung, O. Acevedo, C. R. W. Guimaraes, W. L. Jorgensen and B. F. Cravatt, J. Med. Chem., 2005, 48, 1849–1856 CrossRef CAS.
- G. Tarzia, A. Duranti, A. Tontini, G. Piersanti, M. Mor, S. Rivara, P. V. Plazzi, C. Park, S. Kathuria and D. Piomelli, J. Med. Chem., 2003, 46, 2352–2360 CrossRef CAS.
- M. Mor, S. Rivara, A. Lodola, P. V. Plazzi, G. Tarzia, A. Duranti, A. Tontini, G. Piersanti, S. Kathuria and D. Piomelli, J. Med. Chem., 2004, 47, 4998–5008 CrossRef CAS.
- S. G. Kinsey, J. Z. Long, S. T. O'Neal, R. A. Abdullah, J. L. Poklis, D. L. Boger, B. F. Cravatt and A. H. Lichtman, J. Pharmacol. Exp. Ther., 2009 DOI:10.1124/jpet.109.155465.
- M. D. Jhaveri, D. Richardson and V. Chapman, Br. J. Pharmacol., 2007, 152, 624–632 CrossRef CAS.
- B. N. Dhawan, F. Cesselin, R. Raghubir, T. Reisine, P. B. Bradley, P. S. Portoghese and M. Hamon, Pharmacol. Rev., 1996, 48, 567–592 CAS.
- M. Satoh and M. Minami, Pharmacol. Ther., 1995, 68, 343–364 CrossRef CAS.
- A. Pfeiffer, V. Brantl, A. Herz and H. M. Emrich, Science, 1986, 233, 774–776 CrossRef CAS.
- J. L. Wilson, V. Nayanar and J. S. Walker, Br. J. Pharmacol., 1996, 118, 1754–1760 CAS.
- W. Binder and J. S. Walker, Br. J. Pharmacol., 1998, 124, 647–654 CrossRef CAS.
- M.-C. Ko, E. R. Butelman and J. H. Woods, J. Pharmacol. Exp. Ther., 1999, 289, 378–385 CAS.
- M. C. H. Ko, K. J. Willmont, A. Burritt, V. J. Hruby and J. H. Woods, Eur. J. Pharmacol., 2000, 402, 69–76 CrossRef CAS.
- J. Cami and M. Farre, N. Engl. J. Med., 2003, 349, 975–986 CrossRef CAS.
- E. J. Nestler, Nat. Rev. Neurosci., 2001, 2, 119–128 CrossRef CAS.
- T. S. Shippenberg, A. Zapata and V. I. Chefer, Pharmacol. Ther., 2007, 116, 306–321 CrossRef CAS.
- S. D. Glick, I. M. Maisonneueve, J. Raucci and S. Archer, Brain Res., 1995, 681, 147–152 CrossRef CAS.
- N. K. Mello and S. S. Negus, J. Pharmacol. Exp. Ther., 1998, 286, 812–824 CAS.
- S. S. Negus, N. K. Mello, P. S. Portoghese and C.-E. Lin, J. Pharmacol. Exp. Ther., 1997, 282, 44–55 CAS.
- S. Schenk, B. Partridge and T. S. Shippenberg, Psychopharmacology, 1999, 144, 339–346 CrossRef CAS.
- C. A. Crawford, S. A. McDougall, C. A. Bolanos, S. Hall and S. P. Berger, Psychopharmacology, 1995, 120, 392–399 CrossRef CAS.
- S. Schenk, B. Partridge and T. S. Shippenberg, Psychopharmacology, 2000, 151, 85–90 CrossRef CAS.
- S. Suwanlert, Bull. Narc., 1975, 27, 21–27 Search PubMed.
- M. A. M. Moklas, A. R. Nurul Raudzah, M. Taufik Hidayat, F. Sharida, N. Farah Idayu, A. Zulkhairi and A. R. Shamima, Adv. Med. Dent. Sci., 2008, 2, 56–60 Search PubMed.
- S. Thongpradichote, K. Matsumoto, M. Tohda, H. Takayama, N. Aimi, S.-I. Sakai and H. Watanabe, Life Sci., 1998, 62, 1371–1378 CrossRef CAS.
- H. Takayama, H. Ishikawa, M. Kurihara, M. Kitajima, N. Aimi, D. Ponglux, F. Koyama, K. Matsumoto, T. Moriyama, L. T. Yamamoto, K. Watanabe, T. Murayama and S. Horie, J. Med. Chem., 2002, 45, 1949–1956 CrossRef CAS.
- K. M. Babu, C. R. McCurdy and E. W. Boyer, Clin. Toxicol., 2008, 46, 146–152 Search PubMed.
- E. W. Boyer, K. M. Babu, G. E. Macalino and W. Compton, Am. J. Addictions (American Academy of Psychiatrists in Alcoholism and Addictions), 2007, 16, 352–356 Search PubMed.
- E. W. Boyer, K. M. Babu, J. E. Adkins, C. R. McCurdy and J. H. Halpern, Addiction (Abingdon, England), 2008, 103, 1048–1050 Search PubMed.
- H. Takayama, Chem. Pharm. Bull., 2004, 52, 916–928 CrossRef CAS.
- H. Takayama, K. Misawa, N. Okada, H. Ishikawa, M. Kitajima, Y. Hatori, T. Murayama, S. Wongseripipatana, K. Tashima, K. Matsumoto and S. Horie, Org. Lett., 2006, 8, 5705–5708 CrossRef CAS.
- K. Matsumoto, H. Takayama, H. Ishikawa, N. Aimi, D. Ponglux, K. Watanabe and S. Horie, Life Sci., 2006, 78, 2265–2271 CrossRef CAS.
- J. Ma, W. Yin, H. Zhou and J. M. Cook, Org. Lett., 2007, 9, 3491–3494 CrossRef CAS.
- J. Ma, W. Yin, H. Zhou, X. Liao and J. M. Cook, J. Org. Chem., 2008, 74, 264–273.
- K. Matsumoto, H. Takayama, M. Narita, A. Nakamura, M. Suzuki, T. Suzuki, T. Murayama, S. Wongseripipatana, K. Misawa, M. Kitajima, K. Tashima and S. Horie, Neuropharmacology, 2008, 55, 154–165 CrossRef CAS.
- A. Ortega, J. F. Blount and P. S. Manchand, J. Chem. Soc., Perkin Trans. 1, 1982, 2505–2508 RSC.
- L. J. Valdes, III, W. M. Butler, G. M. Hatfield, A. G. Paul and M. Koreeda, J. Org. Chem., 1984, 49, 4716–4720 CrossRef.
- D. J. Siebert, J. Ethnopharmacol., 1994, 43, 53–56 CrossRef CAS.
- L. J. Valdes, III, J. Psychoact. Drugs, 1994, 26, 277–283 Search PubMed.
- C. Epling and C. D. Jativa-M, Bot. Mus. Leaflets, Harvard Univ., 1962, 20, 75–76 Search PubMed.
- V. E. Tyler, Lloydia, 1966, 29, 275–292 CAS.
- L. J. Valdes, III, Ph.D. Thesis, University of Michigan, 1983.
- L. J. Valdes, III, H. M. Chang, D. C. Visger and M. Koreeda, Org. Lett., 2001, 3, 3935–3937 CrossRef CAS.
- B. L. Roth, K. Baner, R. Westkaemper, D. Siebert, K. C. Rice, S. Steinberg, P. Ernsberger and R. B. Rothman, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11934–11939 CrossRef CAS.
- C. Chavkin, S. Sud, W. Jin, J. Stewart, J. K. Zjawiony, D. J. Siebert, B. A. Toth, S. J. Hufeisen and B. L. Roth, J. Pharmacol. Exp. Ther., 2004, 308, 1197–1203 CAS.
- C. B. Willmore-Fordham, D. M. Krall, C. R. McCurdy and D. H. Kinder, Neuropharmacology, 2007, 53, 481–486 CrossRef CAS.
- E. R. Butelman, T. J. Harris and M. J. Kreek, Psychopharmacology, 2004, 172, 220–224 CrossRef CAS.
- T. F. John, L. G. French and J. S. Erlichman, Eur. J. Pharmacol., 2006, 545, 129–133 CrossRef CAS.
- C. R. McCurdy, K. J. Sufka, G. H. Smith, J. E. Warnick and M. J. Nieto, Pharmacol., Biochem. Behav., 2006, 83, 109–113 CrossRef CAS.
- Y. Zhang, E. R. Butelman, S. D. Schlussman, A. Ho and M. J. Kreek, Psychopharmacology, 2005, 179, 551–558 CrossRef CAS.
- E. H. Chartoff, D. Potter, D. Damez-Werno, B. M. Cohen and W. A. Carlezon, Jr., Neuropsychopharmacology, 2008, 33, 2676–2687 CrossRef CAS.
- E. R. Butelman, M. Mandau, K. Tidgewell, T. E. Prisinzano, V. Yuferov and M. J. Kreek, J. Pharmacol. Exp. Ther., 2007, 320, 300–306 CAS.
- E. R. Butelman, T. E. Prisinzano, H. Deng, S. Rus and M. J. Kreek, J. Pharmacol. Exp. Ther., 2009, 328, 588–597 CrossRef CAS.
- M. N. Pangalos, L. E. Schechter and O. Hurko, Nat. Rev. Drug Discovery, 2007, 6, 521–532 CrossRef CAS.
- B. L. Roth and P. J. Conn, Neuropsychopharmacology, 2008, 33, 2048–2060 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |