Epigenome manipulation as a pathway to new natural product scaffolds and their congeners
Robert H.
Cichewicz
*
Natural Products Discovery Group and Graduate Program in Ecology and Evolutionary Biology, Department of Chemistry and Biochemistry, 620 Parrington Oval, Room 208, University of Oklahoma, Norman, OK 73019, USA. E-mail: rhcichewicz@ou.edu; Fax: +1 (405) 325-6111; Tel: +1 (405) 325-6969
First published on 27th October 2009
Abstract
Covering: up to the end of September 2009
The covalent modification of chromatin is an important control mechanism used by fungi to modulate the transcription of genes involved in secondary metabolite production. To date, both molecular-based and chemical approaches targeting histone and DNA posttranslational processes have shown great potential for rationally directing the activation and/or suppression of natural-product-encoding gene clusters. In this Highlight, the organization of the fungal epigenome is summarized and strategies for manipulating chromatin-related targets are presented. Applications of these techniques are illustrated using several recently published accounts in which chemical-epigenetic methods and mutant studies were successfully employed for the de novo or enhanced production of structurally diverse fungal natural products (e.g., anthraquinones, cladochromes, lunalides, mycotoxins, and nygerones).
 Robert H. Cichewicz | Robert H. Cichewicz received B.S. degrees in biology and anthropology from Grand Valley State University and a M.S. degree in pharmaceutical sciences from the University of Louisiana at Monroe in 1999. He received his Ph.D. degree in 2002 under Muraleedharan G. Nair at Michigan State University and conducted postdoctoral training with Phil Crews at the University of California, Santa Cruz. In 2005, he joined the Department of Chemistry and Biochemistry at the University of Oklahoma as an assistant professor. His research interests include developing therapeutics for neurodegenerative and infectious diseases, as well as creating new approaches for enhancing natural products research. |
1 Introduction
The flow of genetic information from DNA to RNA to proteins is governed by an assemblage of interwoven regulatory networks that are responsible for affording cells control over transcriptional and translational processes. This is important since the capacity to carefully moderate information flow is critical for maintaining homeostasis and cell survival. Unfortunately, these essential control mechanisms also present a unique set of challenges for natural product chemists due to the restrictions they place on secondary metabolite biosynthesis. This is illustrated by the fact that many microorganisms harbor significant numbers of secondary-metabolite-encoding biosynthetic pathways, yet only a fraction of their small-molecule products are detected in the laboratory.1–4 As a result, a variety of techniques have been devised to sidestep transcriptional roadblocks and directly access natural products from silent biosynthetic pathways using heterologous expression systems and in situ gene manipulation strategies.5–9Many of the mechanisms regulating transcriptional and translational processes that are associated with primary metabolism and cell maturation have been intensively studied in microbes.10–16 In contrast, much less is known concerning the molecular-level regulation of secondary metabolite biosynthesis.17,18 Emerging evidence suggests that epigenetic processes are extensively used by cells to control the flow of genetic information including genes involved in secondary metabolite production (Fig. 1).19 The effects of epigenetic suppression are particularly notable in fungi, whose biosynthetic talents have been only partially realized.4,20,21 This review examines how epigenetic mechanisms participate in the silencing of biosynthetic pathways and how new molecular and chemical-based methods are beginning to emerge that are enabling chemists to access a hidden wealth of fungal natural products. The examples presented come exclusively from the fungal natural products literature, since these organisms have been the sole focus of research efforts within this growing field of investigation. However, it is expected that these results will have important applications to other eukaryotic sources of natural products. Given the voluminous nature of the literature pertaining to the field of epigenetics, in-depth review articles have been cited wherever possible, so that interested readers can further explore the details concerning the molecular and biochemical aspects of epigenetic gene regulation.
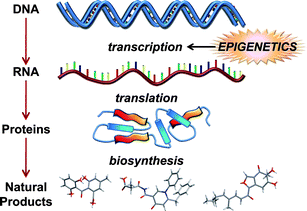 | ||
| Fig. 1 Outline illustrating the flow of genetic information leading to secondary metabolite biosynthesis in fungi and how these events are impacted by epigenetic processes. | ||
2 Epigenome features
2.1 Histones and their modifications
Histones are highly abundant chromatin-associated proteins that serve two important roles.22–24 First, histones function as scaffolds for nucleosome assembly by providing a substrate for DNA binding. Second, histones participate in the control of transcriptional regulation, which is essential for proper cell function.25 There are two types of histones: linker histones and core histones.26 Although several variants are reported to exist,27 histone H1 is the predominant linker histone in fungi.26 Histone H1 strongly associates with linker DNA (stretches of DNA that join adjacent nucleosome complexes) and they help facilitate its role directing the binding of transcriptional proteins to chromatin.28–30 Linker histones fulfil many essential functions in higher eukaryotes, which include influencing the addition/removal of epigenetic marks on chromatin (e.g., DNA methylation).27,31–33 However, most of the roles that linker H1 histones play in higher eukaryotes have not been confirmed in fungi. Instead, fungal H1 histones exhibit considerable interspecies diversity,34 with deletion mutants (e.g., Ascobolus,35Aspergillus,36Neurospora,37 and Saccharomyces38–40) demonstrating variable non-lethal phenotypes.33,41Core histones are composed of paired subunits that coalesce into octamers.42 The primary core histone proteins found in fungi consist of highly conserved43 H2A, H2B, H3, and H4 subunits (Fig. 2). The unstructured N-terminal tails are important features of the core histones since they serve as the primary regulatory sites by virtue of their susceptibility to an extensive array of covalent modifications.44 A number of nuclear proteins are known to interact with amino acid residues along the N-terminal tails of linker and core histones,45–48 allowing for modifier enzymes to add/remove acetate,43,49–56 ADP-ribose,57 ubiquitin,58–60 biotin,61,62 phosphate,63–65 SUMO,66 methyl,67–71 ethyl,72 propyl,72 and butyl72 groups, along with other known45,73 and yet-to-be-deciphered72 posttranslational marks (Fig. 3). Most of these reactions target the addition/deletion of modifiers to conserved lysine and arginine residues; however, other amino acid side-chains are also known to serve as substrates for modification (e.g., phosphorylation of serine and threonine).72
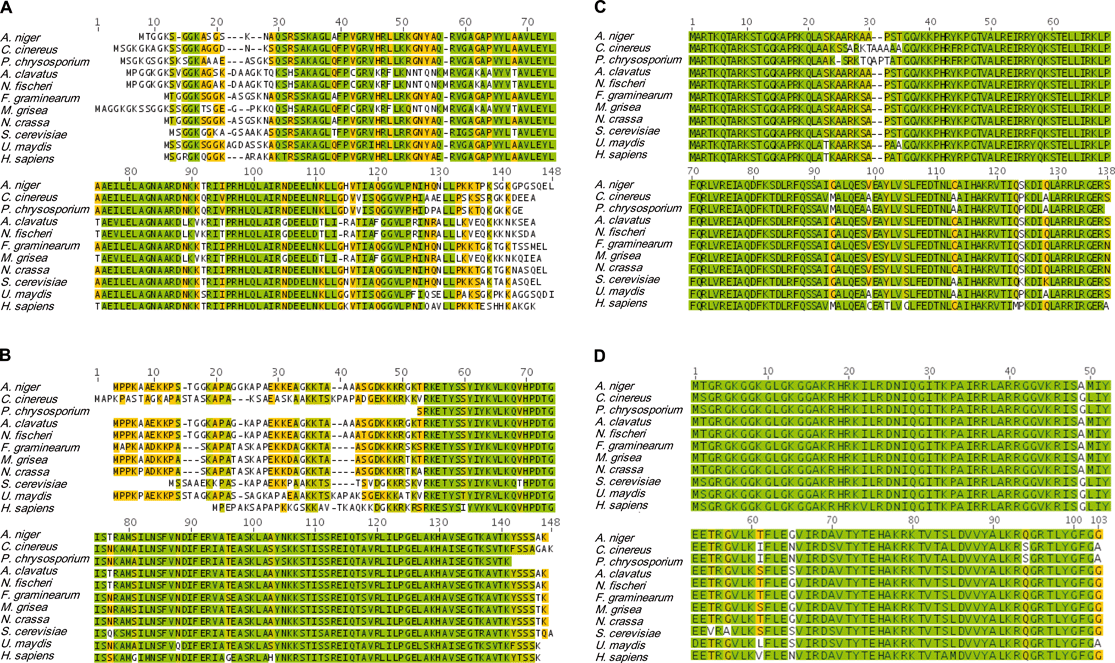 | ||
| Fig. 2 Amino acid sequences of histone subunits H2A (A), H2B (B), H3 (C), and H4 (D) for the fungi (from top to bottom) Aspergillus niger, Coprinus cinereus, Phanerochaete chrysosporium, Aspergillus clavatus, Neosartorya fischeri, Fusarium graminearum, Magnaporthe grisea, Neurospora crassa, Saccharomyces cerevisiae, and Ustilago maydis. The sequences of human histones were included for comparisons to illustrate the substantial conservation of their primary structures. Pair-wise identities for the listed histone subunit sequences are 69%, 80%, 92%, and 94% for H2A, H2B, H3, and H4, respectively. Color-coding of the amino acid residues is as follows: bright green = identical residues; olive-green = highly conserved, but non-identical residues; orange = modest conservation of residues, and white = residue conservation is low or the residue is not present. Alignments were performed in Geneious v4.7 using sequence data available from GenBank and the Broad Institute Fungal Genome Initiative database. | ||
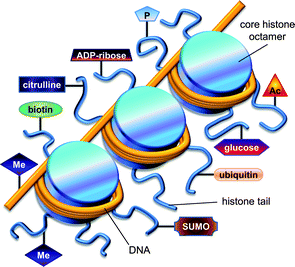 | ||
| Fig. 3 Illustration showing the association of DNA with histones, as well as common posttranslational modifications that influence these interactions. Several epigenetic modifications that alter DNA and histones include (clockwise from the bottom left): methylation of histones and DNA, biotinylation, citrullination, ADP-ribosylation, phosphorylation, acetylation, glycosylation, ubiquitination, and sumoylation. | ||
Acetylation of the lysine ε-amino group is by far the most thoroughly studied histone posttranslational modification in fungi.26 The process of acetylating and deacetylating lysine is carried out by two sets of enzymes: histone acetyltransferases (HATs) and histone deacetylases (HDACs), respectively (Fig. 4).74,75 Under physiological conditions, lysine's ε-amino group (pKa ∼ 10.4) exists in a predominately protonated state and this facilitates the electrostatic binding of lysine's positively charged side-chain to negatively charged DNA. In contrast, acetylation of the ε-amino group forms the corresponding amide, which remains neutral at physiological pH and does not preferentially bind to DNA. These changes in lysine's affinity for DNA contribute to the transitioning between ‘open’ and ‘closed’ chromatin states that are involved in regulating gene activation and silencing, respectively.76 In addition, HATs and HDACs are subject to a variety of posttranslational modifications that afford cells control over their catalytic functions.77–79
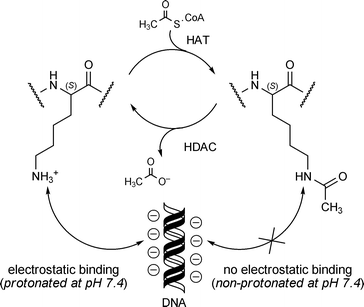 | ||
| Fig. 4 Acetylation of lysine residues found in histone tails and their interactions with DNA. | ||
Methylation of lysine and arginine residues represent another set of posttranslational modifications that are frequently encountered.26,74,75,80 Whereas lysine acetylation typically leads to the inhibition of transcription, the effects of methylation are varied and range from gene activation to silencing. The methylation of lysine and arginine residues is carried out by different sets of enzymes exhibiting distinct specificities for ε-amino or δ-guanidino functionalities. Both lysine and arginine histone methyltransferases catalyze the addition of one or more methyl groups to amino acid side-chains using S-adenosylmethionine as the methyl group donor.74
Lysine methylation is a reversible process, and several enzymes have been identified that participate in the sequence-specific addition or removal of methyl groups.26,80–82 Methylation of lysines is carried out by two major families of histone lysine methyltransferases (HKMTs): SET domain-containing methyltransferases (SUV39, SET1, SET2, and RIZ)83–86 and non-SET domain-containing methyltransferases (DOT1).87 The opposing reaction consisting of methyl group removal is accomplished by at least two different groups of demethylases. Lysine-specific demethylase 1 (LSD1) enzymes carry out the FAD-dependent removal of methyl groups from mono- and di-methylated lysines, resulting in the generation of formaldehyde as a byproduct. Similarly, members from the jumonji class (e.g., JMJC) of demethylases have been shown to catalyze the Fe(II)-dependent removal of methyl groups from mono-, di, and tri-methylated lysines in the presence of various cofactors.26,88–91 Lysines embedded within non-tail portions of histones are also subject to posttranslational modification, and these changes are likely to have important epigenetic roles.92
Arginine methylation is performed by protein arginine methyltransferases (PRMTs) that are grouped into two major classes, type I and type II PRMTs, depending on whether they catalyze asymmetric (type I) or symmetric (type II) methyl addition reactions. An analysis of fungal PRMT gene sequence data indicates that this family of methyltransferases exhibit significant diversity in their structures and functions.93 Evidence suggests that the demethylation of arginine residues can occur by means of jumonji-type demethylases, but this pathway has yet to be confirmed in fungi.80 Another group of enzymes known as the protein arginine deiminases (PADs) are also capable of carrying out the demethylation of monomethyl arginine residues. However, the final product of this reaction is not a free guanidinium group, but rather a carbamide derivative known as citrulline. Notably, PADs also accommodate the deimination of unmethylated arginine to yield citrulline in a similar fashion.80
2.2 DNA modification
DNA is posttranslationally modified in fungi by DNA methyltransferases (DNMTs), resulting in the conversion of cytosine to its corresponding 5-methylcytosine product (Fig. 3). DNA methylation has been shown to serve a number of vital functions in fungi,94–96 which include altering gene transcription97,98 and modulating transcript elongation.99 Typical methylation levels ranging from 0.25% to 1.5% of cytosines in Aspergillus flavus100 and Neurospora crassa,101 respectively, have been reported in metabolically active fungi. However, these levels tend to be lower than the 5-methylcytosine content observed in plants and vertebrates.102 It has been proposed that this variation could be an artifact of a highly dynamic DNA methylation process in fungi, which fluctuates according to an organism's life-stage.103 For example, it was shown that DNA methylation in Phycomyces blakesleeanus occurred minimally in three-day-old mycelia (as low as ∼0.48% of the genomic cytosines), but was highly prevalent (up to 2.9% of the genomic cytosine content) in spores.95 DNA methylation is a heritable trait, but this epigenetic mark may have limited stability during continuous subculturing.104,105 Genetic manipulations resulting in altered DNA methylation levels have been studied and these changes tend to result in dramatically modified phenotypes including altered sexual development106 and aberrant chromosomal behavior (e.g., abnormal DNA replication or chromosome segregation).101 Interestingly, a genome protection role for DNA methylation has been proposed in fungi, since many of the modified cytosine residues are associated with ‘foreign’ DNA107,108 and sequences subject to repeat-induced point mutations (e.g., transposons).1092.3 Interactions among epigenetic features
Histone modifications and DNA methylation operate in tandem to bring about changes in chromatin status and gene expression or silencing in fungi71,110,111 and other organisms.112–116 A substantial body of literature is evolving, which demonstrates that epigenetic processes function collectively to regulate the activity of variously sized genomic regions, which range from individual genes to entire domains and chromosomes.117,118 Epigenetic marks also interact with other nuclear proteins, and together they serve to shape chromatin architecture and create discrete regions of genomic functionalization.119 A substantial repertoire of interacting epigenetic elements has been described, and this list will assuredly grow as more links between chromatin features emerge.70,71,84,99,110,111,120–123 Given the incredible breadth of protein–DNA interactions that have been reported and hypothesized,124–129 a detailed discussion of these features is not included here. However, an understanding of these signaling networks will be necessary to ultimately appreciate how genes involved in secondary metabolite biosynthesis are actively regulated in fungi.3 Epigenome manipulation
3.1 Chemical epigenetics – using small molecules to manipulate the fungal epigenome
With so many well established epigenome-modifying enzyme targets described, an array of small-molecule tools have emerged that are capable of selectively or semi-selectively inhibiting DNA and histone posttranslational modifying proteins. While the majority of chemical probe development has centered on creating inhibitors that are effective at altering human-disease-related processes (e.g., cancer),24,130–135 these compounds have also shown considerable efficacy against many of their fungal counterparts. The scientific community's accessibility to a growing assortment of small-molecule tools (Fig. 5) has enabled the development of chemical epigenetic techniques21 that are geared toward probing how epigenome features regulate biological processes. This includes the regulation of biosynthetic mechanisms involved in secondary metabolite generation.4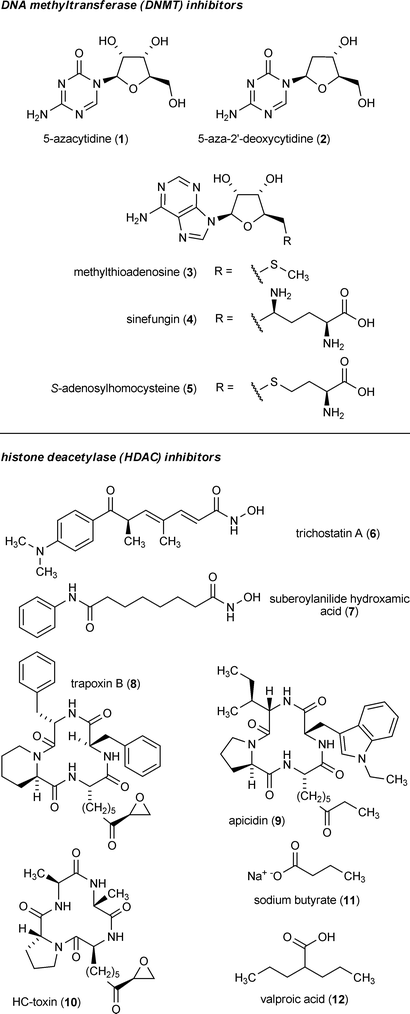 | ||
| Fig. 5 Example small-molecule inhibitors and their respective epigenetic targets. | ||
A substantial body of knowledge has accumulated pertaining to the effects of epigenetic probes on fungi. Most examples have focused on the use of DNMT inhibitors such as 5-azacytidine (1) and 5-aza-2′-deoxycytidine (2), which have demonstrated the ability to reduce the DNA-methylation-mediated silencing of hygromycin-resistance genes in Schizophyllum commune136 and N. crassa137 and a phleomycin-resistance gene in Phanerochaete chrysosporium.138 Chemical inhibition of fungal DNMTs has also been shown to impact a variety of developmental and other cellular processes in Candida albicans,139Aspergillus spp.,140–142Fusarium oxysporum,143,144 and N. crassa.145–147 Some of these data support a role for 1 being capable of inducing heritable epigenetic modifications in fungi, resulting in the acquisition of new and mitotically stable phenotypic characteristics.140,143 This is similar to reports in which 1 has been shown to cause heritable changes in DNA methylation patterns and give rise to new phenotypic traits in plants.148–151 However, a mutation-inducing role for 1 as a result of its incorporation into DNA cannot be completely ruled out under some circumstances.152,153
Several reports have described the variable effects of HDAC inhibitors on fungi. Most studies concern the application of hydroxamic-acid-containing compounds such as trichostatin A (6) or cyclic peptides including trapoxin B (8). Interestingly, tests concerning the use of 6 on N. crassa revealed a critical link between histone and DNA modifications, since histone hypoacetylation induced by 6 led to a substantial reduction in DNA methylation within selected portions of the fungal genome.123 A chemical genetics approach employing 6 has also been used to explore HDAC functions and global transcriptional control mechanisms in Saccharomyces cerevisiae.154 It is noteworthy that the fungal pathogen Cochliobolus caronum, which is responsible for the production of HC-toxin (a potent HDAC inhibitor155) (10), produces a structurally modified HDAC that is insensitive to both 10 and 6. It is presumed that this uniquely adapted HDAC serves to protect C. caronum from the autotoxic effects of 10 during the fungus’s invasion and chemical attack upon its maize host.156
3.2 Chemical epigenetic manipulation of fungal secondary metabolite production
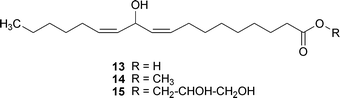
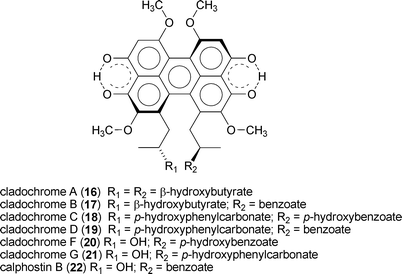
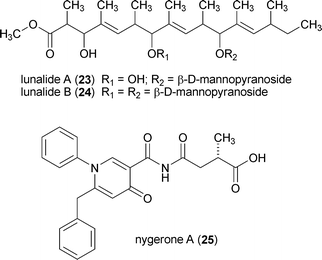
The potential for using epigenetic modifying agents to induce changes in fungal secondary metabolism have only recently come to light. In one example, the addition of low doses of 1 (≤30 µM) to N. crassa cultures resulted in the increased production of carotenoids; however, higher doses (100 and 300 µM) led to depressed carotenoid levels, as well as altered reproductive structures.145 In another example, the addition of 6 (1 µM) to cultures of Alternaria alternata and Penicillium expansum was shown to significantly increase the concentrations of several unidentified natural products.157 Based on the encouraging evidence presented in these and related studies,158 the author's group was motivated to test the feasibility of using small-molecule epigenetic modifiers to activate the production of new natural products from fungi.Our group's initial endeavor to test the applicability of a chemical epigenetic approach for accessing secondary metabolites consisted of screening nine DNMT and HDAC inhibitors alone or in combination against 12 fungal species (Aspergillus flavus, Aspergillus westerdijkiae, Cladosporium cladosporioides, Clonostachys sp., Diatrype sp., Penicillium chrysogenum, Penicillium citrinum, Rhizopus sp., Verticillium psalliotae, and three unidentified filamentous fungi).20 The results of this investigation confirmed that the majority of fungi (11 of 12 organisms) were responsive to one or more of the chemical epigenetic modifiers, as demonstrated by the production of new (or enhanced levels of) secondary metabolites. Two of the fungi were subsequently selected for scale-up fermentation and characterization of the newly generated compounds. In one set of experiments, cultures of C. cladosporioides were treated with either 1 or 7 resulting in the generation of two distinct secondary metabolite profiles. Purification of three of the natural products induced by 1 led to the identification of oxylipins (13–15) that were not detected in any of the non-epigenetically treated cultures. Likewise, incubation of C. cladosporioides with 7 provided a series of unique perylenequinone metabolites (16–22). Both sets of natural products are hypothesized to facilitate distinct functional roles during discrete stages in the life-history of C. cladosporioides (i.e., oxylipins are presumed to fulfil important developmental and host invasion roles in fungi,159 whereas the perylenequinones are likely involved in pathogenesis160). Therefore, overcoming the biosynthetic suppression of these metabolites in a controlled manner is quite remarkable. Similarly, we observed that treatment of a Diatrype sp. with 1 yielded two new galactose-conjugated polyunsaturated polyketides (23 and 24). It is notable that production of the polyketides was restricted to only two of the many experimental conditions that were investigated: induction with 1 and elicitation with heat-inactivated Escherichia coli.
During the course of follow-up studies, Aspergillus niger was screened for its response to epigenetic modifiers and this fungus was determined to be highly affected by the administration of 7 (10 µM) (Fig. 6). Purification and characterization performed on one of its many new secondary metabolites yielded the novel compound nygerone A (25), which was distinguished by its highly unusual 1-phenylpyridin-4(1H)-one core.21 Subsequent studies were undertaken to examine the genome-wide effects of 1 and 7 on secondary metabolite gene transcription in A. niger.4 We noted that <30% of the 31 polyketide (PKS), 15 non-ribosomal peptide (NRPS), and 9 hybrid PKS-NRPS (HPN) biosynthetic gene clusters in A. niger were transcribed when the organism was grown using a variety of traditional laboratory culture techniques and conditions. However, data compiled from experiments testing the effects of adding 1 or 7 to A. niger demonstrated increased transcriptional rates among all, but seven (i.e., PKSs 2, 30, 32, and 33; NRPS 14, and HPNs 7 and 31) of its PKS, NRPS, and HPN gene clusters in response to one or both epigenetic modifiers. The reason why the expression of these seven gene clusters was negatively impacted by epigenetic modifiers is not known, but it is possible that epigenetic treatments could have activated the production of endogenous transcriptional suppressors. The remarkable epigenetic-inhibitor-induced changes in secondary metabolite gene cluster expression illustrate the incredible potential that a chemical epigenetic approach holds for accessing structurally diverse natural products from silent biosynthetic pathways.
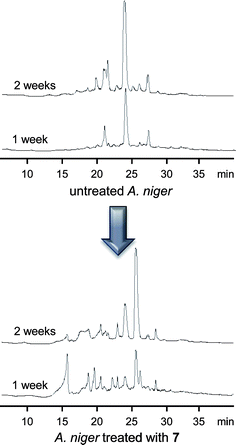 | ||
| Fig. 6 Chromatograms of the reverse-phase-separated dichloromethane extracts of A. niger ATCC 1015 cultures treated with 7 showing the impact of HDAC inhibition on secondary metabolite diversity (UV traces at 210 nm were collected one and two weeks after addition of 10 µM of 7 or vehicle alone). | ||
3.3 Molecular methods for altering the epigenome
The application of molecular techniques to manipulate the performance of fungal genes (e.g., deletion or promotion) has been a challenging pursuit;17 however, significant advances have been made within this field resulting in novel opportunities for directly probing the genetic basis by which fungi control secondary metabolite production.9,18,161,162 In 2004, the nuclear localized protein LaeA was reported from Aspergillus spp. and its cellular function was linked to the transcriptional control of several secondary metabolite families.163 The global effects of LaeA were demonstrated by TLC and/or gene expression analyses to involve a wide range of secondary metabolite pathways, including those encoding for sterigmatocystin (26), lovastatin (27), a penicillin, gliotoxin (28), as well as many yet-undefined natural products.158,163 The presence of a putative S-adenosylmethionine binding motif in LaeA164 and its exclusive occurrence in the nuclei of cells led to the speculation that this protein might serve as a histone methylating agent;163 however, the specific mechanism by which LaeA facilitates its control over such a wide range of natural products has not been determined.165,166 Despite this limitation, the discovery of LaeA offered striking evidence that multiple natural product pathways could be activated/deactivated by a single regulatory gene and that manipulation of cellular targets endowed with this degree of global control held exceptional promise for mining the secondary metabolite diversity of fungal genomes.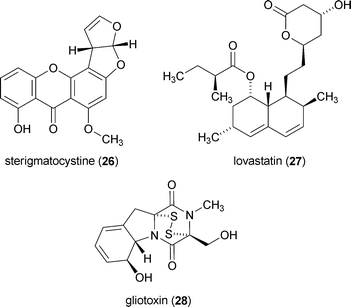
The role of LaeA in the transcriptional control of fungal natural products has now been extended to other groups of fungi167,168 and manipulation of laeA has been applied to defining biosynthetic gene cluster boundaries,169–171 promoting virulence,172,173 and altering fungal morphology and development.174–176 The intricacies concerning LaeA-facilitated transcriptional control of secondary metabolic pathways have recently been expanded to include a role for alternatively spliced forms of this protein.167 This was illustrated in Monascus pilosus wherein differentially spliced LaeA transcripts consisting of 1810 and 1532 base pairs (d-MpLaeA and l-MpLaeA, respectively) were produced at varying levels dependent on media conditions. Furthermore, changes in the production of alternatively spliced forms of LaeA were shown to be correlated with varying production levels of 27. This adds a new and exciting dimension to the mechanisms with which fungi exert control over natural product production; however, further confirmation of these findings is needed to establish a clear link between alternatively spliced mRNA species and modulation of secondary metabolite biosynthetic control.
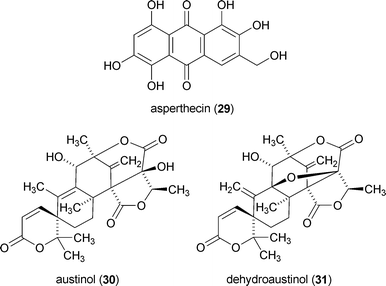
The effects of sumoylation on secondary metabolite production have also been examined in Aspergillus nidulans.177 SUMO is a ubiquitin-like protein, which is posttranslationally added to many proteins including histones. Deletion of sumO from the A. nidulans genome was shown to significantly change secondary metabolite production, which included a nearly 200-fold increase in the level of asperthecin (29).177 However, the amounts of austinol (30), dehydroaustinol (31), and 26 declined substantially (21-, 10-, and 54-fold, respectively). In contrast, the ΔsumO mutant exhibited almost no change in production of the cyclic depsipeptides emericellamides A, C, D, E, and F. Unfortunately, it is impossible at this time to discern whether loss of sumO had a direct impact on epigenetic targets (e.g., histones) or if changes in secondary metabolome profiles arose indirectly through posttranslational modifications to other yet-undefined protein targets.
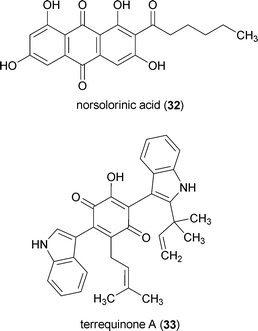
Strong evidence for an epigenetic role in secondary metabolite biosynthesis was put forward employing a molecular genetics approach in which genes encoding for three types of HDACs (hdaA, hosB, and hstA) were deleted from A. nidulans.157 Inactivation of hosB (HOS3-like HDAC family unique to fungi) and hstA (homolog of sirtuins) had modest or no significant effects on secondary metabolite production. However, inclusion of an inactivated hdaA (conserved class II HDAC) caused a dramatic increase in norsolorinic acid (32) and penicillin production. A mutant strain deficient for all three HDACs exhibited even greater norsolorinic acid accumulation (but no change in penicillin levels) and this trend was maintained even in a ΔlaeA background. Despite being carefully scrutinized, none of the HDAC deletion mutants demonstrated appreciable changes in terraquinone A (33) levels. This was rationalized as being due to the relative differences in the chromosomal positioning of the gene clusters encoding for each of the three investigated pathways. Whereas the gene clusters for penicillin and 32 (the same cluster produces 26) are located within 100 kb of the telomeres of chromosomes VI and IV, respectively, the gene cluster responsible for biosynthesizing 33 is more distally located on chromosome V (∼700 kb from the chromosome tip). Moreover, loci of the gene clusters responsible for production of penicillin and 32 are surrounded by regions of conserved DNA repeats, which are considered hallmarks of subtelomeric portions of A. nidulans chromosomes.157
Although these data suggested a strong preference for the influence of epigenetic processes on secondary metabolite expression to be manifested at subtelomeric gene clusters,157 our own observations suggest a more complex pattern of epigenetic control is at play.4 Our examination of the genome-wide effects of 1 and 7 on secondary-metabolite-encoding biosynthetic pathway expression revealed that although the majority of highly upregulated gene clusters were proximal to telomeres, several internally positioned pathways were also highly affected.4 In addition, the gene clusters most strongly down-regulated by epigenetic modifiers were also located near chromosome termini. A recent report examining NRPS gene cluster expression in Aspergillus fumigatus failed to detect a definitive correlation between the proximity of NRPS gene clusters to chromosome termini and their transcriptional activation following hdaA deletion.178 Additional studies using other fungal model systems to examine the genome-wide effects of epigenetic modifiers and deletion mutants will be needed to help determine if there is a positional bias for which gene clusters are normally regulated by epigenetic processes. Another critical question for future investigations will be deciphering which of the epigenetically up-regulated genes are triggered due to direct manipulation of epigenetic pathways versus those that are activated by the upregulation of transcriptional activators (e.g., epigenetically-facilitated induction of transcriptional promoters).
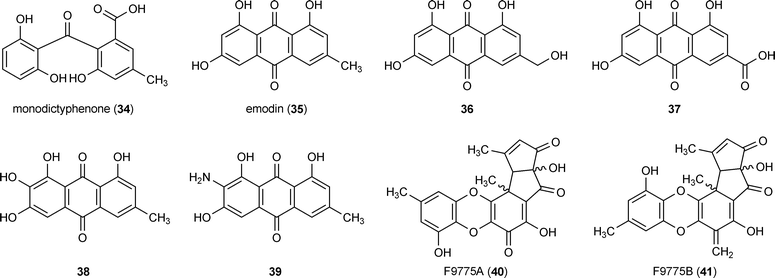
In another study investigating the activation of secondary-metabolite-encoding pathways in A. nidulans, researchers targeted the deletion of cclA, which is orthologous to S. cerevisiae bre2. The bre2 gene encodes for a protein component of the Set1-containing COMPASS complex that is responsible for the methylation of lysine 4 of histone H3 (H3K4). Using a stcJ deletion mutant that was deficient for the biosynthesis of 26, at least eight previously undetected metabolites were observed, including mondictyphenone (34), several emodin analogs (35–39), and the aromatic polyketides F9775A (40) and F9775B (41). Examination of histone H3 methylation in the ΔcclA strain showed a marked decrease in the di- and tri-methylation of H3K4 along with an associated decline in H3K9 di- and tri-methylation of genes involved in emodin biosynthesis. These data demonstrate that chromatin-level changes are involved in the suppression/activation of biosynthetic gene clusters.
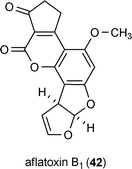
Further evidence demonstrating the participation of epigenome markers on natural product biosynthesis were provided by directly probing the acetylation and methylation status of histones associated with the aflatoxin B1 (42) gene cluster throughout the process of its upregulation.53 Using a chromatin immunoprecipitation method, researchers tracked levels of histone H4 acetylation in Aspergillus parasiticus across the gene cluster responsible for the production of 42. A wave-like pattern of histone H4 acetylation was observed to radiate from the promoter region and encompass the entire gene cluster over a 40 h period.53 Moreover, this effect was linked to a sequential rise in levels of biosynthetic proteins associated with the production of 42, as well as the extracellular accumulation of this mycotoxin. This simple mapping of posttranslation histone modification across a natural-product-encoding gene cluster provides an exciting glimpse into the dynamism that is hypothesized to characterize much of the chromatin landscape involved in generating fungal secondary metabolites.
4 Conclusions and future prospects
Targeting the epigenome represents an incredible opportunity for obtaining new secondary metabolites from fungi and other organisms. The two approaches currently in use are chemical epigenetic methods and molecular-based gene manipulation. The most impressive advantage of using small-molecule inhibitors that target epigenetic features is that this low-cost technique is relatively easy to apply in high-throughput screening operations. This approach can be effectively used to sample both well-studied and new fungi without a priori knowledge of a fungus's genomic composition. Unfortunately, this method is not flawless, since the diversity of epigenome-related targets occurring throughout the fungal kingdom suggests that a portion of fungal isolates might be resistant to currently available small-molecule inhibitors. In contrast, molecular-based epigenome manipulation strategies provide opportunities for preparing stable mutants that have altered secondary metabolite expression profiles. Regrettably, this technique requires substantial knowledge of the epigenome targets uniquely encoded within each fungal strain. More importantly, this more costly and laborious approach is not well-suited for rapidly screening microbial libraries.Perhaps the best solution to accessing natural products from silent biosynthetic pathways will require a marriage of both approaches in which chemical epigenetic screening is used to rapidly identify promising fungal strains and subsequent molecular manipulation techniques are employed to provide long-term control over metabolite production. However, certain questions must be answered before this type of secondary metabolite discovery paradigm can be judiciously used. For example, current understanding of epigenetic processes in fungi is highly dependent on discoveries derived from a limited number of model organisms. Further studies will be needed to delineate how fungal interspecies diversity is reflected in epigenetic processes that impact natural product biosynthesis. Addressing this question will help provide a predictive evidence-based approach for rationally activating silent biosynthetic pathways in new fungal systems. In addition, it will be important to identify other regulatory features that operate in conjunction with epigenetic mechanisms so that we are able to better understand how natural product biosynthesis is controlled.
5 Acknowledgements
Work in the author's laboratory was supported by the University of Oklahoma College of Arts and Sciences and the University of Oklahoma Department of Chemistry and Biochemistry. The assistance of M. Joyner and J. Henrikson for providing data used in Fig. 2 and Fig. 6, respectively, is appreciated.6 References
- J. E. Galagan, S. E. Calvo, C. Cuomo, L.-J. Ma, J. R. Wortman, S. Batzoglou, S.-I. Lee, M. Basturkmen, C. C. Spevak, J. Clutterbuck, V. Kapitonov, J. Jurka, C. Scazzocchio, M. Farman, J. Butler, S. Purcell, S. Harris, G. H. Braus, O. Draht, S. Busch, C. D'Enfert, C. Bouchier, G. H. Goldman, D. Bell-Pedersen, S. Griffiths-Jones, J. H. Doonan, J. Yu, K. Vienken, A. Pain, M. Freitag, E. U. Selker, D. B. Archer, M. A. Penalva, B. R. Oakley, M. Momany, T. Tanaka, T. Kumagai, K. Asai, M. Machida, W. C. Nierman, D. W. Denning, M. Caddick, M. Hynes, M. Paoletti, R. Fischer, B. Miller, P. Dyer, M. S. Sachs, S. A. Osmani and B. W. Birren, Nature, 2005, 438, 1105–1115 CrossRef CAS.
- J. E. Galagan, S. E. Calvo, K. A. Borkovich, E. U. Selker, N. D. Read, D. Jaffe, W. FitzHugh, L.-J. Ma, S. Smirnov, S. Purcell, B. Rehman, T. Elkins, R. Engels, S. Wang, C. B. Nielsen, J. Butler, M. Endrizzi, D. Qui, P. Ianakiev, D. Bell-Pedersen, M. A. Nelson, M. Werner-Washburne, C. P. Selitrennikoff, J. A. Kinsey, E. L. Braun, A. Zelter, U. Schulte, G. O. Kothe, G. Jedd, W. Mewes, C. Staben, E. Marcotte, D. Greenberg, A. Roy, K. Foley, J. Naylor, N. Stange-Thomann, R. Barrett, S. Gnerre, M. Kamal, M. Kamvysselis, E. Mauceli, C. Bielke, S. Rudd, D. Frishman, S. Krystofova, C. Rasmussen, R. L. Metzenberg, D. D. Perkins, S. Kroken, C. Cogoni, G. Macino, D. Catcheside, W. Li, R. J. Pratt, S. A. Osmani, C. P. C. DeSouza, L. Glass, M. J. Orbach, J. A. Berglund, R. Voelker, O. Yarden, M. Plamann, S. Seiler, J. Dunlap, A. Radford, R. Aramayo, D. O. Natvig, L. A. Alex, G. Mannhaupt, D. J. Ebbole, M. Freitag, I. Paulsen, M. S. Sachs, E. S. Lander, C. Nusbaum and B. Birren, Nature, 2003, 422, 859–868 CrossRef CAS.
- S. Kroken, N. L. Glass, J. W. Taylor, O. C. Yoder and B. G. Turgeon, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15670–15675 CrossRef CAS.
- K. M. Fisch, A. F. Gillaspy, M. Gipson, J. C. Henrikson, A. R. Hoover, L. Jackson, F. Z. Najar, H. Wägele and R. H. Cichewicz, J. Ind. Microbiol. Biotechnol., 2009, 36, 1199–1213 CrossRef CAS.
- C. Corre and G. L. Challis, Nat. Prod. Rep., 2009, 26, 977–986 RSC.
- H. Gross, Appl. Microbiol. Biotechnol., 2007, 75, 267–277 CrossRef CAS.
- G. L. Challis, Microbiology, 2008, 154, 1555–1569 CrossRef CAS.
- A. A. Brakhage, J. Schuemann, S. Bergmann, K. Scherlach, V. Schroeckh and C. Hertweck, Prog. Drug Res., 2008, 66, 3–12 Search PubMed.
- K. Scherlach and C. Hertweck, Org. Biomol. Chem., 2009, 7, 1753–1760 RSC.
- D. L. Spector, Annu. Rev. Biochem., 2003, 72, 573–608 CrossRef CAS.
- J. Mata, S. Marguerat and J. Bähler, Trends Biochem. Sci., 2005, 30, 506–514 CrossRef CAS.
- R. D. Hawkins and B. Ren, Hum. Mol. Genet., 2006, 15, R1–R7 CrossRef CAS.
- A. Shilatifard, Annu. Rev. Biochem., 2006, 75, 243–269 CrossRef CAS.
- S. I. S. Grewal and S. Jia, Nat. Rev. Genet., 2007, 8, 35–46 CrossRef CAS.
- M. Bühler, Chromosoma, 2009, 118, 141–151 CrossRef CAS.
- H.-J. Schüller, Curr. Genet., 2003, 43, 139–160.
- N. P. Keller, G. Turner and J. W. Bennett, Nat. Rev. Microbiol., 2005, 3, 937–947 Search PubMed.
- D. Hoffmeister and N. P. Keller, Nat. Prod. Rep., 2007, 24, 393–416 RSC.
- G. Felsenfeld and M. Groudine, Nature, 2003, 421, 448–453 CrossRef.
- R. B. Williams, J. C. Henrikson, A. R. Hoover, A. E. Lee and R. H. Cichewicz, Org. Biomol. Chem., 2008, 6, 1895–1897 RSC.
- J. C. Henrikson, A. R. Hoover, P. M. Joyner and R. H. Cichewicz, Org. Biomol. Chem., 2009, 7, 435–438 RSC.
- J. M. Harp, B. L. Hanson and G. J. Bunick, in New Comprehensive Biochemistry, eds. J. Zlatanova and S. H. Leuba, Elsevier, Amsterdam, 2004, pp. 13–44 Search PubMed.
- E. M. Bradbury and K. E. van Holde, in New Comprehensive Biochemistry, eds. J. Zlatanova and S. H. Leuba, Elsevier, Amsterdam, 2004, pp. 1–11 Search PubMed.
- M. Biel, V. Wascholowski and A. Giannis, Angew. Chem., Int. Ed., 2005, 44, 3186–3216 CrossRef CAS.
- R. Allshire and E. U. Selker, in Epigenetics, ed. C. D. Allis, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, 2007, pp. 101–125 Search PubMed.
- G. Brosch, P. Loidl and S. Graessle, FEMS Microbiol. Rev., 2008, 32, 409–439 CrossRef CAS.
- N. Happel and D. Doenecke, Gene, 2009, 431, 1–12 CrossRef CAS.
- K. Luger and J. C. Hansen, Curr. Opin. Struct. Biol., 2005, 15, 188–196 CrossRef CAS.
- M. Bustin, F. Catez and J.-H. Lim, Mol. Cell, 2005, 17, 617–620 CrossRef CAS.
- H. E. Kasinsky, J. D. Lewis, J. B. Dacks and J. Ausio, FASEB J., 2001, 15, 34–42 CrossRef CAS.
- Y. Fan, T. Nikitina, J. Zhao, T. J. Fleury, R. Bhattacharyya, E. E. Bouhassira, A. Stein, C. L. Woodcock and A. I. Skoultchi, Cell, 2005, 123, 1199–1212 CrossRef CAS.
- J. Zlatanova, C. Seebart and M. Tomschik, Trends Biochem. Sci., 2008, 33, 247–253 CrossRef CAS.
- A. Izzo, K. Kamieniarz and R. Schneider, Biol. Chem., 2008, 389, 333–343 CrossRef CAS.
- A. Ralph-Edwards and J. C. Silver, Exp. Cell Res., 1983, 148, 363–376 CrossRef CAS.
- J. L. Barra, L. Rhounim, J.-L. Rossignol and G. Faugeron, Mol. Cell. Biol., 2000, 20, 61–69 CrossRef CAS.
- A. Ramón, M. I. Muro-Pastor, C. Scazzocchio and R. Gonzalez, Mol. Microbiol., 2000, 35, 223–233 CrossRef CAS.
- H. D. Folco, M. Freitag, A. Ramon, E. D. Temporini, M. E. Alvarez, I. Garcia, C. Scazzocchio, E. U. Selker and A. L. Rosa, Eukaryotic Cell, 2003, 2, 341–350 CrossRef CAS.
- M. Veron, Y. Zou, Q. Yu, X. Bi, A. Selmi, E. Gilson and P.-A. Defossez, Genetics, 2006, 173, 579–587 CrossRef CAS.
- G. Schäfer, C. R. E. McEvoy and H.-G. Patterton, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14838–14843 CrossRef CAS.
- Q. Yu, H. Kuzmiak, Y. Zou, L. Olsen, P.-A. Defossez and X. Bi, J. Biol. Chem., 2009, 284, 740–750 CAS.
- J. Ausió, BioEssays, 2000, 22, 873–877 CrossRef CAS.
- V. Ramakrishnan, Annu. Rev. Biophys. Biomol. Struct., 1997, 26, 83–112 CrossRef CAS.
- H. Nishida, Comp. Funct. Genomics, 2009, 2009, 379317 Search PubMed.
- J. R. Davie, inNew Comprehensive Biochemistry, eds. J. Zlatanova and S. H. Leuba, Elsevier, Amsterdam, 2004, pp. 205–240 Search PubMed.
- C. L. Peterson and M.-A. Laniel, Curr. Biol., 2004, 14, R546–R551 CrossRef CAS.
- C. B. Millar and M. Grunstein, Nat. Rev. Mol. Cell Biol., 2006, 7, 657–666 CrossRef CAS.
- M. Wiren, R. A. Silverstein, I. Sinha, J. Walfridsson, H.-m. Lee, P. Laurenson, L. Pillus, D. Robyr, M. Grunstein and K. Ekwall, EMBO J., 2005, 24, 2906–2918 CrossRef CAS.
- J. E. Krebs, Mol. BioSyst., 2007, 3, 590–597 RSC.
- W. Peng, C. Togawa, K. Zhang and S. K. Kurdistani, Genetics, 2008, 179, 277–289 CrossRef CAS.
- Y. Reyes-Dominguez, F. Narendja, H. Berger, A. Gallmetzer, R. Fernandez-Martin, I. Garcia, C. Scazzocchio and J. Strauss, Eukaryotic Cell, 2008, 7, 656–663 CrossRef CAS.
- B. Grimaldi, P. Coiro, P. Filetici, E. Berge, J. R. Dobosy, M. Freitag, E. U. Selker and P. Ballario, Mol. Biol. Cell, 2006, 17, 4576–4583 CrossRef CAS.
- T. Tsubota, C. E. Berndsen, J. A. Erkmann, C. L. Smith, L. Yang, M. A. Freitas, J. M. Denu and P. D. Kaufman, Mol. Cell, 2007, 25, 703–712 CrossRef CAS.
- L. V. Roze, A. E. Arthur, S.-Y. Hong, A. Chanda and J. E. Linz, Mol. Microbiol., 2007, 66, 713–726 CrossRef CAS.
- M. Vogelauer, J. Wu, N. Suka and M. Grunstein, Nature, 2000, 408, 495–498 CrossRef CAS.
- J. H. Waterborg, J. Biol. Chem., 2000, 275, 13007–13011 CrossRef CAS.
- J. R. Davie, C. A. Saunders, J. M. Walsh and S. C. Weber, Nucleic Acids Res., 1981, 9, 3205–3216 CrossRef CAS.
- P. O. Hassa, S. S. Haenni, M. Elser and M. O. Hottiger, Microbiol. Mol. Biol. Rev., 2006, 70, 789–829 CrossRef CAS.
- F. Geng and W. P. Tansey, Mol. Biol. Cell, 2008, 19, 3616–3624 CrossRef CAS.
- J. Kim and R. G. Roeder, J. Biol. Chem., 2009, 284, 20582–20592 CrossRef CAS.
- A. L. Haas, P. M. Bright and V. E. Jackson, J. Biol. Chem., 1988, 263, 13268–13275 CAS.
- Y. I. Hassan and J. Zempleni, J. Nutr., 2006, 136, 1763–1765 CAS.
- S. Healy, T. D. Heightman, L. Hohmann, D. Schriemer and R. A. Gravel, Protein Sci., 2009, 18, 314–328 CrossRef CAS.
- T. Krishnamoorthy, X. Chen, J. Govin, W. L. Cheung, J. Dorsey, K. Schindler, E. Winter, C. D. Allis, V. Guacci, S. Khochbin, M. T. Fuller and S. L. Berger, Genes Dev., 2006, 20, 2580–2592 CrossRef CAS.
- M. Fink, D. Imholz and F. Thoma, Mol. Cell. Biol., 2007, 27, 3589–3600 CrossRef CAS.
- A. Hammet, C. Magill, J. Heierhorst and S. P. Jackson, EMBO Rep., 2007, 8, 851–857 CrossRef CAS.
- D. Nathan, K. Ingvarsdottir, D. E. Sterner, G. R. Bylebyl, M. Dokmanovic, J. A. Dorsey, K. A. Whelan, M. Krsmanovic, W. S. Lane, P. B. Meluh, E. S. Johnson and S. L. Berger, Genes Dev., 2006, 20, 966–976 CrossRef CAS.
- B. E. Bernstein, E. L. Humphrey, R. L. Erlich, R. Schneider, P. Bouman, J. S. Liu, T. Kouzarides and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8695–8700 CAS.
- A. Kirmizis, H. Santos-Rosa, C. J. Penkett, M. A. Singer, R. D. Green and T. Kouzarides, Nat. Struct. Mol. Biol., 2009, 16, 449–451 CrossRef CAS.
- K. Verzijlbergen, A. Faber, I. Stulemeijer and F. van Leeuwen, BMC Mol. Biol., 2009, 10, 76 CrossRef.
- K. Smith, G. Kothe, C. Matsen, T. Khlafallah, K. Adhvaryu, M. Hemphill, M. Freitag, M. Motamedi and E. Selker, Epigenetics & Chromatin, 2008, 1, 5 Search PubMed.
- H. Tamaru, X. Zhang, D. McMillen, P. B. Singh, J. Nakayama, S. I. Grewal, C. D. Allis, X. Cheng and E. U. Selker, Nat. Genet., 2003, 34, 75–79 CrossRef CAS.
- K. Zhang, Y. Chen, Z. Zhang and Y. Zhao, J. Proteome Res., 2009, 8, 900–906 CrossRef CAS.
- J. A. Latham and S. Y. R. Dent, Nat. Struct. Mol. Biol., 2007, 14, 1017–1024 CrossRef CAS.
- B. C. Smith and J. M. Denu, Biochim. Biophys. Acta, 2009, 1789, 45–57 CAS.
- R. Marmorstein and R. C. Trievel, Biochim. Biophys. Acta, 2009, 1789, 58–68 CAS.
- T. Nakayama and Y. Takami, J. Biol. Chem., 2001, 129, 491–499 CAS.
- G. Legube and D. Trouche, EMBO Rep., 2003, 4, 944–947 CrossRef CAS.
- N. Sengupta and E. Seto, J. Cell. Biochem., 2004, 93, 57–67 CrossRef CAS.
- A. Brandl, T. Heinzel and O. H. Krämer, Biol. Cell, 2009, 101, 193–205 CrossRef CAS.
- R. J. Klose and Y. Zhang, Nat. Rev. Mol. Cell Biol., 2007, 8, 307–318 CrossRef CAS.
- X. Cheng and X. Zhang, Mutat. Res., 2007, 618, 102–115 CrossRef CAS.
- S. Nakanishi, B. W. Sanderson, K. M. Delventhal, W. D. Bradford, K. Staehling-Hampton and A. Shilatifard, Nat. Struct. Mol. Biol., 2008, 15, 881–888 CrossRef CAS.
- S. Dillon, X. Zhang, R. Trievel and X. Cheng, Genome Biol., 2005, 6, 227 CrossRef.
- K. K. Adhvaryu, S. A. Morris, B. D. Strahl and E. U. Selker, Eukaryotic Cell, 2005, 4, 1455–1464 CrossRef CAS.
- X. Zhang, Z. Yang, S. I. Khan, J. R. Horton, H. Tamaru, E. U. Selker and X. Cheng, Mol. Cell, 2003, 12, 177–185 CrossRef CAS.
- J. Sollier, W. Lin, C. Soustelle, K. Suhre, A. Nicolas, V. Geli and C. de La Roche Saint-Andre, EMBO J., 2004, 23, 1957–1967 CrossRef CAS.
- K. Sawada, Z. Yang, J. R. Horton, R. E. Collins, X. Zhang and X. Cheng, J. Biol. Chem., 2004, 279, 43296–43306 CrossRef CAS.
- R. J. Klose, E. M. Kallin and Y. Zhang, Nat. Rev. Genet., 2006, 7, 715–727 CrossRef CAS.
- T. Takeuchi, Y. Watanabe, T. Takano-Shimizu and S. Kondo, Dev. Dyn., 2006, 235, 2449–2459 CrossRef CAS.
- M. G. Lee, R. Villa, P. Trojer, J. Norman, K.-P. Yan, D. Reinberg, L. Di Croce and R. Shiekhattar, Science, 2007, 318, 447–450 CrossRef CAS.
- K. Ingvarsdottir, C. Edwards, M. G. Lee, J. S. Lee, D. C. Schultz, A. Shilatifard, R. Shiekhattar and S. L. Berger, Mol. Cell. Biol., 2007, 27, 7856–7864 CrossRef CAS.
- H. H. Ng, Q. Feng, H. Wang, H. Erdjument-Bromage, P. Tempst, Y. Zhang and K. Struhl, Genes Dev., 2002, 16, 1518–1527 CrossRef CAS.
- P. Trojer, M. Dangl, I. Bauer, S. Graessle, P. Loidl and G. Brosch, Biochemistry, 2004, 43, 10834–10843 CrossRef CAS.
- M. Hosny, J.-P. Pais de Barros, V. Gianinazzi-Pearson and H. Dulieu, Fungal Genet. Biol., 1997, 22, 103–111 CrossRef CAS.
- F. Antequera, M. Tamame, J. R. Villanueva and T. Santos, Nucleic Acids Res., 1985, 13, 6545–6558 CrossRef CAS.
- C. Goyon, J. L. Rossignol and G. Faugeron, Nucleic Acids Res., 1996, 24, 3348–3356 CrossRef CAS.
- M. M. Suzuki and A. Bird, Nat. Rev. Genet., 2008, 9, 465–476 CrossRef CAS.
- A. Bird, Genes Dev., 2002, 16, 6–21 CrossRef CAS.
- M. R. Rountree and E. U. Selker, Genes Dev., 1997, 11, 2383–2395 CrossRef CAS.
- H. Gowher, K. C. Ehrlich and A. Jeltsch, FEMS Microbiol. Lett., 2001, 205, 151–155 CrossRef CAS.
- H. Foss, C. Roberts, K. Claeys and E. Selker, Science, 1993, 262, 1737–1741 CrossRef CAS.
- F. Antequera, M. Tamame, J. R. Villanueva and T. Santos, J. Biol. Chem., 1984, 259, 8033–8036 CAS.
- E. R. Jupe, J. M. Magill and C. W. Magill, J. Bacteriol., 1986, 165, 420–423 CAS.
- M. E. Zolan and P. J. Pukkila, Mol. Cell. Biol., 1986, 6, 195–200 CAS.
- J. T. Irelan and E. U. Selker, Genetics, 1997, 146, 509–523 CAS.
- D. W. Lee, M. Freitag, E. U. Selker and R. Aramayo, PLoS One, 2008, 3, e2531 CrossRef.
- T. A. Schuurs, E. A. M. Schaeffer and J. G. H. Wessels, Genetics, 1997, 147, 589–596 CAS.
- A. Codon, Y. Lee and V. Russo, Nucleic Acids Res., 1997, 25, 2409–2416 CrossRef CAS.
- E. U. Selker, N. A. Tountas, S. H. Cross, B. S. Margolin, J. G. Murphy, A. P. Bird and M. Freitag, Nature, 2003, 422, 893–897 CrossRef CAS.
- H. Tamaru and E. U. Selker, Nature, 2001, 414, 277–283 CrossRef CAS.
- K. K. Adhvaryu and E. U. Selker, Genes Dev., 2008, 22, 3391–3396 CrossRef CAS.
- J. Zlatanova, I. Stancheva and P. Caiafa, in New Comprehensive Biochemistry, eds. J. Zlatanova and S. H. Leuba, Elsevier, Amsterdam, 2004, pp. 309–341 Search PubMed.
- B. M. Turner, in New Comprehensive Biochemistry, eds. J. Zlatanova and S. H. Leuba, Elsevier, Amsterdam, 2004, pp. 291–308 Search PubMed.
- I. Stancheva, Biochem. Cell Biol., 2005, 83, 385–395 Search PubMed.
- T. Vaissière, C. Sawan and Z. Herceg, Mutat. Res., 2008, 659, 40–48 CAS.
- A. C. D'Alessio and M. Szyf, Biochem. Cell Biol., 2006, 84, 463–476 Search PubMed.
- W. Fischle, Genes Dev., 2008, 22, 3375–3382 CrossRef CAS.
- W. Fischle, Y. Wang and C. D. Allis, Curr. Opin. Cell Biol., 2003, 15, 172–183 CrossRef CAS.
- K. A. Gelato and W. Fischle, Biol. Chem., 2008, 389, 353–363 CrossRef CAS.
- A. Vitaliano-Prunier, A. Menant, M. Hobeika, V. Geli, C. Gwizdek and C. Dargemont, Nat. Cell Biol., 2008, 10, 1365–1371 CrossRef CAS.
- F. Martino, S. Kueng, P. Robinson, M. Tsai-Pflugfelder, F. van Leeuwen, M. Ziegler, F. Cubizolles, M. M. Cockell, D. Rhodes and S. M. Gasser, Mol. Cell, 2009, 33, 323–334 CrossRef CAS.
- H. Santos-Rosa, A. Kirmizis, C. Nelson, T. Bartke, N. Saksouk, J. Cote and T. Kouzarides, Nat. Struct. Mol. Biol., 2009, 16, 17–22 CrossRef CAS.
- E. U. Selker, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 9430–9435 CrossRef CAS.
- M. A. Hall, A. Shundrovsky, L. Bai, R. M. Fulbright, J. T. Lis and M. D. Wang, Nat. Struct. Mol. Biol., 2009, 16, 124–129 CrossRef CAS.
- I. Sinha, M. Wirén and K. Ekwall, Chromosome Res., 2006, 14, 95–105 CrossRef CAS.
- D. K. Pokholok, C. T. Harbison, S. Levine, M. Cole, N. M. Hannett, T. I. Lee, G. W. Bell, K. Walker, P. A. Rolfe, E. Herbolsheimer, J. Zeitlinger, F. Lewitter, D. K. Gifford and R. A. Young, Cell, 2005, 122, 517–527 CrossRef CAS.
- S. M. Fuchs, R. N. Laribee and B. D. Strahl, Biochim. Biophys. Acta, 2009, 1789, 26–36 CAS.
- Z. Yang, C. Zheng and J. J. Hayes, J. Biol. Chem., 2007, 282, 7930–7938 CrossRef CAS.
- C. Zheng and J. J. Hayes, Biopolymers, 2003, 68, 539–546 CrossRef CAS.
- M. Paris, M. Porcelloni, M. Binaschi and D. Fattori, J. Med. Chem., 2008, 51, 1505–1529 CrossRef CAS.
- P. A. Cole, Nat. Chem. Biol., 2008, 4, 590–597 CrossRef CAS.
- S. Minucci and P. G. Pelicci, Nat. Rev. Cancer, 2006, 6, 38–51 CrossRef CAS.
- M. Dokmanovic, C. Clarke and P. A. Marks, Mol. Cancer Res., 2007, 5, 981–989 CrossRef CAS.
- J.-P. J. Issa, H. M. Kantarjian and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2005, 4, 275–276 CrossRef CAS.
- J. Goffin and E. Eisenhauer, Ann. Oncol., 2002, 13, 1699–1716 CrossRef CAS.
- H. Mooibroek, A. G. J. Kuipers, J. H. Sietsma, P. J. Punt and J. G. H. Wessels, Mol. Gen. Genet., 1990, 222, 41–48 CAS.
- J. C. Cheng, C. B. Matsen, F. A. Gonzales, W. Ye, S. Greer, V. E. Marquez, P. A. Jones and E. U. Selker, J. Nat. Cancer Inst., 2003, 95, 399–409 Search PubMed.
- P. R. J. Birch, P. F. G. Sims and P. Broda, J. Appl. Microbiol., 1998, 85, 417–424 CrossRef CAS.
- S. Pancaldi, L. Del Senno, M. P. Fasulo, F. Poli and G. L. Vannini, Cell Biol. Int. Rep., 1988, 12, 35–40 CrossRef CAS.
- M. Tamame, F. Antequera, J. R. Villanueva and T. Santos, J. Gen. Microbiol., 1983, 129, 2585–2594 CAS.
- M. Tamame, F. Antequera, J. R. Villanueva and T. Santos, Mol. Cell. Biol., 1983, 3, 2287–2297 CAS.
- M. Tamame, F. Antequera and E. Santos, Mol. Cell. Biol., 1988, 8, 3043–3050 CAS.
- K. Akiyama, T. Ohguchi and K. Takata, Nippon Shokubutsu Byori Gakkaiho, 1997, 63, 385–387 Search PubMed.
- K. Akiyama, H. Katakami and R. Takata, Plant Biotechnol., 2007, 24, 345–348 CAS.
- M. S. Kritsky, S. Y. Filippovich, T. P. Afanasieva, G. P. Bachurina and V. E. A. Russo, Appl. Biochem. Microbiol., 2001, 37, 243–247 Search PubMed.
- M. S. Kritsky, V. E. A. Russo, S. Y. Filippovich, T. P. Afanasieva and G. P. Bachurina, Photochem. Photobiol., 2002, 75, 79–83 CrossRef CAS.
- S. Y. Filippovich, G. P. Bachurina and M. S. Kritsky, Appl. Biochem. Microbiol., 2004, 40, 398–403 Search PubMed.
- T. Kakutani, J. A. Jeddeloh, S. K. Flowers, K. Munakata and E. J. Richards, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 12406–12411 CrossRef CAS.
- M. Fieldes and L. Amyot, J. Hered., 1999, 90, 199–206 CrossRef CAS.
- S. P. Kumpatla and T. C. Hall, Plant Mol. Biol., 1998, 38, 1113–1122 CrossRef CAS.
- M. A. Fields, Genome, 1994, 37, 1–11 CrossRef.
- M.d. F. P. S. Machado and M. A. A. de Castro-Prado, Biochem. Genet., 2001, 39, 357–368 CrossRef CAS.
- M. A. Costa, N. C. G. Silva and M. A. A. Castro-Prado, Biol. Res., 2001, 34, 91–98 Search PubMed.
- B. E. Bernstein, J. K. Tong and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13708–13713 CrossRef CAS.
- J. D. Walton, Phytochemistry, 2006, 67, 1406–1413 CrossRef CAS.
- G. Brosch, M. Dangl, S. Graessle, A. Loidl, P. Trojer, E.-M. Brandtner, K. Mair, J. D. Walton, D. Baidyaroy and P. Loidl, Biochemistry, 2001, 40, 12855–12863 CrossRef CAS.
- E. K. Shwab, J. W. Bok, M. Tribus, J. Galehr, S. Graessle and N. P. Keller, Eukaryotic Cell, 2007, 6, 1656–1664 CrossRef CAS.
- R. M. Perrin, N. D. Fedorova, J. W. Bok, R. A. Cramer, J. R. Wortman, H. S. Kim, W. C. Nierman and N. P. Keller, PLoS Pathog., 2007, 3, e50 Search PubMed.
- A. Andreou, F. Brodhun and I. Feussner, Prog. Lipid Res., 2009, 48, 148–170 CrossRef CAS.
- J. C. Overeem, A. K. Sijpesteijn and A. Fuchs, Phytochemistry, 1967, 6, 99–105 CrossRef CAS.
- J. Schumann and C. Hertweck, J. Biotechnol., 2006, 124, 690–703 CrossRef.
- J.-H. Yu and N. Keller, Annu. Rev. Phytopathol., 2005, 43, 437–458 Search PubMed.
- J. W. Bok and N. P. Keller, Eukaryotic Cell, 2004, 3, 527–535 CrossRef CAS.
- J. W. Bok, D. Noordermeer, S. P. Kale and N. P. Keller, Mol. Microbiol., 2006, 61, 1636–1645 CrossRef CAS.
- N. Keller, J. Bok, D. Chung, R. M. Perrin and E. K. Shwab, Med. Mycol., 2006, 44, 83–S85 Search PubMed.
- C. L. Schardl, Chem. Biol., 2006, 13, 5–6 CrossRef CAS.
- M.-Y. Zhang and T. Miyake, J. Agric. Food Chem., 2009, 57, 4162–4167 CrossRef CAS.
- K. Kosalková, C. García-Estrada, R. V. Ullán, R. P. Godio, R. Feltrer, F. Teijeira, E. Mauriz and J. F. Martín, Biochimie, 2009, 91, 214–225 CrossRef CAS.
- S. Bouhired, M. Weber, A. Kempf-Sontag, N. P. Keller and D. Hoffmeister, Fungal Genet. Biol., 2007, 44, 1134–1145 CrossRef CAS.
- J. W. Bok, D. Hoffmeister, L. A. Maggio-Hall, R. Murillo, J. D. Glasner and N. P. Keller, Chem. Biol., 2006, 13, 31–37 CrossRef CAS.
- S. Lodeiro, Q. Xiong, W. K. Wilson, Y. Ivanova, M. L. Smith, G. S. May and S. P. T. Matsuda, Org. Lett., 2009, 11, 1241–1244 CrossRef CAS.
- A. McDonagh, N. D. Fedorova, J. Crabtree, Y. Yu, S. Kim, D. Chen, O. Loss, T. Cairns, G. Goldman, D. Armstrong-James, K. Haynes, H. Haas, M. Schrettl, G. May, W. C. Nierman and E. Bignell, PLoS Pathog., 2008, 4, e1000154 Search PubMed.
- J. W. Bok, S. A. Balajee, K. A. Marr, D. Andes, K. F. Nielsen, J. C. Frisvad and N. P. Keller, Eukaryotic Cell, 2005, 4, 1574–1582 CrossRef CAS.
- J. A. Sugui, J. Pardo, Y. C. Chang, A. Mullbacher, K. A. Zarember, E. M. Galvez, L. Brinster, P. Zerfas, J. I. Gallin, M. M. Simon and K. J. Kwon-Chung, Eukaryotic Cell, 2007, 6, 1552–1561 CrossRef CAS.
- O. Bayram, S. Krappmann, M. Ni, J. W. Bok, K. Helmstaedt, O. Valerius, S. Braus-Stromeyer, N.-J. Kwon, N. P. Keller, J.-H. Yu and G. H. Braus, Science, 2008, 320, 1504–1506 CrossRef CAS.
- S. P. Kale, L. Milde, M. K. Trapp, J. C. Frisvad, N. P. Keller and J. W. Bok, Fungal Genet. Biol., 2008, 45, 1422–1429 CrossRef CAS.
- E. Szewczyk, Y.-M. Chiang, C. E. Oakley, A. D. Davidson, C. C. C. Wang and B. R. Oakley, Appl. Environ. Microbiol., 2008, 74, 7607–7612 CrossRef CAS.
- I. Lee, J.-H. Oh, E. K. Shwab, T. R. T. Dagenais, D. Andes and N. P. Keller, Fungal Gen. Biol., 2009, 46, 782–790 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |