Biosynthesis and biomimetic synthesis of alkaloids isolated from plants of the Nitraria and Myrioneuron genera: an unusual lysine-based metabolism
Edmond
Gravel
and
Erwan
Poupon
*
Laboratoire de Pharmacognosie associé au CNRS, UMR 8076 (BioCIS), Centre d'Études Pharmaceutiques, Université Paris-Sud 11, 5 rue Jean-Baptiste Clément, 92296, Châtenay-Malabry, France. E-mail: erwan.poupon@u-psud.fr
First published on 17th November 2009
Abstract
Covering: up to November 2009
This review describes a wide panel of alkaloids isolated from plants of the Nitraria genus, focusing on their biosynthesis and discussing the resulting biomimetic chemistry in relevant cases. The scope is purposely limited to alkaloids derived at least to some extent from L-lysine, considering that most of these molecules have unique structures and are specific to the genus. Some of the biosynthetic pathways described are taken from the literature, but others are proposed here for the first time. The latter are mostly hypotheses justified by the fact that they are based on metabolic routes frequently encountered for other Nitraria alkaloids, and thus permit unification of the biosynthesis around common pivotal biosynthetic intermediates. Myrioneuron alkaloids are also presented as a newly discovered class with striking similarities to Nitraria alkaloids.
 Edmond Gravel | Edmond Gravel studied pharmacy at the Paris-Sud 11 University, where he obtained his M.Sc. degree in 2005 and received his Ph.D. under the guidance of Prof. Erwan Poupon in 2008. He is now working at the CEA (Commissariat à l'Énergie Atomique, Service de Chimie Bioorganique et de Marquage – iBiTec-S) in Saclay, France. His research interests focus on syntheses of natural products, molecular self-constructions and supramolecular self-assemblies. |
 Erwan Poupon | Erwan Poupon studied pharmacy at the University of Rennes and completed his Ph.D. under the supervision of Prof. Henri-Philippe Husson and Dr Nicole Kunesch at Université Paris–Descartes. In 2000 he became a postdoctoral fellow of Prof. Emmanuel Theodorakis at the University of California at San Diego. He then moved to the School of Pharmacy at Université Paris-Sud, where he became a full professor of pharmacognosy and natural product chemistry in 2007. His research interests focus on natural product chemistry, biomimetic synthesis and chemical biology. |
1 Introduction
Plants of the Nitraria genus (Nitrariaceae) are shrubs that can grow up to 2 meters high. They bear fleshy leaves and small flowers, with five petals and fifteen stamens, arranged in scorpioid inflorescences. These plants also produce small edible fruits that can be consumed raw or after cooking.1 Well adapted to arid environments, Nitraria species are found in the plains of south-eastern Europe (Nitraria komarovii, Nitraria sibirica) and in the Middle-East (Nitraria schoberi = Nitraria billardieri) but also in Australia and northern Africa (Nitraria tridentata = Nitraria retusa) in northern and western parts of the Saharan desert and in Mauritania. The alkaloids discussed in this review are all obtained by extraction of the aerial parts of the plants.Many unique alkaloids have been extracted from the Nitraria genus, and the study of these molecules will be presented from an historical point of view. The vast majority of the compounds have been published in the Russian journal Khimiya Prirodnykh Soedinenii (with the English translation in Chemistry of Natural Compounds) by a team based today at the Institute of the Chemistry of Plant Substances at the Academy of Sciences, Tashkent, Republic of Uzbekistan. It is also worth mentioning that, apparently, few plant-gathering excursions have been undertaken in the last 40 years. Most studies published have therefore been conducted on existing batches of dried plants. It should be noted that some structures have been assigned on the basis of spectral data (1H-NMR, IR and UV). Some of them have later been reassigned, either after total synthesis or relying on better analysis (X-ray), which is not surprising considering the molecular complexity of some of the compounds concerned. Some other structures remain in doubt, and one of the aims of this review is to clarify this issue.
In 1996, a series of review articles were published in memory of S. Yu. Yunusov. These were compiled from reports on the investigations of all the alkaloids published in journals from the former USSR up to 1994. These reviews therefore included the Nitraria alkaloids isolated up to this point, and contain descriptions of the physical and chemical properties.2,3
Most Nitraria alkaloids have been extracted by classical alkaloid acid/base extraction, usually from a chloroform, benzene or ether phase followed by crystallization or silica gel chromatography. An interesting study was published in 1977 relating the Polybuffer® separation of alkaloids with an adaptation of the historical Craig apparatus.4 Several publications have also studied the total amount of alkaloids in different plant organs of three Nitraria species (N. komarovii,5N. sibirica,6 and N. schoberi7), and according to the location. The most common species (N. schoberi) is the richest in alkaloids, both in the quantitative and (as we will show) qualitative respects; usually, the leaves contain the highest quantities of alkaloids.
2 Biosynthetic hypotheses: reactive units from lysine
Gerrit-Jan Koomen and Martin J. Wanner, two researchers responsible for a large part of the chemical studies of Nitraria alkaloids,8 have split these molecules into three groups: spiro-alkaloids, tripiperidine alkaloids and indole alkaloids. The biosynthesis of compounds belonging to the first two groups can be explained by the assembly of C5 units derived from L-lysine, as we will show in this review. For the indole group an additional unit derived from L-tryptophan is required (Scheme 1).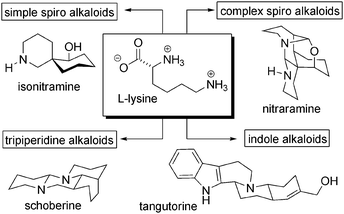 | ||
| Scheme 1 Classification of Nitraria alkaloids according to Koomen. | ||
For the biosynthesis of Nitraria alkaloids, L-lysine first undergoes decarboxylation and yields cadaverine, a C5 linear symmetric diamine (Scheme 2). After an oxidative deamination process at one of its extremities, cadaverine gives rise to 5-aminopentanal, which is unstable and cyclizes into Δ2-piperidine (1) that exists either as an enamine (Δ2-piperidine) or as an imine (Δ1-piperidine) depending on the acidity of the medium. Another oxidative deamination could in principle give rise to the formation of glutaraldehyde (2). This latter is a known protein-reticulating agent, and its presence as a free molecule in a living cell is of course unlikely, but we will see that equivalents in terms of oxidation state can be put forward (vide infra).
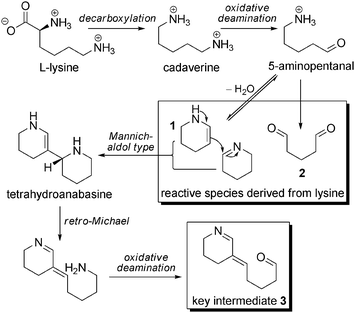 | ||
| Scheme 2 Key intermediates from lysine. | ||
Δ2-Piperidine (1) can dimerize into tetrahydroanabasine, which can lead to compound 39 through a retro-Michael mechanism followed by an oxidative deamination step. The importance of 3 will be highlighted throughout this article, and will be referred as to the “key intermediate” in the Nitraria metabolism.
3 Simple spiranic alkaloids
At first, a perhydroquinoline type skeleton was proposed as a key-element of both (+)-nitramine10 and (−)-isonitramine (Fig. 1).11 The actual spiranic structure was disclosed a decade later based on 1H, 13C and X-ray analysis.12 An absolute configuration was proposed according to the X-ray analysis (Scheme 2),13 which concluded that nitramine and isonitramine were diastereomers at the 7-position. A few years later, chemical synthesis brought a clear answer to the confusion concerning the absolute stereochemistry, which was then settled (Scheme 3 – box) by Schultz et al.14 and Husson et al.15 Several analogs with various N-substitutions have also been isolated: (−)-sibirine (an N-methyl isonitramine),16 nitrabirine,17 nitrabirine N-oxide18,19 and sibirinine.20 Despite their relatively simple structure, careful analysis of the literature can cause great confusion. As already pointed out by different authors and discussed by Koomen,8 controversy about the sign of optical rotation of these alkaloids still exists. Even though optical rotation values for sibirine are in agreement, the same cannot be said for nitramine and isonitramine: the relevant figures are summarized in Table 1 (for the synthetic samples, only records from the three first enantioselective syntheses have been collected14,15). We also cast doubt onto the absolute configuration studies by circular dichroism published in 1986, which established incorrect configurations.21 The influence of various proportions of stabilizing methanol in chloroform and intramolecular hydrogen bonds could explain these discrepancies.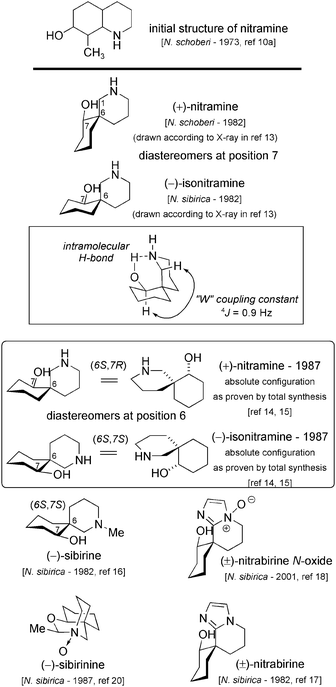 | ||
| Fig. 1 Simple spiranic alkaloids. | ||
 | ||
| Scheme 3 Biosynthetic hypotheses for spiro-alkaloids. | ||
| Compound | [α]D | c | Solvent | Source | Ref. |
|---|---|---|---|---|---|
| a np = not provided. | |||||
| Natural nitramine | +16.5 | 4.85 | CHCl3 | N. schoberi | 10a |
| −8 | np | MeOH | N. schoberi | 10b | |
| 0 | np | CHCl3 | N. sibirica | 26 | |
| Synthetic (+)-nitramine | +23 | 1.58 | CH2Cl2 | — | 14a |
| +21.3 | 1.52 | CH2Cl2 | — | 14b | |
| Natural isonitramine | −30 | 1.36 | CHCl3 | N. sibirica | 11 |
| −37 | np | MeOH | N. sibirica | 10b | |
| Synthetic (−)-isonitramine | −5 | 1.2 | CHCl3 | — | 15 |
| −3.5 | 1.61 | CHCl3 | — | 14a | |
| Synthetic (+)-isonitramine | +5 | 1.2 | CHCl3 | — | 15 |
| Natural sibirine | −22.5 | 0.81 | CHCl3 | N. sibirica | 16 |
| Synthetic (−)-sibirine from synthetic (−)-isonitramine | −22.5 | 0.8 | CHCl3 | — | 15 |
| Synthetic (+)-sibirine | +25 | 0.73 | CHCl3 | — | 14a |
| Natural nitrabirine | 0 | 2 | CHCl3 | N. sibirica | 17 |
| Natural nitrabirine N-oxide | 0 | np | np | N. sibirica | 18 |
| Natural sibirinine | −9.4 | 0.53 | CHCl3 | N. sibirica | 20 |
The best agreement concerning optical rotation values appears to be for sibirine, and in consequence this ought to be reasonably certain. In reality, a striking disorder is observed upon reading the literature. Natural sibirine is described as laevorotatory ((−)-sibirine: [α]D= −22.5 (0.81, CHCl3)). In the same publication, Yunusov et al. methylated both natural nitramine and isonitramine using methyl iodide: they apparently obtained (+)-sibirine from (−)-isonitramine, and logically concluded that natural (−)-sibirine is in fact N-methylated (+)-isonitramine (this latter being the non-natural antipode of natural (−)-isonitramine). Husson et al. repeated the experiment with both synthetic (−)- and (+)-isonitramine, and obtained natural (−)-sibirine from (−)-isonitramine: this result is in agreement with sibirine being simply the N-methylated derivative of isonitramine.22 But the conclusion in Husson's paper is ambiguous because they ultimately say that natural isonitramine is dextrorotatory.23 We therefore believe that isonitramine is laevorotatory,24 with an absolute configuration as represented in Fig. 1 (6S,7S25). The optical rotation for nitramine varies from +16.510a to −8,10b, as shown in Table 1, with even no optical rotation for a sample from N. sibirica.26 However, total synthesis by Schultz et al. established the absolute configuration of (+)-nitramine as (6S,7R) – the natural enantiomer.27
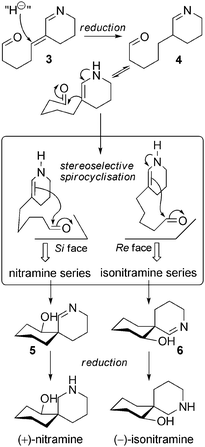
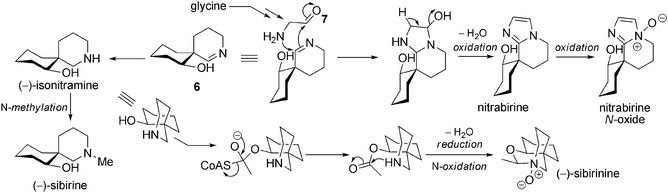
Biosynthetic hypotheses. The biosynthesis of simple spiro-alkaloids, as proposed by Koomen et al., is initiated by the formation of pivotal achiral intermediate 3. Reduction could afford 4, a compound ready for the spirocyclization step. In this crucial step, the two stereogenic centres are fixed, leading to the nitramine or isonitramine skeletons. A second reduction of the remaining imines 5 and 6 gives rise to nitraramine or isonitramine depending on the stereochemical outcome of the reaction (Scheme 3).
The analogs that have been isolated seem to derive from isonitramine, and their biosynthesis can be explained with simple functionalizations (Scheme 4). Indeed, like isonitramine, they have all been found in N. sibirica. A single N-methylation provides sibirine, while reaction with acetylCoA followed by N-oxidation can explain the biosynthesis of (−)-sibirinine. Yunusov et al. described the hemisynthesis of sibirinine from isonitramine with acetaldehyde/oxygen and degradative conditions, which allowed them to establish a link between the two molecules.
 | ||
| Scheme 4 Biosynthetic hypotheses of isonitramine congeners. | ||
Nitrabirine and nitrabirine N-oxide are optically inactive. In their 1983 paper,17 Yunosov et al. drew nitrabirine with an isonitramine configuration, but since then no chemical synthesis has been reported that could give information concerning the stereochemistry of nitrabirine and nitrabirine N-oxide.28 Their formation may be rationalized when considering the condensation of imine 6 with a C2 unit 7, which may come from glycine.
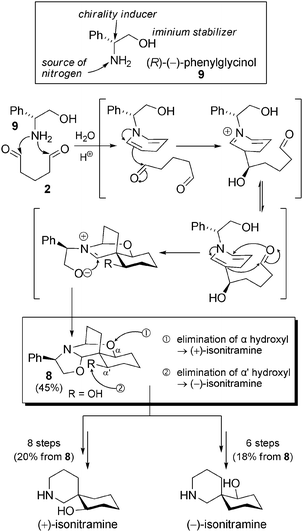
Biomimetic synthesis of simple spiro-alkaloids. Numerous total syntheses, racemic or enantioselective, of nitramine or isonitramine and related compounds have been published, but we will focus only on biomimetic approaches. The pioneering work of the Husson group in the 1980s was decisive for the establishment of absolute configurations of these alkaloids. Their elegant synthesis of (+)-isonitramine and (−)-isonitramine,29 using a chiral pool approach, paved the way for the development of biosynthetic hypotheses30 in the Nitraria genus. Taking advantage of the exceptional contribution of this group to the chemistry of 1,4-dihydropyridines, a creative synthetic route was developed. The key aspect rested in the formation of compound 8 from (R)-(−)-phenylglycinol (9) and glutaraldehyde (2) through an efficient one-pot trimolecular reaction (Scheme 5). Exploitation of iminium/enamine and Mannich-type reactions give rise to a single enantiopure intermediate featuring the entire carbon spiro-skeleton of the natural compounds. With this building block readily available (45%, one step), the opportunity to access (+)-isonitramine or (−)-isonitramine was at hand. Indeed, the total synthesis of both enantiomers of isonitramine (20% and 18% starting from 8) was possible. The synthesis of (−)-sibirine was also possible, but required 6–8 additional steps.
 | ||
| Scheme 5 Husson's first approach to spiro-alkaloids. | ||
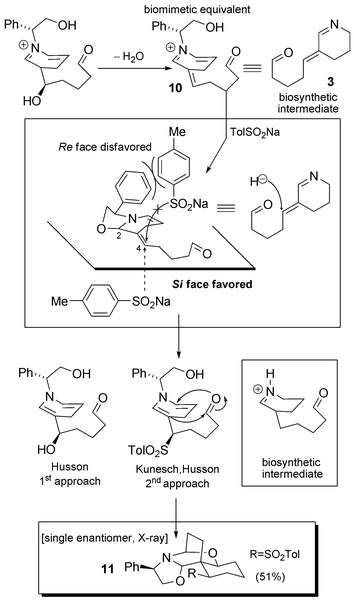
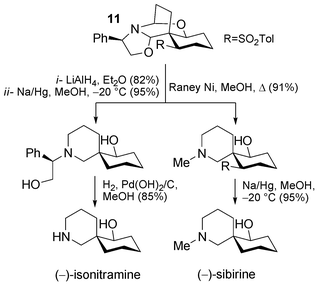
Ten years later, a slight but crucial modification was reported by Kunesch and Husson (Scheme 6).31 In fact, according to the biosynthetic hypotheses postulated (see Scheme 3), reduction of intermediate 3 converts a 1,4-Michael-type system into a simple enamine. This step is not taken into account in Husson's first report. In the course of the cascade reaction described above in Scheme 5, intermediate 10 can be postulated. This is a direct synthetic equivalent of pivotal biosynthetic intermediate 3. If a correctly chosen nucleophile could reduce this intermediate in the same manner as hydride does in Nature, a perfect biomimetic cascade could be designed. The use of para-toluene sodium sulfinate proved to be the perfect means towards this end. A trimolecular reaction between phenylglycinol (9), 2.5 equivalents of glutaraldehyde (2) and 2.2 equivalents of sulfinate in aqueous citric acid buffer permitted the isolation of the spiro-compound 11 in 51% yield. Compound 11 is the most thermodynamically stable outcome of a highly efficient cascade of equilibration reactions. The addition of sulfinate anion occurs only at position 4 of the α,β-unsaturated iminium system of 10, according to the principle of hard/soft acids/bases. This addition is diastereoselective due to the presence of a chiral appendage on the nitrogen, one face of the planar Michael system being sterically less accessible. Having now reached this advanced intermediate and starting from (R)-(−)-phenylglycinol, access to natural (−)-isonitramine (33% from 9) and (−)-sibirine (44% from 9) was possible in only a couple of steps (compare this with the first approach). In the case of sibirine, these included reductive removal of the sulfone with Na(Hg) and a novel debenzylation–N-methylation sequence with Raney® nickel in MeOH (Scheme 7). When using (S)-(+)-phenylglycinol, access to the enantiomeric series was possible, as was illustrated by the authors by the synthesis of (+)-isonitramine.32
 | ||
| Scheme 6 Husson's second approach to spiro-alkaloids (part 1). | ||
 | ||
| Scheme 7 Husson's second approach to spiro-alkaloids (part 2). | ||
Between the publication of these two syntheses, Koomen et al. also published a biomimetic synthesis of (±)-nitramine (Scheme 8).33 They chose compound 12 as a synthetic stable equivalent of key intermediate 3. This glutarimide-type alternative to 3 has been exploited throughout the years by this group to access numerous Nitraria alkaloids (vide infra). The major drawback of the strategy is the need to adjust the oxidation state by reduction at a late stage in the synthesis. In the case of nitramine, building block 12 was able to undergo the biomimetic spirocyclization with thiophenol–trimethylaluminium complex in THF, giving a single syn diastereomer 13. An explanation for the high stereoselectivity can be put forward by considering a chelated intermediate such as 14. Chemical manipulations then allow the preparation of nitramine from 13 in three steps (global yield: 45% from 12).
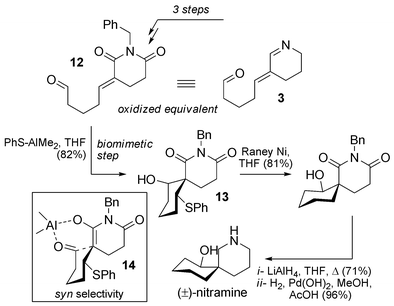 | ||
| Scheme 8 Biomimetic synthesis of nitramine by Koomen. | ||
4 Complex spiranic alkaloids: nitraramine and analogs
Isolated from Nitraria schoberi, N. komarovii and N. sibirica, nitraramine and its congeners are complex spiro-alkaloids with intricate cage-like structures that require an additional Δ2-piperidine unit for their biosynthesis (Fig. 2). Consisting of a total of six rings, three in a chair-like conformation and three in a boat-like conformation, these compounds bear an amino-ether group, an aminal function and six contiguous stereogenic centres, one of which is the quaternary spiro-centre common to all alkaloids of this group.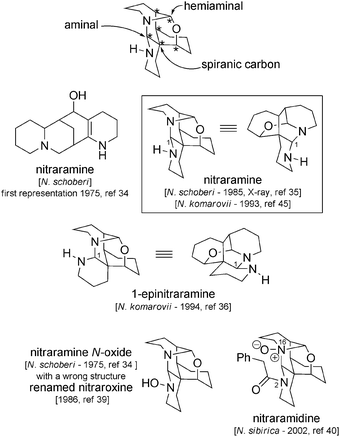 | ||
| Fig. 2 Complex spiranic alkaloids. | ||
Of these alkaloids, nitraramine was the first to be isolated, but was at that time assigned the wrong structure (see Fig. 2).34 Ten years later, the actual structure of the molecule was determined by X-ray analysis.35 In 1994, epinitraramine was isolated from N. komarovii (along with nitraramine itself) from samples collected in Tasmania.36
Biosynthesis. The Koomen team37 was the first to propose that these molecules might come from the assembly of L-lysine derivatives through a cascade of simple reactions. The first steps of the biosynthetic pathway postulated for these complex spiro-alkaloids are identical to those encountered in the case of the simpler spiro-alkaloids leading to the formation of key intermediate 3 from tetrahydroanabasine (Scheme 9). However, intermediate 3, instead of being reduced before spirocyclization, undergoes attack by a new molecule of Δ2-piperidine (1), which can take place at two different positions (path a or b, Scheme 9). The spirocyclization process then takes place through an intramolecular 1,4-conjugate addition, which gives rise to the quaternary centre of the molecule. A ring inversion is necessary to permit the cascade reduction of the two remaining iminiums initiated by the free hydroxyl group.
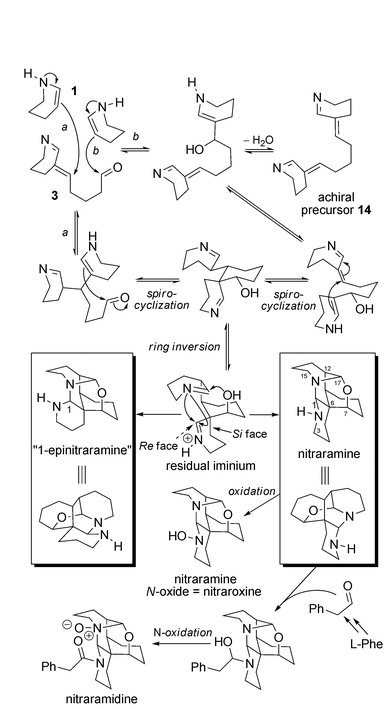 | ||
| Scheme 9 Biosynthetic hypotheses for nitramine and congeners. | ||
It is noteworthy that the last reduction of the cascade might occur by attack on the Si or the Re face of the iminium ion, resulting in the formation of nitraramine or epinitraramine, respectively. The oxidation of nitraramine can give rise to nitraramine N-oxide, first described in 197538 with an incorrect structure and later named nitraroxine.39 Another compound bearing an N-phenylacetyl group was isolated in 200240 from Nitraria sibirica and named nitraramidine. In this case, the N-oxidation took place at the N16 nitrogen. Nucleophilic attack of the secondary amine of nitraramine on phenylacetaldehyde (which results from the decarboxylation and oxidative deamination of L-phenylalanine) permits the formation of nitraramidine after a final oxidation step.
Biomimetic syntheses. Two syntheses of nitraramine have been published so far, both being based on biosynthetic considerations. From reagents that can be seen as analogs of Δ2-piperidine (1) and aminopentanal, Koomen and Wanner chose to synthesize compound 14, which bears all the atoms required for the construction of the target carbon skeleton. This biomimetic intermediate then served as a starting point for the reaction cascade leading to nitraramine. In our team, we instead chose to investigate the biosynthetic process from an earlier stage by forming compound 3 through the condensation of Δ2-piperidine and glutaraldehyde (which can arguably be seen as an oxidated form of aminopentanal), from which the entire cascade would take place and eventually give rise to nitraramine.
Synthesis by Koomen et al. In 1995, the Koomen team published the first synthesis of nitraramine, in a dozen steps from precursors 15 and 16 and with a global yield of 0.5% (Scheme 10). The researchers carried out the synthesis of their precursors by an aldol reaction under alkaline conditions followed by a dehydration process which was initiated by mesylation of the product followed by heating in the presence of triethylamine. Hydrolysis of the acetal afforded the corresponding aldehyde. Then, the attack of a second equivalent of 15 permitted the formation of compound 17, which was treated with lithium triethylborohydride and trifluoroacetic acid, resulting in the formation of compound 14. In presence of water at pH 7, the latter compound then spontaneously underwent the final steps of the reaction cascade postulated for the biosynthesis of nitraramine (17–22% yield for the final cascade). This synthetic route, directly inspired by the biosynthetic hypothesis, was an elegant approach, but the choice of precursors resulted in numerous steps (activation, deprotection, reduction) that decreased the overall yield.
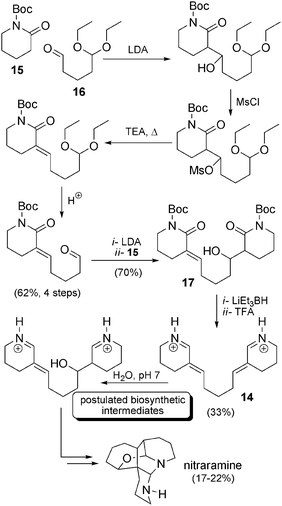 | ||
| Scheme 10 Koomen’s biomimetic synthesis of nitraramine. | ||
Synthesis by Poupon et al. 41 It is possible to imagine that a simple condensation between a Δ2-piperidine (1) unit and a molecule of glutaraldehyde could induce the formation of a compound which would then only require the attack of a second molecule of Δ2-piperidine followed by the reaction cascade described in the biosynthetic hypothesis, thus yielding nitraramine (Scheme 11). With this in mind we decided to carry out a total synthesis of nitraramine in a single step by reacting 1 and 2 in appropriate proportions. In fact, from such simple and inexpensive starting materials, no oxidation or reduction steps are required to complete the postulated reaction cascade.
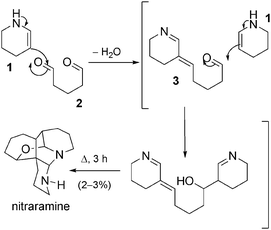 | ||
| Scheme 11 A totally biomimetic synthesis of nitraramine. | ||
By reacting two equivalents of Δ2-piperidine with one equivalent of glutaraldehyde in ethanol under reflux, we were able to obtain nitraramine in a reproducible manner. Even though the overall yield was low (2–3%), it was better than that of the only other published synthesis of this natural product. Therefore, only after a few hours of reaction and a relatively simple purification process, we were able to obtaine quantities of nitraramine sufficient for biological evaluation. This was especially significant because the extraction of nitraramine from the plant material is rather inefficient.
It is interesting to note that this total synthesis of nitraramine showed that the product known as epinitraramine was actually an artefact resulting from the protonation of nitraramine in the NMR tube (Scheme 12). Fig. 3 shows the evolution of the 1H-NMR spectrum of nitraramine at different concentrations. The spectra obtained for concentrations of 24 and 6 mg mL−1 are those attributed to nitraramine and epinitraramine respectively. The absence of epinitraramine in the crude reaction mixture reflects the high stereoselectivity of the reaction cascade, without any enzymatic assistance.
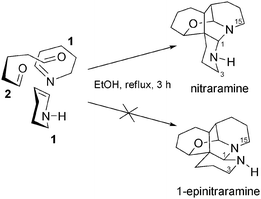 | ||
| Scheme 12 One-pot synthesis of nitraramine. | ||
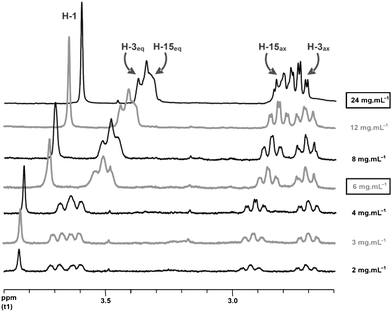 | ||
| Fig. 3 1H-NMR signals as a function of nitraramine concentrations. | ||
As shown in Scheme 9 for nitraramine, most of the reactions involved in the biosynthesis of Nitraria alkaloids are potentially reversible, suggesting that the process probably has a dynamic behavior in vivo.
Degradation reactions of nitraramine. Yunusov et al. described many simple derivatizations (acetylation,35 reduction,35 oxidation into N-oxide,39 and acetylation of N-oxide39). In 1986, a publication appeared focussing on an extensive conformational analysis of nitraramine,42 and interesting data concerning the stability of the compound were collected. Nitraramine is a remarkably stable molecule that decomposes neither in 10–40% sulfuric acid solution at room temperature, nor upon heating for 10 h at 80 °C. However, upon longer exposures (i.e. 100 days at 30 °C) or when acidic solutions were heated under reflux, the addition of water occurred. The paper also discusses in detail the tautomeric possibilities of compound 18 (Scheme 13).
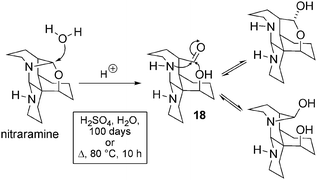 | ||
| Scheme 13 Hydrolytic degradation of nitraramine. | ||
5 Tripiperidine alkaloids
Schoberine43 was the first member of this group to be disclosed (isolated from Nitraria schoberi first44 and then from N. komarovii45 and N. sibirica46) in the late 1980s (Fig. 4). The molecule is composed of the basic units described for nitraramine, but its structure is far simpler and displays an analogy to that of matrine (isolated from plants of the Lupinus genus). Dehydroschoberine,45 sibiridine46 and dihydroschoberine18 were later isolated from N. komarovii and N. sibirica.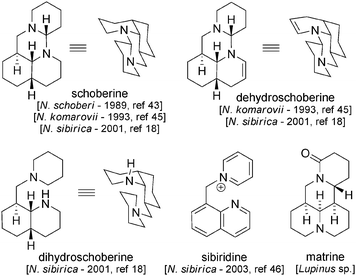 | ||
| Fig. 4 Overview of tripiperidine alkaloids. | ||
Biosynthesis. The biosynthesis of tripiperidine alkaloids requires the condensation of a Δ2-piperidine unit with a molecule of key precursor 3, which does not take place viaC-alkylation (as seen with complex spiro-alkaloids) but rather viaN-alkylation (Scheme 14). After this condensation, an intramolecular reduction of the imine by the enamine followed by total or partial reduction can lead to either dihydroschoberine or intermediate compound 19. The latter can either be aromatized into sibiridine or undergo an intramolecular cyclization to give rise to schoberine, which can be oxidized into dehydroschoberine.
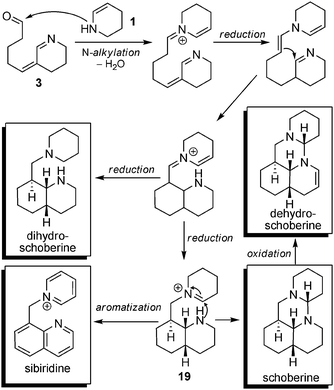 | ||
| Scheme 14 Biosynthetic hypotheses of tripiperidinic alkaloids. | ||
Chemical interconversions of tripiperidinic alkaloids. Oxidations and reductions were carried out mainly for structure confirmation purposes by the authors who first isolated the different alkaloids (Scheme 15). Starting from dehydroschoberine, reduction furnished schoberine,45 which could also be reduced to dihydroschoberine.43,47 This latter product was converted to sibiridine via palladium-catalyzed oxidation at high temperature.46
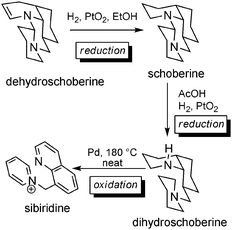 | ||
| Scheme 15 Reductive and oxidative conversions in the schoberine series. | ||
6 Indole alkaloids
6.1 Monopiperidine β-carbolines
The simplest of the Nitraria indole alkaloids are monopiperidine β-carbolines, consisting of the assembly of a tryptamine and a piperidine unit (Fig. 5). In this subgroup, indoloquinolizidine 20,48 its N-oxide equivalent 21 (Nitraria komarovii)49 and nazlinine (Nitraria schoberi)50 are found. For the latter, structure 22 was first proposed, but this was shown to be incorrect, and on the basis of the biosynthetic pathway (see Scheme 16) structure 23 was preferred.51 The authors initially had to rely on spectral data (1H-NMR, IR and UV), from which it was virtually impossible to distinguish between the two structures. All monopiperidine β-carbolines were isolated as racemates.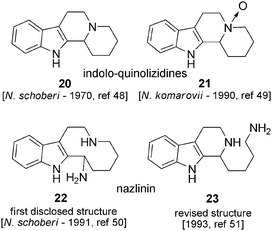 | ||
| Fig. 5 | ||
Three new indole alkaloids, schobericine, komaroidine and acetylkomaroidine, were recently isolated from Nitraria schoberi and Nitraria komarovii.52 These are simple β-carbolines bearing a propyl chain at position 2 (Fig. 6).
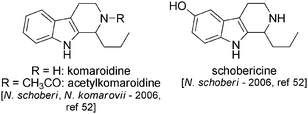 | ||
| Fig. 6 | ||
Biosynthesis. The biosynthesis of monopiperidine β-carbolines probably begins by the reaction of tryptamine with a Δ2-piperidine unit (1), to give rise to nazlinin. Then, an oxidative deamination occurs, followed by cyclization with loss of water, which, after reduction, leads to indolo-quinolizidine 20. Oxidation of 20 leads to its N-oxide derivative 21, which is also found in plants of the Nitraria genus (Scheme 16).
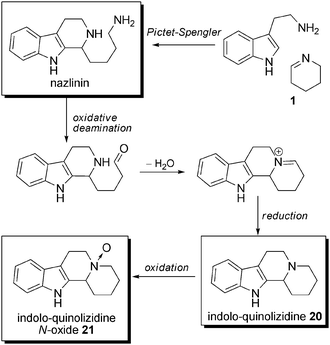 | ||
| Scheme 16 Biosynthetic proposals for nazlinin and indoloquinolizidines 20 and 21. | ||
Concerning the biosynthesis of komaroidine and close analogs, even though the authors proposed a polyacetic metabolism, it is easy to postulate a pathway that would have nazlinin as a starting point (see Scheme 17). Under oxidative conditions, nazlinin will first lead to 24 which can be decarboxylated to komaroidine. This can then be N-acetylated or oxidized.
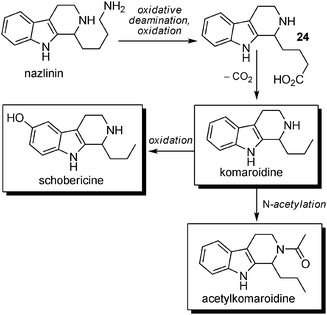 | ||
| Scheme 17 Biosynthetic proposals for komaroidine (and analogs) from nazlinin. | ||
Biomimetic syntheses. When the Koomen group proposed an alternative structure of nazlinin, they synthetically verified this assumption51 by reacting tryptamine with Δ2-piperidine (1) with aqueous acid (Scheme 18). They also studied biomimetic oxidations of nazlinin and converted it into 20 (51% yield), demonstrating a clear biosynthetic link between these structures. In this case, ortho-quinone 25 was cleverly utilized as a mimic of the topaquinone cofactor of amine oxidases, such as those involved in deaminative oxidations.53
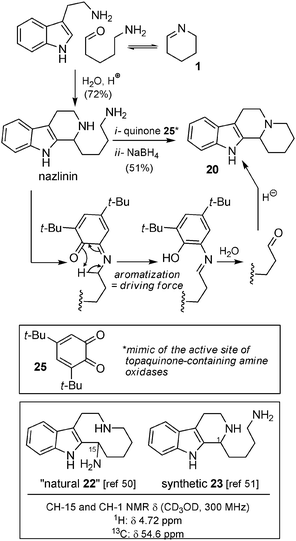 | ||
| Scheme 18 Biomimetic synthesis of nazlinin and 20 by the Koomen group. | ||
The case of komavine. In 2001, the structure of two new indolic alkaloids, found in both N. komarovii and N. schoberi, was communicated. Komavine and acetylkomavine have a common and unprecedented spirocyclohexane ring (Scheme 19).54 No obvious biosynthetic pathway implicating the “Nitraria lysine metabolism” can be put forward to explain the formation of such compounds (komavine is formally the result of a Pictet–Spengler reaction between tryptamine and a cyclohexanone55).
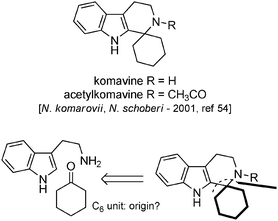 | ||
| Scheme 19 Komavine and acetylkomavine. | ||
6.2 Dipiperidinic β-carbolines: nitrarine and related molecules
In N. schoberi and N. komarovii racemic dipiperidinic structures can be found, which have an additional piperidine ring system. They also have a more complex bridged structure (formally the juxtaposition of an isoquinuclidine and a piperidine) which can possess up to 5 stereogenic centres. We will consider nitrarine to be the representative compound in this series (Fig. 7). An initial structure, namely an indoloquinolizidine substituted with a piperidine ring, was proposed in 196956 for this new alkaloid (based on mass and UV spectra), and the true structure, based upon X-ray analysis, was published in 1975.57 The same year, zwitterionic analogs with higher oxidation states were isolated from the same plant and were given the name nitramidine58 and schoberidine.59 Another compound of N. schoberi apparently isomeric with nitrarine was attributed the structure of 3-epinitrarine and named isonitrarine.60 More recently, N-substituted derivatives of nitrarine (N-methyl-nitrarine,61N-allyl-nitrarine62), isonitrarine (N-allyl-isonitrarine63) and schoberidine (N-allyl-schoberidine,64 schoberidine N-oxide = nitraricine65) have been characterized, along with analogs with higher oxidation state (dehydronitramidine,64 komarine66 and nitrarizine65) (Fig. 8).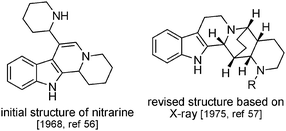 | ||
| Fig. 7 Revision of the nitrarine structure. | ||
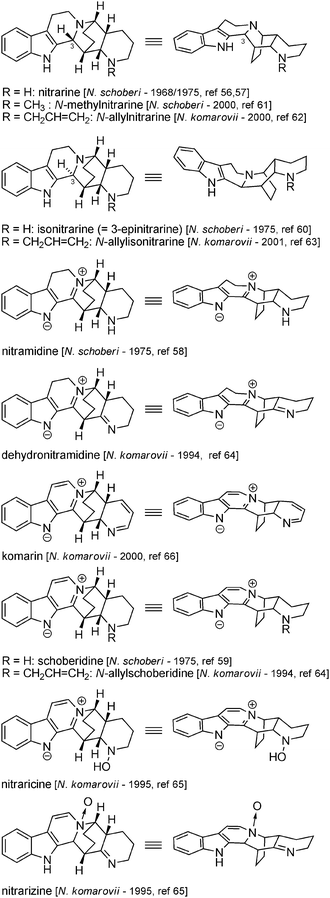 | ||
| Fig. 8 Structures of dipiperidinic β-carbolines. | ||
Biosynthesis.
The Uzbek chemists who have isolated most of Nitraria alkaloids never suggested a possible biosynthesis of nitrarine (as they did for other alkaloids),67 and Wanner and Koomen were the first to propose a biosynthetic route to explain the formation of nitrarine and related compounds (Scheme 20). The two pathways rely on oxidation steps. Starting either from simple β-carboline 20/Δ2-piperidine (1) (pathway  ) or tryptamine/precursor 3 (pathway
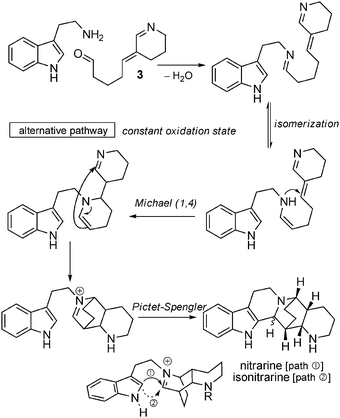
) or tryptamine/precursor 3 (pathway  ), the common intermediate 26 needs to be oxidized to give rise to the formation of the central tricyclic core. Through this pathway, nitramidine is the central compound from which the analogs can be envisioned through oxidations (dehydronitramidine, komarine, schoberidine) or reduction (nitrarine, isonitrarine) and N-alkylation (N-allylschoberidine, N-methylnitrarine, N-allylnitrarine, N-isonitrarine). To avoid the oxidation state issue, we propose an alternative pathway leading, with constant oxidation state throughout, to nitrarine starting from tryptamine and 3 (Scheme 21). By simply inverting the sequence of reactions, i.e. considering first the Michael addition and then the Pictet–Spengler reaction, a direct biosynthesis of nitrarine is possible.
), the common intermediate 26 needs to be oxidized to give rise to the formation of the central tricyclic core. Through this pathway, nitramidine is the central compound from which the analogs can be envisioned through oxidations (dehydronitramidine, komarine, schoberidine) or reduction (nitrarine, isonitrarine) and N-alkylation (N-allylschoberidine, N-methylnitrarine, N-allylnitrarine, N-isonitrarine). To avoid the oxidation state issue, we propose an alternative pathway leading, with constant oxidation state throughout, to nitrarine starting from tryptamine and 3 (Scheme 21). By simply inverting the sequence of reactions, i.e. considering first the Michael addition and then the Pictet–Spengler reaction, a direct biosynthesis of nitrarine is possible.
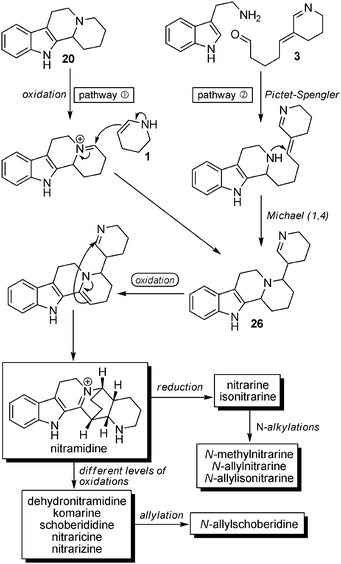 | ||
| Scheme 20 Biosynthesis of nitrarine and congeners as proposed by Wanner and Koomen. | ||
 | ||
| Scheme 21 Alternative biosynthesis of nitrarine and congeners. | ||
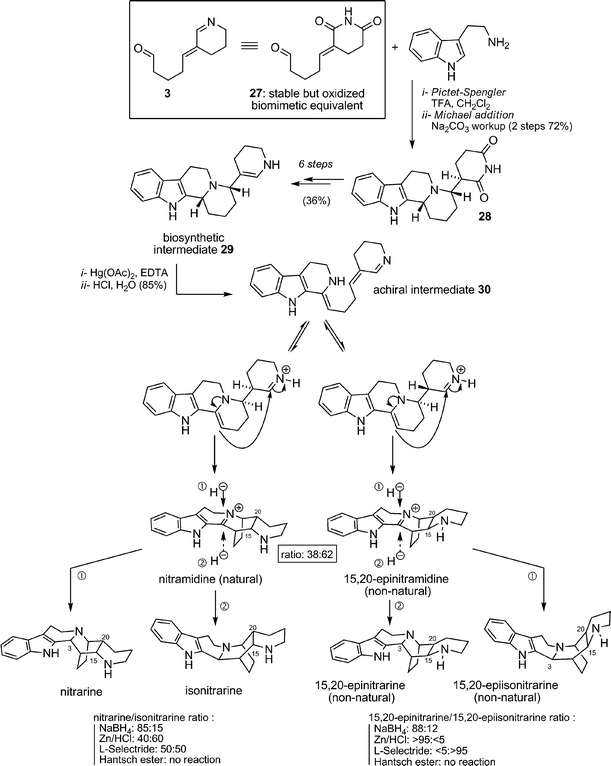
Biomimetic synthesis of nitrarine. In 1994 Wanner and Koomen disclosed the only total synthesis of nitrarine and nitramidine published so far (Scheme 22).68 In this pioneering and beautiful synthesis, glutarimide aldehyde 27 was used as a stable equivalent of pivotal intermediate 3 (similar to 12, already exploited in their synthesis of nitramine). After a Pictet–Spengler condensation with tryptamine leading to 28, delicate chemical modifications permitted the preparation of postulated intermediate 29. From there, and according to Koomen's biosynthetic proposals, an oxidation step was required: Hg(OAc)2-catalyzed oxidation of the β-carboline gave biomimetic achiral precursor 30. Subsequent cyclization led to the formation of the E-ring and to the formation of natural nitramidine along with diastereomeric non-natural 15,20-epinitramidine (separable compounds). Reduction of the iminium was then studied with various reductive agents (NaBH4, Zn/HCl, L-Selectride®, Hantsch ester), giving access to the nitrarine series (nitrarine and isonitrarine) but also to the 15,20-epinitrarine series (15,20-epinitrarine, 15,20-epiisonitrarine) in different ratios (see Scheme 22). Despite the necessary use of oxidation/reduction manipulations, this synthesis is doubtlessly a milestone in the biomimetic chemistry of complex alkaloids.
 | ||
| Scheme 22 Koomen's total synthesis of nitrarine and congeners. | ||
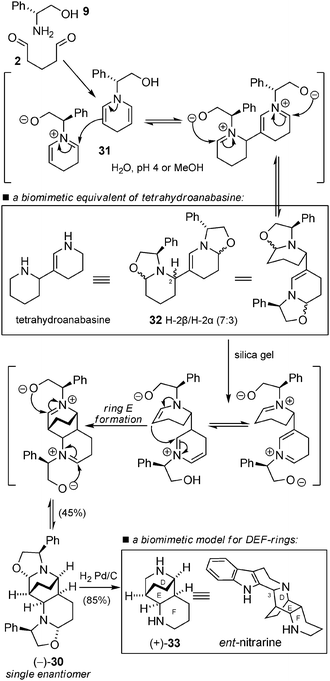
Later, Kunesch and Husson gave a striking demonstration of the power of biomimetic synthesis and cascade reactions in organic synthesis with the one step synthesis of the complete tricyclic aliphatic ring system of nitrarine (Scheme 23).69 The equimolar reaction of (−)-phenylglycinol (9) and glutaraldehyde (2) in a polar medium (aqueous pH 4 buffer or MeOH) gave compound 30, which is directly related to nitrarine, in a 45% yield in a one-pot reaction. The first intermediate 31 (as already postulated in the biomimetic synthesis of spiro-alkaloids – see Scheme 5) was in this case involved in a self-condensation reaction, giving rise to 32 – a biomimetic equivalent of tetrahydroanabasine. The authors noted the low stability of 32 and its spectacular spontaneous conversion into 30. The cyclization occurred during prolonged contact with silica gel via an iminium/enamine cascade. The remarkable stereochemical outcome of the reaction (formation of a single enantiomer) was explained as the result of a series of equilibrated reactions leading to the more stable stereoisomer. Hydrogenolysis of the chiral auxiliary permitted the synthesis of 33, a model compound towards the biomimetic synthesis of nitrarine. This work is a beautiful example of the power of iminium ions as intermediates in cascade reactions.
 | ||
| Scheme 23 Biomimetic approach of nitrarine by Kunesch and Husson. | ||
6.3 Tangutorine and nitraraine
Nitraraine and tangutorine are two indolic molecules closely related in terms of structure (they share the same mass and formula). Nitraraine was isolated from Nitraria schoberi collected in the Kyzyl-Kum (Uzbekistan) in 1985 (Fig. 9).70 A yohimbane-type structure was proposed supported by 1H-NMR, MS, UV and IR data. Dihydronitraraine was also isolated from the same plant.71 A total synthesis of nitraraine was published in 1989 by Takano et al.,72 but the physical and spectral data did not match the data available for natural nitraraine, and since then, no revised structure of nitraraine has been proposed.73 It is therefore not impossible that nitraraine might in fact be closely related to the more logical (in terms of biosynthesis) structure of tangutorine.74 This latter alkaloid was isolated in 1999 from the leaves of Nitraria tangutorum collected in China by Che et al.75,76 A complete set of data (including X-ray analysis) ascertained the structure,77 which has been confirmed by a series of total syntheses.78,79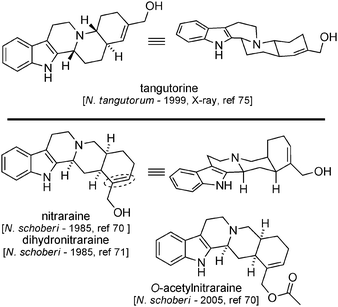 | ||
| Fig. 9 Tangutorine and nitraraine: two closely related indole alkaloids. | ||
Biosynthetic hypotheses. The biosynthetic proposals for tangutorine made by Jokela et al. in the introduction of their pioneering total synthesis paper established a possible connection between a yohimbine-type precursor and the new skeleton of tangutorine. A series of rearrangements explains the conversion, but the scheme is not in agreement with the fact that tangutorine is extracted as a racemate in Nature, which would imply the total racemization of all stereogenic centres in yohimbine. A close look at all publications related to the isolation of “Nitraria alkaloids” did not reveal, to the best of our knowledge, either any yohimbane-type precursors or analogs nor any terpenoid alkaloids in general in the Nitraria genus. Having in mind the homogeneity of metabolism in the Nitraria genus, we can put forward an alternative biosynthetic scheme,80 implicating intermediate 3 as shown in Scheme 24. In fact, a direct precursor of tangutorine such as 34 can be seen as the product of hydrolysis followed by oxidative deamination of achiral precursor 3. An E/Z-isomerization and intramolecular aldolization/crotonization could generate a cyclohexadiene 35 (the E-ring of tangutorine). This could then undergo condensation with tryptamine involving a Pictet–Spengler reaction and a 1,6-Michael addition to form the D-ring of tangutorine. Finally, tangutorine might be biosynthesized after reduction of the remaining aldehyde of intermediate 36 into a primary alcohol. Alternatively, 35 may formally be considered as arising directly from two molecules of dialdehyde 2 by aldolization/crotonization.
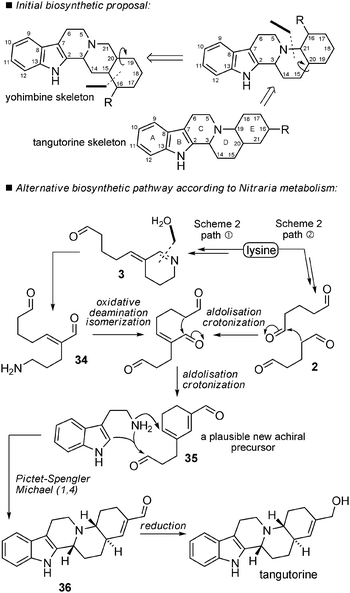 | ||
| Scheme 24 A possible biosynthetic pathway to tangutorine. | ||
Biomimetic synthesis of tangutorine. Soon after its disclosure as a new natural compound, tangutorine became an interesting target for total synthesis, and we have recently described the first biomimetic total synthesis of tangutorine (Scheme 25). Up to now, several total syntheses have been published,81 but only one can be regarded as being biomimetically inspired. In 2009 we reported a straightforward total synthesis starting from glutaraldehyde 2.82 Under basic conditions, 2 can undergo dimerization, giving access to bicyclic intermediate 37 – an equivalent of biosynthetic intermediate 35 (see Scheme 24). Treatment of 37 with tryptamine in mild acid directly afforded 36 (25–30% yield), which was reduced to give tangutorine after recrystallization (50% yield). Biosynthetic intermediate 35 is postulated as an intermediate in the course of the reaction with tryptamine, but it could not be isolated.
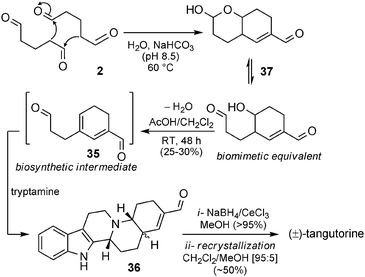 | ||
| Scheme 25 Biomimetic synthesis of tangutorine. | ||
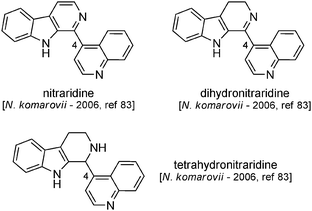
6.4 Nitraraidine
Nitraraidine was isolated in 200240 from N. sibirica (Kyrgyzstan), its structure being found to be identical to that of a compound hemisynthesized in 1985 from natural dihydronitraraine (Scheme 26).83 Having in mind that the actual structures of nitraraine/dihydronitraraine themselves might be wrong, investigations (including total synthesis efforts) are needed before we can reach any conclusion about the true structure of nitraraidine.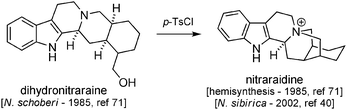 | ||
| Scheme 26 Hemisynthesis of nitraraidine and its discovery as a natural compound. | ||
6.5 Aromatic β-carbolines
A number of aromatic alkaloids are found in plants of the Nitraria genus such as N. komarovii. These compounds bear a β-carboline region (in various oxidation states) that is attached to a quinoline in different positions, thus generating the molecular diversity observed for this group (Fig. 10). | ||
| Fig. 10 General structure of aromatic β-carbolines. | ||
On the basis of the possible biosynthetic pathways, these molecules can be classified into three subgroups. The metabolisms of L-lysine and L-tryptophan can account for the biosynthesis of the alkaloids composing three subgroups: (i) molecules with a quinoline unit attached at positions 8 or 6; (ii) molecules with a quinoline unit attached at position 5; (iii) molecules with a quinoline unit attached at position 2. Recently, metabolites of the same type have been described with a β-carboline region attached at position 4 of the quinoline moiety.
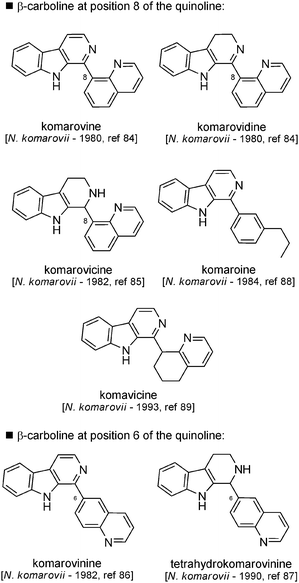 | ||
| Fig. 11 Structures of β-carbolines with quinolines attached at C-6 or C-8. | ||
Biosynthetic hypotheses. A biosynthetic pathway (involving early steps similar to those encountered for the biosynthesis of dipiperidinic indole alkaloids such as nitrarine) was proposed by Koomen and Wanner8 (Scheme 27). These compounds could arise from the condensation of tryptamine unit with precursor 3 followed (after oxidation) by a 1,2-addition to the conjugated system to give rise to compound 38. This latter step is where the two pathways diverge, since in the case of nitrarine it is a 1,4-conjugated addition. Compound 38 can either undergo an aromatization process and furnish komarovine, komarovidine and komarovicine, or undergo a retro-Michael reaction to give 39, which displays a 1,6-conjugated system and a free propylamine chain. At this point, an oxidative deamination step followed by the reduction of the residual aldehyde and aromatization will lead to komaroine, whereas a 1,6-conjugated addition followed by an aromatization process will lead to komarovinine and tetrahydrokomarovinine. Komavicine formation can also be explained by selective oxidation of intermediate 39.
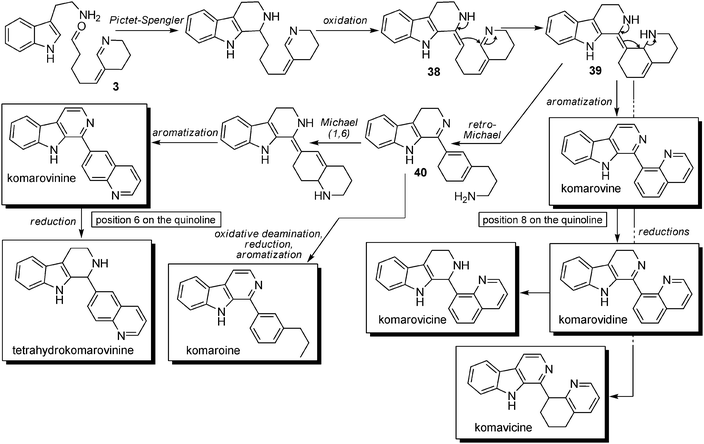 | ||
| Scheme 27 Possible biosynthetic pathway for aromatic β-carbolines with quinolines attached at C-6 or C-8 (aromatization and reduction sequences may be inverted). | ||
An alternative pathway involving intermediate 35 (seen in the biosynthetic hypotheses of tangutorine) can also be put forward here (Scheme 28).
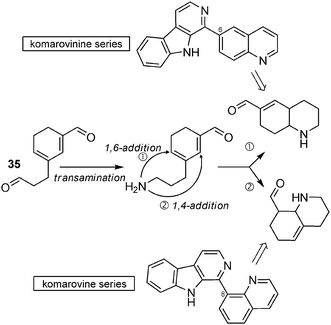 | ||
| Scheme 28 Alternative pathway towards aromatic β-carbolines. | ||
This latter proposal highlights that 35 might be, besides key intermediate 3, another pivotal intermediate in the “Nitraria metabolism” (Scheme 29).
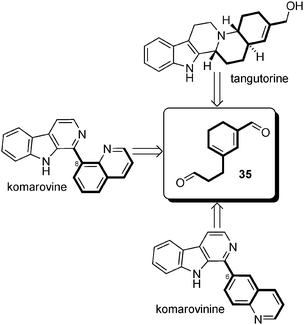 | ||
| Scheme 29 Intermediate 35 as a pivotal intermediate. | ||
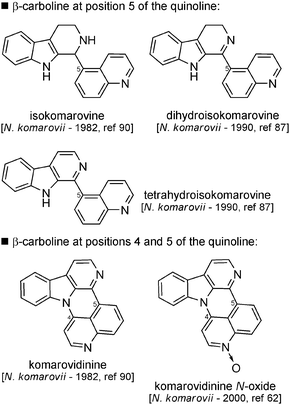 | ||
| Fig. 12 Structures of β-carbolines with quinolines substituted at C-5. | ||
Biosynthesis. No biosynthetic hypothesis has been reported for these molecules in the literature. However, it is possible to hypothesize the biosynthesis of these compounds from nazlinin, which could, after oxidative deamination and reduction of the terminal aldehyde, give rise to compound 40 (Scheme 30). Dehydration followed by an oxidation process could then lead to compound 41 bearing a dienic side chain that could be engaged in a formal [4 + 2] cycloaddition with a molecule of Δ2-piperidine (1). The resulting compound 42 could then either undergo aromatization to give rise to isokomarovine or be partly oxidized. This product can undergo further cyclization via an intramolecular 1,4-Michael addition followed by aromatization to form komarovidinine and its N-oxide form. Isokomarovine could also be reduced to dihydro- or tetrahydroisokomarovine.
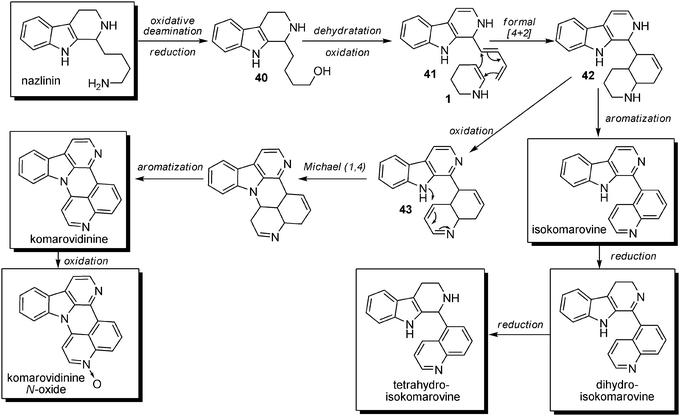 | ||
| Scheme 30 Possible biosynthetic pathway for aromatic β-carbolines with quinolines attached at C-5 and C-4. | ||
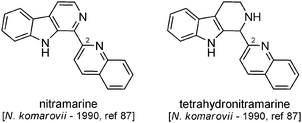 | ||
| Fig. 13 Structures of β-carbolines with quinolines attached at C-2. | ||
Biosynthetic hypotheses. As for the previous subgroup, no biosynthetic hypothesis is reported in the literature for these compounds. Nonetheless, the first steps towards the formation of these alkaloids can be hypothesized in a manner similar to that seen in the case of isokomarovine and its derivatives (Scheme 31). Here, intermediate 41 would once again play the role of a diene engaged in a formal [4 + 2] cycloaddition but this time with a molecule of Δ1-piperidine (1) (imine form) as the dienophile. The resulting compound 44 would then undergo an oxidative deamination process followed by oxidation to generate an enamine, which would perform a nucleophilic addition on the newly generated terminal aldehyde. After dehydration into 45, aromatization would lead to the formation of nitramarine, which could then be reduced to tetrahydronitramarine.
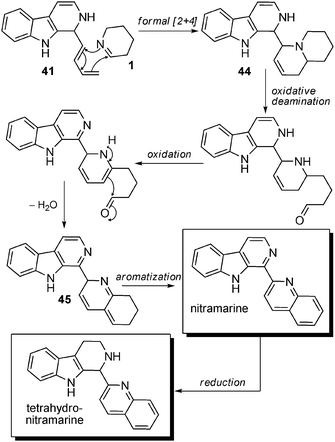 | ||
| Scheme 31 Possible biosynthetic pathway for aromatic β-carbolines with quinolines attached at C-2. | ||
 | ||
| Fig. 14 Structures of aromatic β-carbolines with quinolines attached at C-4. | ||
6.6 Chemical interconversions in the complex β-carboline series
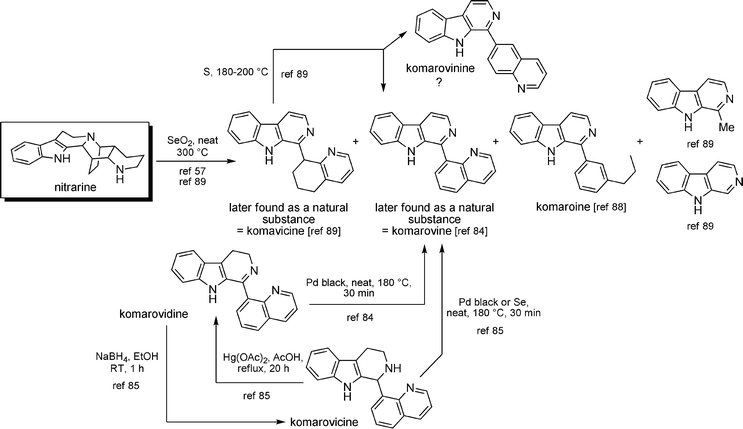
Many chemical transformations in the nitrarine series have been conducted for structure correlation purposes. Some of them are listed in Scheme 32. In general, harsh conditions (high temperatures, strong oxidants, etc.) are needed to convert nitrarine-type metabolites into aromatic β-carbolines, casting doubt on a possible direct biosynthetic link between both families. Furthermore, correlations were commonly established only by TLC and/or mass analysis.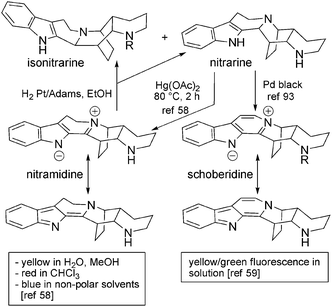 | ||
| Scheme 32 Conversions in the nitrarine series keeping the nitrarine skeleton unaltered. | ||
Conversions of nitramidine into nitrarine and vice versa were accomplished in the work describing the structure of nitramidine.58,92 The catalytic dehydrogenation of nitrarine with palladium-black in the presence of maleic acid enabled schoberidine to be obtained.93
Many oxidative/reductive transformations in the aromatic carboline series were also performed, again for the purpose of confirming the structure of the different members of this family. The main results are depicted in Scheme 33. The “komarovine series” was also accessible from nitrarine. Nitrarine could be oxidized in the presence of selenium at 300 °C and gave several compounds, one of which was identified a few years later as being komarovine.84 It was also prepared from reduced analogs komarovidine and komarovicine. Komaroine was also detected in the course of the oxidation of nitrarine.94 Dehydrogenation of komavicine with sulfur afforded komarovine (a 6-substituted quinoline), as expected, but also komarovinine (an 8-substituted quinoline) – the author did not give any explanation for this latter transformation.95
 | ||
| Scheme 33 Interconversions in the β-carboline series. | ||
7 Miscellaneous alkaloids
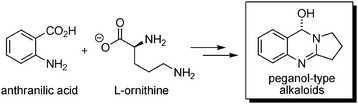
Alkaloids derived from peganol will be mentioned briefly here. Four of these, viz. deoxypeganine,96 peganine,66 vasicinone97 and deoxyvasicinone,97 have also been found in Nitraria komarovii (Fig. 15), all being isolated as their N-oxides. They are biosynthetically derived from anthranilic acid and L-ornithine (Scheme 34). Additionally, tryptamine and serotonin were also isolated from N. schoberi.50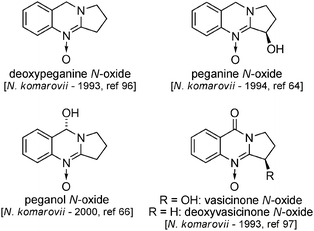 | ||
| Fig. 15 Peganol derivatives in N. komarovii. | ||
 | ||
| Scheme 34 Biosynthetic origin of peganol-type alkaloids. | ||
8 Alkaloids from the Myrioneuron genus
Myrioneuron is a small genus belonging to the Rubiaceae family which is distributed across south-east Asia. Of the 15 described species, Myrioneuron nutans, a small native tree in north Vietnam, was studied at the Muséum National d'Histoire Naturelle in Paris by Bernard Bodo and coworkers. Unusual structures have been isolated that are closely related to the Nitraria alkaloids (Fig. 16). This new family of alkaloids (“the Myrioneuron alkaloids” as proposed by Bodo et al.) is characterized by a decahydroquinoline (cis-DHQ) system often attached to an oxazine ring. All alkaloids known to date have been isolated from M. nutans.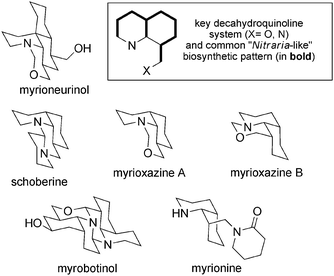 | ||
| Fig. 16 The Myrioneuron alkaloids. | ||
8.1 Tricyclic alkaloids
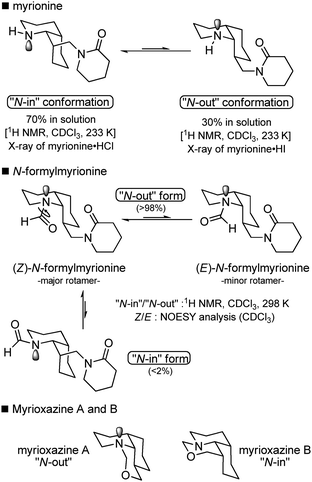
Myrionine and myrioxazines A and B are the simplest structures of this type (Scheme 35).98 Once again, their biosynthesis can be explained from intermediate 3 (Scheme 35), linking the Myrioneuron metabolism to the Nitraria one. After reduction of 3, Mannich-type cyclization on the remaining imine could afford isomeric intermediates 46 and 47, which can be reduced to give hypothetical alcohols. These latter could react which a formaldehyde equivalent to give rise to myrioxazines A and B. The formation of myrionine from intermediate 46 is also easy to imagine with the intervention of a piperidine equivalent. Chemical syntheses have been conducted by the authors that confirmed the proposed structures.99 In the case of myrionine, a conformational equilibrium resulting from the ring inversion of the cis-DHQ and the consequent formation of two chair–chair conformers was observed and carefully studied (Scheme 36).99 In solution, a 70:30 ratio of “N-in” to “N-out” conformers was measured by 1H-NMR at low temperature in CDCl3. Interestingly, X-ray analysis permitted the complete analysis of both conformers, because myrionine hydrochloride crystallized exclusively in the “N-in” form, and myrionine hydroiodide crystallised in the “N-out” form.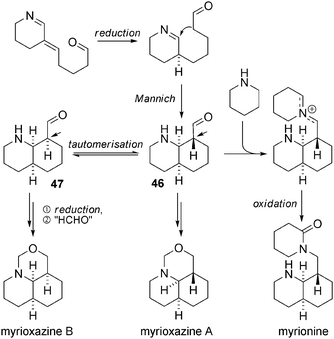 | ||
| Scheme 35 Possible biosynthesis of tricyclic Myrioneuron alkaloids. | ||
 | ||
| Scheme 36 Conformational analysis of myriorine and myrioxazines. | ||
N-Formylmyrionine was described recently.100 Bodo et al. showed that in solution the two conformers equilibrated (a 6:4 mixture in CDCl3). Analysis of nOe data clearly determined that (Z)-N-formylmyrionine predominated over the (E)-isomer (Scheme 36). In addition, only the “N-out” form is observed by 1H-NMR. In turn, the above-cited myrioxazines A and B are respectively “N-out” and “N-in” conformers, probably because of a strong constraint due to the additional oxazine ring (Scheme 36).
8.2 Tetracyclic alkaloids
Three interconnected tetracyclic alkaloids have also been isolated from M. nutans: (−)-myrionidine, (+)-myrionamide, and (noteworthily) optically active (−)-schoberine.101 Oxidation and/or reduction steps easily explain the possible link between these new products. A possible pathway, starting from myrionine, places myrionidine as a central intermediate in terms of oxidation state (Scheme 37).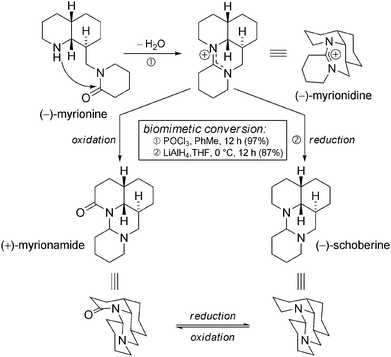 | ||
| Scheme 37 Myrioneuron tetracyclic alkaloids. | ||
With the isolation of myrioneurinol,102 more evidence for the close connection between Nitraria and Myrioneuron metabolism could be collected. Myrioneurinol combines the spiranic C15 core encountered in nitraramine with the cis-DHQ ring of the Myrioneuron alkaloids. A biosynthetic proposal to account for this unusual alkaloid is shown in Scheme 38 in which a dehydroxylation step is highlighted.
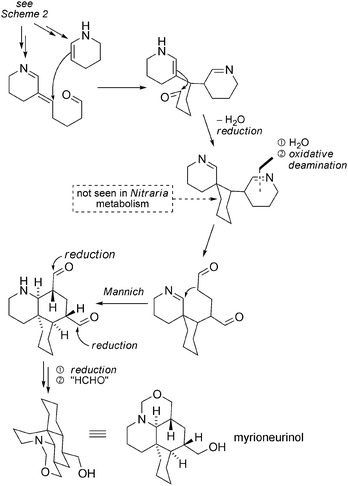 | ||
| Scheme 38 Plausible biosynthetic pathway to myrioneurinol. | ||
8.3 A hexacyclic alkaloid: (+)-myrobotinol
The structure of myrobotinol, a hexacyclic alkaloid, was disclosed in 2007.103 It is likely that it could be the result of a dimerization process between two units of postulated intermediate 46 in the biosynthesis of myrioxazines (vide supra – Scheme 35). One of these units requires specific hydroxylation and oxidation to form an imine-activated system that subsequently results in dimerization cascade (Scheme 39).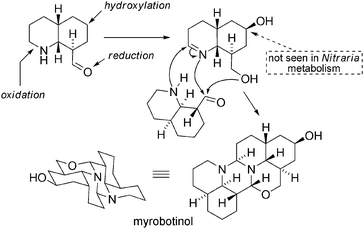 | ||
| Scheme 39 A dimerization cascade can explain the formation of myrobotinol. | ||
Another hexacyclic alkaloid is dehydronitraramine.100 Once again, it is striking to note the similarity with the Nitraria genus. A similar biosynthetic pathway to that described above (Scheme 9), with an additional oxidation step (Scheme 40), may explain the formation of dehydronitraramine, but it is isolated as an optically active substance ([α]D = +9.3 (c = 0.5, MeOH)) whereas natural nitraramine was racemic.
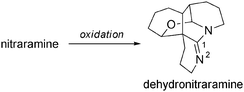 | ||
| Scheme 40 Formation of dehydronitraramine. | ||
8.4 Nitraria versus Myrioneuron: enamine versus imine
The key steps for the two routes from intermediate 3 are presented in Scheme 41. Whereas the enamine form of reduced intermediate 3 will explain Nitraria-like spirocyclization, the imine tautomeric form can undergo a Mannich-type reaction and thereby explain the formation of Myrioneuron quinolic alkaloids.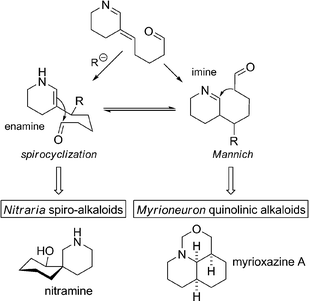 | ||
| Scheme 41 Spirocyclization vs. Mannich reaction from intermediate 3. | ||
8.5 Biomimetic synthesis of Myrioneuron alkaloids
The unique and challenging molecular motifs within this new family of alkaloids will certainly provide new opportunities for biomimetic endeavours. The inventive tactics developed so far with the biomimetic access to Nitraria alkaloids may constitute a good starting point towards this aim. Up to now, myrioxazines A and B,98 (−)-myrionidine and schoberine have been synthesized in enantiomerically pure form.101 The strategy is not biomimetic, yet, in a final stage, a biomimetic conversion of (−)-myrionine to (−)-myrionidine and finally (−)-schoberine was achieved.1019 Chirality of Nitraria and Myrioneuron alkaloids
It is interesting to note that some of the molecules discussed in this article are isolated from Nature as racemic mixtures. It appears that simple spiro-alkaloids are the only Nitraria alkaloids to be found in enantiomerically pure form. Furthermore, we have seen that the biosynthesis of these particular alkaloids requires the reduction of key intermediate 3 followed by an asymmetric spirocyclization. This enantioselective control might be seen as the result of the chiral environment induced by the enzyme responsible for the previous reduction step. All other groups of Nitraria alkaloids are collected as racemic mixtures. Their biosyntheses sometimes also feature reduction of 3, but when this is the case it is performed by the attack of a nucleophile other than a hydride, which would require enzymatic control.Starting from achiral precursor 3, two different cases are possible, as depicted in Scheme 42. When, on the one hand, the required reduction is assured by a hydride ion (viz. from NADH), the process affords enantiomerically pure alkaloids, despite the epimerization that might result from the imine–enamine equilibrium (a step that may arise spontaneously within the active site of the hydrogenase). On the other hand, when the “reducing agent” seems to be a carbon nucleophile (such as Δ2-piperidine 1), a racemic alkaloid is formed in the Nitraria genus, whereas in the Myrioneuron genus the pathway seems to be enantiocontrolled. This is an interesting issue, and the resulting questions of chemotaxonomy and evolutionary convergence remain unanswered.
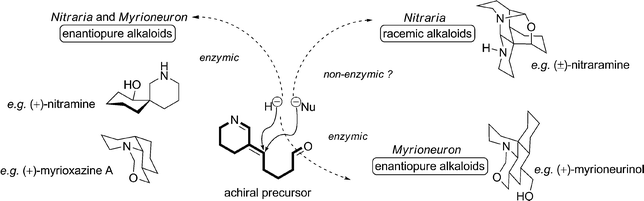 | ||
| Scheme 42 The chirality issue in Nitraria and Myrioneuron metabolisms. | ||
10 A unified scenario for Nitraria and Myrioneuron alkaloids
Trying to unify the biosynthetic pathways of a wide array of alkaloids isolated from plants of the Nitraria genus, we have highlighted a number of key biosynthetic intermediates. Among these are compounds containing 5 or 10 carbon atoms that all appear to result from L-lysine metabolism. All these intermediates are closely related to one another.The pattern consisting in the association of two linear C5 units is of particular interest and is found in many different molecules with different degrees of oxidation. Considering the probable origin of such motifs, it seems very unlikely that any of the Nitraria alkaloids discussed herein might be of indolomonoterpene nature, even though such a hypothesis has been published for tangutorine. It is our belief that the L-lysine hypothesis is the most probable (Schemes 43 and 44).
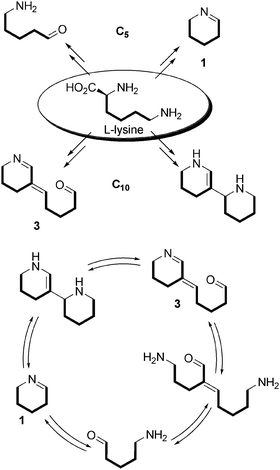 | ||
| Scheme 43 Interconnecting C10 units. | ||
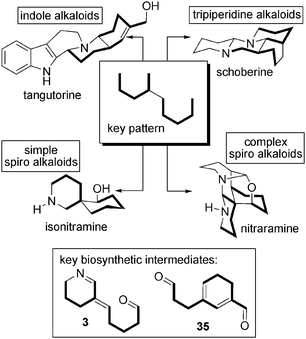 | ||
| Scheme 44 Key pattern within the Nitraria alkaloids. | ||
It is noteworthy that an analogy between the biosynthetic pathways of Nitraria and Lupinus alkaloids exists (Scheme 45). These two alkaloid families are derived from L-lysine and share a common biosynthetic intermediate: tetrahydroanabasine. Depending on the how it rearranges, it will give rise to either Lupinus alkaloids ( ) or Nitraria/Myrioneuron alkaloids (
) or Nitraria/Myrioneuron alkaloids ( ).
).
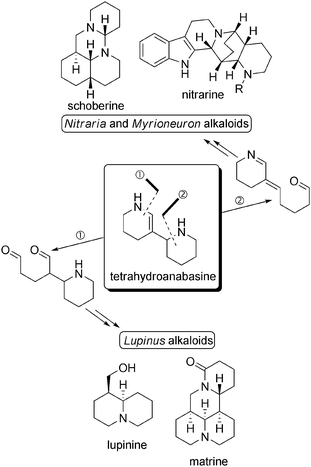 | ||
| Scheme 45 The two metabolic pathways from tetrahydroanabasine. | ||
Scheme 46 displays the amino-acid logic in the Nitraria metabolism. It is apparent that the diversity and (sometimes) complexity of the alkaloids all seems to be derived from ornithine or lysine, with the addition of anthranilic acid or tryptophan.
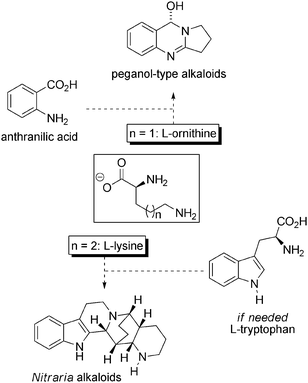 | ||
| Scheme 46 The amino acid structures within the Nitraria alkaloids. | ||
11 Biological activities
Not much is known concerning the biological properties of the Nitraria and Myrioneuron alkaloids.For the Nitraria alkaloids, a little biological data is available for nitramine,104 nitramarine,105 nitrarine,106 schoberidine,107 komarovine,108 komarovinine,109 komarovicine,110 and nazlinin.111 Tangutorine was shown to possess a cell cycle modulation activity involving the induction of p21 expression.112
For the Myrioneuron alkaloids, myrioneurinol,102 dehydronitraramine,100 myrionidine and schoberine101 showed weak inhibition on KB cell proliferation but stronger antimalarial activities, thus suggesting that the latter was not due to their cytotoxicity.
12 Conclusions
In this review, we have tried to give an overview of the various structures encountered in the Nitraria and Myrioneuron genera. In particular, we have tried to formulate a common biosynthetic scenario that is in agreement with their hypothesized common origin, viz. a lysine/tetrahydroanabasine metabolism. From the synthetic standpoint, we have focussed on biomimetic total syntheses, as it is clear that in many cases they are able to shed light on possible biosynthetic pathways.13 References and notes
- E. L. Sturtevant, in Sturtevant's edible plants of the world, ed. U. P. Hedrick; Dover Publications: New York, 1972 Search PubMed.
- (a) R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Khim. Prir. Soedin., 1996, 118–119 CAS; R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Chem. Nat. Compd., 1996, 32, 102–103 CrossRef; (b) R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Khim. Prir. Soedin., 1996, 244–342 CAS; R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Chem. Nat. Compd., 1996, 32, 216–334 CrossRef; (c) R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Khim. Prir. Soedin., 1996, 410–512 CAS; R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Chem. Nat. Compd., 1996, 32, 386–512 CrossRef; (d) R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Khim. Prir. Soedin., 1996, 615–681 CAS; R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Chem. Nat. Compd., 1996, 32, 596–675 CrossRef; (e) R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Khim. Prir. Soedin., 1996, 761–863 CAS; R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Chem. Nat. Compd., 1996, 32, 737–858 CrossRef; (f) R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Khim. Prir. Soedin., 1996, 957–1035 CAS; R. Shakirov, M. V. Telezhenetskaya, I. A. Bessonova, S. F. Aripova, I. A. Israilov, M. N. Sultankhodzhaev, V. I. Vinogradova, V. I. Akhmedzhanova, T. S. Tulyaganov, B. T. Salimov and V. A. Tel'nov, Chem. Nat. Compd., 1996, 32, 932–1028 CrossRef.
- As we will discuss in this review, some of the data should be treated with caution.
- A. A. Ibragimov, N. Yu. Novgorodova and Kh. N. Aripov, Khim. Prir. Soedin., 1977, 196–199; A. A. Ibragimov, N. Yu. Novgorodova and Kh. N. Aripov, Chem. Nat. Compd., 1977, 13, 71–74.
- T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1979, 737 CAS; T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1979, 15, 654 CrossRef.
- Z. Osmanov, A. A. Ibragimov, S. Yu. Yunusov, A. Nigmatullaev and K. Taizhanov, Khim. Prir. Soedin., 1982, 372; Z. Osmanov, A. A. Ibragimov, S. Yu. Yunusov, A. Nigmatullaev and K. Taizhanov, Chem. Nat. Compd., 1982, 18, 372 CrossRef.
- A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1983, 261–262; A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1983, 20, 247–248.
- M. J. Wanner and G.-J. Koomen, in Studies in Natural Products Chemistry: Stereoselectivity in Synthesis and Biosynthesis of Lupine and Nitraria Alkaloids, ed. Atta-ur-Rahman; Elsevier: Amsterdam, 1994, vol. 14, pp. 731–768 Search PubMed. See also several other (less important) accounts from the same authors: M. J. Wanner and G. J. Koomen, Pure Appl. Chem., 1994, 66, 2239–2242 Search PubMed; M. J. Wanner and G. J. Koomen, Pure Appl. Chem., 1996, 68, 2051–2056 Search PubMed; M. J. Wanner and G. J. Koomen, J. Indian Chem. Soc., 1997, 74, 891–895 CrossRef CAS ; http://old.iupac.org/symposia/proceedings/phuket97/wanner.html.
- One needs to bear in mind that in plants, reactive functional groups such as aldehydes will most likely be masked in a transitory way. In this review, in order to simplify schemes and figures, these functional groups will be shown in their reactive form.
- (a) N. Yu. Novgorodova, S. Kh. Maekh and S. Yu. Yunusov, Khim. Prir. Soedin., 1973, 196–199 CAS; N. Yu. Novgorodova, S. Kh. Maekh and S. Yu. Yunusov, Chem. Nat. Compd., 1973, 9, 191–193 CrossRef; (b) A. A. Ibragimov, G. P. Moiseeva, Z. Osmanov and S. Yu. Yunusov, Khim. Prir. Soedin., 1986, 727–730; A. A. Ibragimov, G. P. Moiseeva, Z. Osmanov and S. Yu. Yunusov, Chem. Nat. Compd., 1986, 22, 676–679 CrossRef.
- Z. Osmanov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1977, 720–721 CAS; Z. Osmanov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1977, 13, 607–608 CrossRef . Also mentioned in a 2002 paper: see ref. 40.
- A. A. Ibragimov, Z. Osmanov, B. Tashkhodzhaev, N. D. Abdullaev, M. R. Yagudaev and S. Yu. Yunusov, Khim. Prir. Soedin., 1981, 623–629 CAS; A. A. Ibragimov, Z. Osmanov, B. Tashkhodzhaev, N. D. Abdullaev, M. R. Yagudaev and S. Yu. Yunusov, Chem. Nat. Compd., 1981, 17, 458–463 CrossRef.
- B. Tashkhodzhaev, Khim. Prir. Soedin., 1982, 75–80 CAS; B. Tashkhodzhaev, Chem. Nat. Compd., 1982, 18, 70–74 CrossRef.
- (a) P. J. McCloskey and A. G. Schultz, Heterocycles, 1987, 25, 437–447 CAS; (b) D. Tanner and H. M. He, Tetrahedron, 1989, 45, 4309–4316 CrossRef CAS.
- J.-C. Quirion, D. S. Grierson, J. Royer and H.-P. Husson, Tetrahedron Lett., 1988, 29, 3311–3314 CrossRef CAS.
- Z. Osmanov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1982, 225–227 CAS; Z. Osmanov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1982, 18, 206–208 CrossRef.
- A. A. Ibragimov, Z. Osmanov, M. R. Yagudaev and S. Yu. Yunusov, Khim. Prir. Soedin., 1982, 213–216; A. A. Ibragimov, Z. Osmanov, M. R. Yagudaev and S. Yu. Yunusov, Chem. Nat. Compd., 1982, 18, 202–205.
- T. S. Tulyaganov and F. Kh. Allaberdiev, Khim. Prir. Soedin., 2001, 476–478; T. S. Tulyaganov and F. Kh. Allaberdiev, Chem. Nat. Compd., 2001, 37, 556–558 CrossRef CAS.
- Nitrabirine N-oxide and nitrabirine could be interconverted by reduction (Zn/HCl) or oxidation (H2O2).
- A. A. Ibragimov, N. D. Abdullaev, Z. Osmanov and S. Yu. Yunusov, Khim. Prir. Soedin., 1987, 685–689 CAS; A. A. Ibragimov, N. D. Abdullaev, Z. Osmanov and S. Yu. Yunusov, Chem. Nat. Compd., 1987, 23, 569–573 CrossRef . Also mentioned in a 2002 paper – see ref. 40.
- A. A. Ibragimov, G. P. Moiseeva, Z. Osmanov and S. Yu. Yunusov, Khim. Prir. Soedin., 1986, 727–730; A. A. Ibragimov, G. P. Moiseeva, Z. Osmanov and S. Yu. Yunusov, Chem. Nat. Compd., 1986, 22, 676–679 CrossRef . Interestingly, this paper starts by a correction concerning the diastereorelation between nitramine and isonitramine as established a few years before by the same authors by X-ray analysis (“Since the use of the program [of X-ray analysis] did not permit antipodes to be distinguished, the suggested diastereoisomerism of nitramine and isonitramine at the C(7) asymmetric centre proved to be erroneous”).
- In fact, both are found in the same plant (N. sibirica).
- Page 3314 of ref. 15: “As the (+)-enantiomer of sibirine corresponds to the natural product [which is wrong: [α]D= −22.5 (0.81, CHCl3)] it follows that natural isonitramine is dextrorotatory”.
- This is consistent with all measurements of optical rotation (see Table 1).
- Not (6R,7R) as represented by Koomen et al. (see ref. 8).
- Z. Osmanov, A. A. Ibragimov and S. Yu Yunusov, Khim. Prir. Soedin., 1982, 126–127 CAS; Z. Osmanov, A. A. Ibragimov and S. Yu Yunusov, Chem. Nat. Compd., 1982, 18, 121–122 CrossRef . Nitraramine was isolated again from N. sibirica in 2002, but no references to optical rotation were given – see ref. 40.
- The authors also converted synthetic (+)-nitramine to N-methylnitramine in order to compare with published data: [α]D= +17.6 (1.23, CHCl3), lit. [α]D = +17.0 (0.43, CHCl3, ref. 16), because they thought that “unfortunately, a rotation for (nitramine) has not been reported” (see ref. 14a, p. 440) which is, in fact, not the case (see Table 1).
- Are sibirinine and sibirinine N-oxide racemic in Nature? Knowing that alkaloids such as isonitramine and sibirine are optically active, this would be a strange statement. Interestingly, nitramine was found to be optically inactive when extracted from N. sibirica (ref. 26, no solvent given) and optically active in N. schoberi (ref. 10a and Table 1), but significant discrepancies are observed, as already discussed.
- J.-C. Quirion, D. S. Grierson, J. Royer and H.-P. Husson, Tetrahedron Lett., 1988, 29, 3311–3314 CrossRef CAS.
- Strangely, in this publication, words such as biosynthesis and biomimetic do not appear.
- D. François, M.-C. Lallemand, M. Selkti, A. Tomas, N. Kunesch and H.-P. Husson, Angew. Chem., Int. Ed., 1998, 37, 104–105 CrossRef CAS.
- As mentioned in the publication, (+)-isonitramine is not the natural isomer..
- M. J. Wanner and G.-J. Koomen, Tetrahedron, 1992, 48, 3935–3944 CrossRef CAS.
- Yu. Novgorodva, S. Kh. Maekh and S. Yu. Yunusov, Khim. Prir. Soedin., 1975, 435–437; Yu. Novgorodva, S. Kh. Maekh and S. Yu. Yunusov, Chem. Nat. Compd., 1975, 11, 455–456 CrossRef.
- B. Tashkodzhaev, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1985, 692–698; B. Tashkodzhaev, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1985, 21, 649–655 CrossRef.
- M. Y. Shen, J. A. Zuanazzi, C. Kan, J.-C. Quirion, H.-P. Husson and I. R. C. Bick, Nat. Prod. Lett., 1995, 6, 119–125 CrossRef CAS.
- Wanner and Koomen published their biosynthetic proposals (see ref. 8) before their total synthesis of nitraramine, see: M. J. Wanner and G.-J. Koomen, J. Org. Chem., 1995, 60, 5634–5637 Search PubMed.
- N. Yu. Novgorodova, S. Kh. Maekh and S. Yu. Yunusov, Khim. Prir. Soedin., 1975, 529–530 CAS; N. Yu. Novgorodova, S. Kh. Maekh and S. Yu. Yunusov, Chem. Nat. Compd., 1975, 11, 562 CrossRef.
- A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1986, 655–656 CAS; A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1986, 22, 620–621 CrossRef.
- T. S. Tulyaganov and F. Kh. Allaberdiev, Khim. Prir. Soedin., 2002, 479–481; T. S. Tulyaganov and F. Kh. Allaberdiev, Chem. Nat. Compd., 2002, 38, 602–604 CrossRef CAS.
- (a) E. Gravel, E. Poupon and R. Hocquemiller, Org. Lett., 2005, 7, 2497–2499 CrossRef CAS; (b) E. Gravel, E. Poupon and R. Hocquemiller, Tetrahedron, 2006, 62, 5248–4253 CrossRef CAS.
- A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1986, 730–735 CAS; A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1986, 22, 680–684 CrossRef.
- B. Tashkodzhaev, A. A. Ibragimov, B. T. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1989, 30–31; B. Tashkodzhaev, A. A. Ibragimov, B. T. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1989, 25, 24–25 CrossRef.
- However, the name schoberine first appeared in the 1977 paper dedicated to the Polybuffer® extractions of Nitraria alkaloids (ref. 4), but no structure was given. The authors referred to the dissertation of N. Yu. Novgorodova (1975). It was also mentioned later as being isolated from N. sibirica (see ref. 6).
- T. S. Tulyaganov, Khim. Prir. Soedin., 1993, 43–34; T. S. Tulyaganov, Chem. Nat. Compd., 1993, 29, 31–34 CrossRef.
- T. S. Tulyaganov and F. Kh. Allaberdiev, Khim. Prir. Soedin., 2003, 228–229; T. S. Tulyaganov and F. Kh. Allaberdiev, Chem. Nat. Compd., 2003, 39, 292–293 CrossRef CAS.
- The hemisynthesis of dihydroschoberine was therefore accomplished before being isolated from a natural source (in 2001).
- B. M. Pakhritdinov, N. Yu. Novgorodova, M. Normatova and S. Yu. Yunusov, Khim. Prir. Soedin., 1970, 641–642 CAS; B. M. Pakhritdinov, N. Yu. Novgorodova, M. Normatova and S. Yu. Yunusov, Chem. Nat. Compd., 1970, 6, 663–664 CrossRef.
- T. S. Tulyaganov and N. N. Shorakhimov, Khim. Prir. Soedin., 1990, 560–561 CAS; T. S. Tulyaganov and N. N. Shorakhimov, Chem. Nat. Compd., 1990, 26, 478–479.
- To the best of our knowledge, this is the only work carried out with a plant sample collected in Turkey (province of Ankara); see: L. Üstünes, A. Özer, G. M. Laekeman, J. Corthout, L. A. C. Pieters, W. Baeten, A. G. Hertman, M. Claeys and A. J. Vlietinck, J. Nat. Prod., 1991, 54, 959–966 Search PubMed.
- M. J. Wanner, A. W. Velzel and G.-J. Koomen, J. Chem. Soc., Chem. Commun., 1993, 174–175 RSC.
- T. S. Tulyaganov, N. M. Kozimova and F. Kh. Allaberdiev, Khim. Prir. Soedin., 2006, 164–166; T. S. Tulyaganov, N. M. Kozimova and F. Kh. Allaberdiev, Chem. Nat. Compd., 2006, 42, 198–200 CrossRef CAS.
- E. Cheng, J. Botzem, M. J. Wanner, B. E. A. Burm and G.-J. Koomen, Tetrahedron, 1996, 52, 6725–6732 CrossRef CAS.
- T. S. Tulyaganov, O. M. Nazarov, M. G. Levkovich and N. D. Abdullaev, Khim. Prir. Soedin., 2001, 56–58; T. S. Tsulalaganov, O. M. Nazarov, M. G. Levkovich and N. D. Abdullaev, Chem. Nat. Compd., 2001, 37, 61–64 CrossRef.
- This reaction was conducted by Tulyaganov et al. and gave a compound “that had properties that were identical in all respects to those of the natural alkaloid komavine” (see ref. 54).
- N. Normatov and S. Yu. Yunusov, Khim. Prir. Soedin., 1968, 139; N. Normatov and S. Yu. Yunusov, Chem. Nat. Compd., 1969, 4, 120.
- (a) A. A. Ibragimov, S.-M. Nasirov, V. T. Andrianov, S. Kh. Maekh, Yu. T. Struchkov and S. Yu. Yunusov, Khim. Prir. Soedin., 1975, 273–274 CAS; A. A. Ibragimov, S.-M. Nasirov, V. T. Andrianov, S. Kh. Maekh, Yu. T. Struchkov and S. Yu. Yunusov, Chem. Nat. Compd., 1975, 11, 293–294 CrossRef; (b) S. M. Nasirov, A. A. Ibragimov, V. G. Andrianov, S. Kh. Maekh, Yu. T. Struchkov and S. Yu. Yunusov, Khim. Prir. Soedin., 1976, 334–345 CAS; S. M. Nasirov, A. A. Ibragimov, V. G. Andrianov, S. Kh. Maekh, Yu. T. Struchkov and S. Yu. Yunusov, Chem. Nat. Compd., 1976, 12, 294–301 CrossRef.
- A. A. Ibragimov, S. Kh. Maekh and S. Yu. Yunusov, Khim. Prir. Soedin., 1975, 275–276; A. A. Ibragimov, S. Kh. Maekh and S. Yu. Yunusov, Chem. Nat. Compd., 1975, 11, 295–296 CrossRef.
- A. A. Ibragimov, S. Kh. Maekh and S. Yu. Yunusov, Khim. Prir. Soedin., 1975, 297; A. A. Ibragimov, S. Kh. Maekh and S. Yu. Yunusov, Chem. Nat. Compd., 1975, 11, 297 CrossRef . Despite an ambiguous title (“The structure of schoberine”), the publication deals with the isolation of schoberidine.
- A. A. Ibragimov, S. Kh. Maekh and S. Yu. Yunusov, Khim. Prir. Soedin., 1975, 276–277 CAS; A. A. Ibragimov, S. Kh. Maekh and S. Yu. Yunusov, Chem. Nat. Compd., 1975, 11, 298 CrossRef.
- T. S. Tulyaganov and O. M. Nazarov, Khim. Prir. Soedin., 2000, 323–324; T. S. Tulyaganov and O. M. Nazarov, Chem. Nat. Compd., 2000, 36, 393–395 CrossRef CAS.
- T. S. Tulyaganov and O. E. Makhmudov, Khim. Prir. Soedin., 2000, 325–326; T. S. Tulyaganov and O. E. Makhmudov, Chem. Nat. Compd., 2000, 36, 396–398 CrossRef CAS.
- T. S. Tulyaganov, O. M. Nazarov, O. E. Makhmudov, A. D. Vdovin and N. D. Abdullaev, Khim. Prir. Soedin., 2001, 400–402; T. S. Tulyaganov, O. M. Nazarov, O. E. Makhmudov, A. D. Vdovin and N. D. Abdullaev, Chem. Nat. Compd., 2001, 37, 470–473 CrossRef.
- T. S. Tulyaganov, Khim. Prir. Soedin., 1994, 780–783 CAS; T. S. Tulyaganov, Chem. Nat. Compd., 1994, 30, 727–729 CrossRef.
- T. S. Tulyaganov and N. D. Abdullaev, Khim. Prir. Soedin., 1995, 95–99 CAS; T. S. Tulyaganov and N. D. Abdullaev, Chem. Nat. Compd., 1995, 31, 76–78 CrossRef.
- T. S. Tulyaganov and O. E. Makhmudov, Khim. Prir. Soedin., 2000, 60–61; T. S. Tulyaganov and O. E. Makhmudov, Chem. Nat. Compd., 2000, 36, 76–78 CrossRef CAS.
- However, in their 1976 paper (ref. 57b), they wondered about the racemic nature of nitrarine: “…the possibility of racemization during isolation and in vivo is excluded and it probably takes place in the early stages of biosynthesis”.
- M. J. Wanner and G.-J. Koomen, J. Org. Chem., 1994, 59, 7479–7484 CrossRef CAS.
- D. François, M.-C. Lallemand, M. Selkti, A. Tomas, N. Kunesch and H.-P. Husson, J. Org. Chem., 1997, 62, 8914–8916 CrossRef CAS.
- Nitraraine: A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1985, 536–544 Search PubMed; A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1985, 21, 502–509 CAS . O-Acetylnitraraine: T. S. Tulyaganov and M. M. Kozimova, Khim. Prir. Soedin., 2005, 472–473 CrossRef; T. S. Tulyaganov and M. M. Kozimova, Chem. Nat. Compd., 2005, 41, 578–579 Search PubMed.
- A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1985, 544–546 CAS; A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1985, 21, 510–512 CrossRef.
- S. Takano, K. Samizu, T. Sugihara, S. Satoh and K. Ogasawara, Chem. Lett., 1989, 1777–1180 CAS . In their conclusion the authors add: “the present synthesis raised considerable doubt to the proposed structures of nitraraine and dihydronitraraine”.
- Another total synthesis was published soon after: R. Yamaguchi, T. Hamasaki, T. Sasaki, S. Kozima and H. Takaya, Synlett, 1991, 719–720 Search PubMed . The authors only refer to ref. 72, with no allusion to the structure of the natural compound.
- We will disclose a full account of this proposal (with experimental proof) in due course.
- J.-A. Duan, I. D. Williams, C.-T. Che, R.-H. Zhou and S.-X. Zhao, Tetrahedron Lett., 1999, 40, 2593–2596 CrossRef CAS.
- The first proof of existence of nitraraine or tangutorine might be found in the 1977 paper by Ibragimov et al. (see ref. 4). Various conditions of separation of crude extracts of Nitraria schoberi using different buffers resulted in the isolation of a compound, named “base B” (mass 308), which was not characterized at this time..
- Readers should be careful: the structure drawn on page 2593 of ref. 75 is wrong – the stereochemistry of carbon 3 should be inverted.
- (a) First total synthesis: T. Putkonen, A. Tolvanen and R. Jokela, Tetrahedron Lett., 2001, 42, 6593–6594 Search PubMed; (b) Chiral HPLC separation of enantiomers: T. Putkonen, A. Tolvanen, R. Jokela, S. Caccamese and N. Parrinello, Tetrahedron, 2003, 59, 8589–8595 Search PubMed; (c) S. Luo, C. A. Zificsak and R. P. Hsung, Org. Lett., 2003, 5, 4709–4712 CrossRef CAS; (d) T.-L. Ho and C.-K. Chen, Helv. Chim. Acta, 2006, 89, 122–126 CrossRef CAS.
- (a) Formal total synthesis: S. Luo, J. Zhao and H. Zhai, J. Org. Chem., 2004, 69, 4548–4550 Search PubMed; (b) Synthetic approach: J. A. Wilkinson, N. Ardes-Guisot, S. Ducki and J. Leonard, Tetrahedron Lett., 2005, 46, 8053–8056 Search PubMed.
- In fact, to the best of our knowledge, no indolomonoterpenic alkaloids have ever been isolated from Nitraria species. The optical property (racemic mixture) of tangutorine is also hardly explained by the indolomonoterpenic pathway.
- Total syntheses of tangutorine: (a) T. Putkonen, A. Tolvanen and R. Jokela, Tetrahedron Lett., 2001, 42, 6593–6594 CrossRef CAS; (b) T. Putkonen, A. Tolvanen, R. Jokela, S. Caccamese and N. Parinello, Tetrahedron, 2003, 59, 8589–8595 CrossRef CAS; (c) S. Luo, C. A. Zificsak and R. P. Hsung, Org. Lett., 2003, 5, 4709–4712 CrossRef CAS; (d) T.-L. Ho and C.-K. Chen, Helv. Chim. Acta, 2006, 89, 122–126 CrossRef CAS.
- R. Salame, E. Gravel and E. Poupon, Org. Lett., 2009, 11, 1891–1894 CrossRef CAS.
- A. A. Ibragimov and S. Y. Yunusov, Khim. Prir. Soedin., 1985, 544–546 CAS; A. A. Ibragimov and S. Y. Yunusov, Chem. Nat. Compd., 1985, 21, 510–512 CrossRef.
- T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1981, 192–195 CAS; T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1981, 17, 149–152 CrossRef . The structures of komarovine and komarovidine were first published in 1980; see: T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1980, 732 Search PubMed . We were unable to find the English translation of this article, and so could not check this reference.
- T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1982, 633–635 CAS; T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1982, 18, 598–600 CrossRef.
- T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1982, 638–640 CAS; T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1982, 18, 604–606 CrossRef.
- T. S. Tulyaganov and S. Yu. Yunusov, Khim. Prir. Soedin., 1990, 61–67 CAS; T. S. Tulyaganov and S. Yu. Yunusov, Chem. Nat. Compd., 1990, 26, 49–53 CrossRef.
- T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1984, 398 CAS; T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1984, 20, 378–379 CrossRef.
- T. S. Tulyaganov, Khim. Prir. Soedin., 1993, 33–39 CAS; T. S. Tulyaganov, Chem. Nat. Compd., 1993, 29, 26–30 CrossRef.
- T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1982, 635–638 CAS; T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1982, 18, 601–603 CrossRef.
- T. S. Tulyaganov, Khim. Prir. Soedin., 2006, 370–372; T. S. Tulyaganov, Chem. Nat. Compd., 2006, 42, 459–461 CrossRef CAS.
- The identity of these substances was based on MS, UV analysis and melting points.
- T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Khim. Prir. Soedin., 1979, 737 CAS; T. S. Tulyaganov, A. A. Ibragimov and S. Yu. Yunusov, Chem. Nat. Compd., 1979, 15, 654 CrossRef . Identity with a natural sample of schoberidine was based on UV and mixed melting points.
- The authors gave the PhD thesis of A. A. Ibragimov (1975) as a reference for this result, but this was not mentioned in their 1976 paper (see ref. 57b).
- See ref. 89: “Komarovinine is apparently the product of the migration of a β-carboline residue from position 8 of the quinoline nucleus to position 6 during the dehydration of nitrarine, since the normal product komarovine, both in the pure form and in admixture with selenium, is stable on heating at 300 °C or is formed from komavicine”.
- T. S. Tulyaganov, Khim. Prir. Soedin, 1993, 87–88 CAS; T. S. Tulyaganov, Chem. Nat. Compd., 1993, 29, 73 CrossRef.
- T. S. Tulyaganov, R. Sh. Atadzhanov, N. D. Abdullaev, É. L. Kristallov and Z. Osmanov, Khim. Prir. Soedin., 1993, 580–582 CAS; T. S. Tulyaganov, R. Sh. Atadzhanov, N. D. Abdullaev, É. L. Kristallov and Z. Osmanov, Chem. Nat. Compd., 1993, 29, 509–511 CrossRef.
- V. C. Pham, A. Jossang, A. Chiaroni, T. Sévenet, V. H. Nguyen and B. Bodo, Tetrahedron Lett., 2002, 43, 7565–7568 CrossRef CAS.
- V. C. Pham, A. Jossang, A. Chiaroni, T. Sévenet, V. H. Nguyen and B. Bodo, Org. Lett., 2007, 9, 3531–3534 CrossRef CAS.
- V. C. Pham, A. Jossang, T. Sévenet, V. H. Nguyen and B. Bodo, Eur. J. Org. Chem., 2009, 1412–1416 CrossRef CAS.
- V. C. Pham, A. Jossang, P. Grellier, T. Sévenet, V. H. Nguyen and B. Bodo, J. Org. Chem., 2008, 73, 7565–7573 CrossRef CAS.
- V. C. Pham, A. Jossang, T. Sévenet, V. H. Nguyen and B. Bodo, Tetrahedron, 2007, 63, 11244–11249 CrossRef CAS.
- V. C. Pham, A. Jossang, T. Sévenet, V. H. Nguyen and B. Bodo, J. Org. Chem., 2007, 72, 9826–9829 CrossRef CAS.
- See ref. 2d, LD50 = 229 mg kg−1 (iv, mice) and hypotensive effects.
- See ref. 2d, LD50 = 232 mg kg−1 (iv, mice), tranquilizing properties.
- See ref. 2d, LD50 = 117, 340 and 524 mg kg−1 (iv, ip and sc, respectively, mice). Nitrarine is a well known hypotensive, antiarrhythmic and spasmolytic agent that acts by stabilizing membranes.
- See ref. 2f; LD50 = 11.9 mg kg−1 (iv, mice), hypotensive action.
- See ref. 2c; LD50 = 171 mg kg−1 (iv, mice), hypotensive action.
- See ref. 2c; LD50 = 146.5 mg kg−1 (iv, mice), hypertensive action. See also: T. S. Tulyaganov, A. A. Ibragimov, S. Yu. Yunusov, A. A. Vakhabov and S. D. Aminov, Khim. Pharm. Zh., 1987, 21, 295–297 Search PubMed; S. Yu. Yunusov, A. A. Vakhabov and S. D. Aminov, Pharm. Chem. J., 1987, 21, 182–184 CAS.
- See ref. 2c; LD50 = 33.7 mg kg−1 (iv, mice), hypotensive action.
- See ref. 50; nazlinin has a vasorelaxant effect upon aortic rings; at higher doses (>40 nmol) a contraction was observed (rabbit).
- B. P. L. Liu, E. Y. Y. Chong, F. W. K. Cheung, J.-A. Duan, C.-T. Che and W. K. Liu, Biochem. Pharmacol., 2005, 70, 287–299 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |