Prenylated indole derivatives from fungi: structure diversity, biological activities, biosynthesis and chemoenzymatic synthesis
Shu-Ming
Li
*
Philipps-Universität Marburg, Institut für Pharmazeutische Biologie, Deutschhausstr. 17a, 35037, Marburg, Germany. E-mail: shuming.li@staff.uni-marburg.de; Fax: +49 6421 2825365; Tel: +49 6421 2822461
First published on 19th November 2009
Abstract
Covering: up to mid-2009
Prenylated indole alkaloids are hybrid natural products derived from prenyl diphosphates and tryptophan or its precursors and widely distributed in filamentous fungi, especially in the genera Penicillium and Aspergillus of ascomycota. These compounds represent a group of natural products with diverse chemical structures and biological activities. Significant progress on their biosynthesis has been achieved in recent years by identification of biosynthetic gene clusters from genome sequences and by molecular biological and biochemical investigations. In addition, a series of prenylated indole derivatives have been produced by chemoenzymatic synthesis using overproduced and purified enzymes.
 Shu-Ming Li | Shu-Ming Li is full professor of Pharmaceutical Biology at the Philipps-University in Marburg, Germany. He received his Ph.D. in 1992 from the Rheinische Friedrich-Wilhelms-University in Bonn, Germany. He has served as associate professor for Pharmaceutical Biology at the Heinrich-Heine-University in Düsseldorf. His research interests focus on the genomics of the biosynthesis of secondary metabolites in fungi, especially of prenylated indole alkaloids. |
1 Introduction
Prenylated indole alkaloids are hybrid natural products containing indole/indoline and isoprenoid moieties or structures derived thereof. They are widely distributed in terrestrial and marine organisms, especially in the genera Penicillium and Aspergillus of ascomycota, and display broad structure diversity. These compounds often carry biological and pharmacological activities distinct from their non-prenylated aromatic precursors.1–3The availability of the genome sequences of bacteria and fungi, which has increased tremendously in recent years,4,5 has accelerated the identification of genes involved in the biosynthesis of secondary metabolites by genome mining.6–8 By using this strategy, biosynthetic genes of several prenylated indole derivatives were identified in the genome sequences of ascomycetes.9–11 The functions of these genes have been proven by molecular biological and biochemical approaches.9,10 Additionally, a number of prenylated simple indole derivatives or tryptophan-containing diketopiperazines were synthesised by using overproduced and purified indole prenyltransferases.12 The aim of this review is to give an overview on the structural characteristics of the main classes of prenylated indole derivatives from fungi, without listing all the structures of known natural products. In addition, this review summarizes briefly the considerable progress on the molecular biological and biochemical investigations on the biosynthesis of prenylated indole alkaloids in recent years and the application of the recombinant proteins for the production of prenylated indole derivatives.
2 Diverse structures of prenylated indole derivatives from fungi with significant pharmacological and biological activities
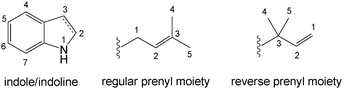
Prenylated indole alkaloids show diverse chemical structures and a wide range of biological and pharmacological activities. With the exception of indole-diterpenes, tryptophan from primary metabolism is a key precursor and acts as the biogenetic origin of indole or indoline rings.3,13,14 Besides L-tryptophan, most of these substances contain a second amino acid and form cyclic dipeptides with a diketopiperazine structure or a derivative thereof. In most cases, the second amino acid is L-proline, L-tryptophan, L-histidine, L-phenylalanine or L-alanine. A further large group within the prenylated indole alkaloids are asterriquinones containing two tryptophan moieties with a bis(indolyl) benzoquinone structure.15In the structures of prenylated indole alkaloids, the prenyl moieties can be connected via its C1 or C3 to an aromatic nucleus, which are referred to as regular or reverse prenyl moieties, respectively (Fig. 1). In prenylated cyclic dipeptides and bis(indolyl) benzoquinones, reverse prenyl moieties are usually attached to position N1, C2 or C3 of the indole or indoline ring, while the regular moieties are normally connected to position N1, C2, C5, C6 or C7, but not C3 of the indole or indoline rings (Fig. 1). Interestingly, prenylation at position C4 was only found in structural skeletons derived exclusively from only one L-tryptophan molecule such as in ergot alkaloids and cyclopiazonic acid or indole-diterpenes, but not in peptides or benzoquinones.
2.1 Prenylated derivatives of the cyclic dipeptide consisting of L-tryptophan and L-proline.
Fungal natural products derived from L-tryptophan and L-proline represent a large group within the prenylated indole alkaloids.3 The common precursor of these compounds is the cyclic dipeptide brevianamide F (1), which is converted to tryprostatin B (2) by attaching a regular prenyl moiety at position C2 or to deoxybrevianamide E (3) by a reverse one. Tryprostatin B (2) is a key intermediate in the biosynthesis of diverse structures such as tryprostatin A (4), spirotryprostatin A (5), fumitremorgins (6–8) and verruculogen (9), while deoxybrevianamide E (3) serves as precursor for brevianamides, e.g. brevianamide A (10), austamide (11) and notoamides (12–14).3,16–18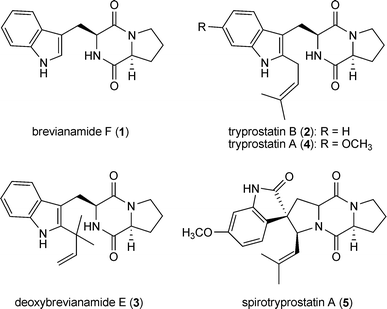
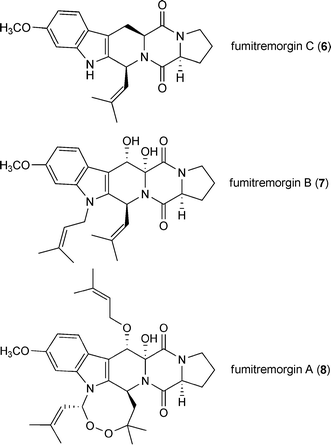
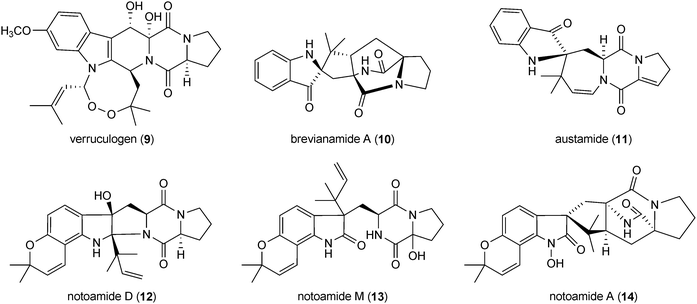
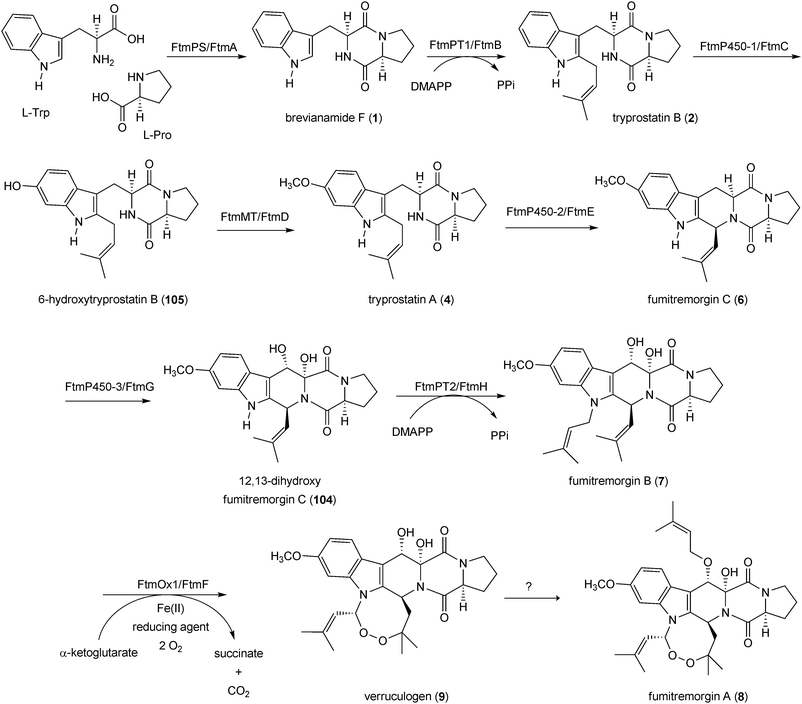
Fumitremorgin B (7) and verruculogen (9), which contain two prenyl moieties at positions N1 and C2 of the indole ring, are mycotoxins from a number of Aspergillus and Penicillium strains.19–23 Verruculogen (9), associated with Aspergillus fumigatus hyphae and conidia, was reported to be able to modify the electrophysiological properties of human nasal epithelial cells and could be involved in the pathogenesis of this fungus.24 Fumitremorgin A (8) is also a mycotoxin from Aspergillus and Penicillium strains20,25 and contains an additional prenyl moiety, in comparison to the structure of verruculogen (9). The biosynthetic intermediates of these compounds containing one prenyl moiety attached to position C2, such as tryprostatin B (2) and its methoxylated derivative tryprostatin A (4) as well as fumitremorgin C (6), showed biological activities of pharmaceutical interest. For example, tryprostatins A (4) and B (2) as well as their diastereomeres were found to exhibit cytotoxicity towards various cancer cell lines, even more potent than etoposide.26–28 Fumitremorgin C (6) is a specific inhibitor of the breast cancer resistance protein and could reverse the resistance of some tumour cell lines.29–31 Tryprostatin A (4) was also able to inhibit the ABC transporter of the resistant tumour cell lines.32,33
Brevianamides and austamides, e.g. brevianamide A (10) and austamide (11), constitute a small group of cyclo-L-Trp-L-Pro derivatives and contain only one reverse prenyl moiety at position C2 of the indole ring or a structure derived thereof. These compounds are produced by Penicillium brevicompactum and Aspergillus ustus, respectively.34–36 The dimeric cyclic dipeptide brevianamide J (15), containing one reverse prenyl moiety at position C2 of the indole ring in each unit, was recently isolated from cultures of Aspergillus versicolor.37
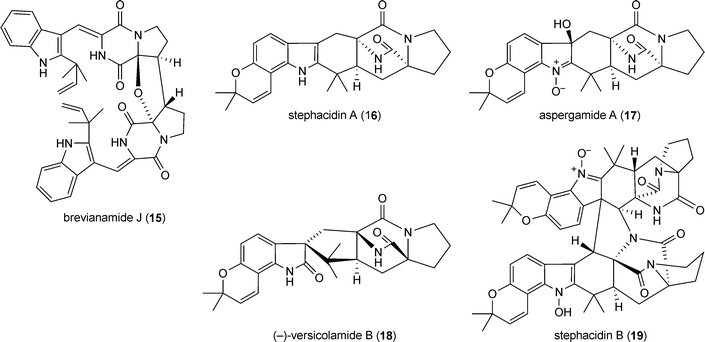
Notoamides (12–14) and related compounds such as aspergamides, staphacidins and (−)-versicolamide B (16–18) are diprenylated derivatives of cyclo-L-Trp-L-Pro. One of the prenyl residues forms a fused dimethyldihydropyran ring via positions C6 and C7 of the indole ring. The second one was found as an intact reverse prenyl moiety at position C2 (notoamide D 12), C3 (notoamide M 13) or as strongly modified structure elements (notoamide A (14), stephacidin A (16), aspergamide A (17) and (−)-versicolamide (18), which could be considered as rearrangement results of the prenyl moiety at position C2 of the indole ring.38 Stephacidin B (19), a dimeric derivative of stephacidin A (16), has also been described.39 Notoamides, (−)-versicolamide and stephacidins are isolated from different Aspergillus strains.17,18,38–40 Notoamide A (14) showed moderate cytotoxicity against HeLa and L1210 cells.17
2.2 Prenylated derivatives of the cyclic dipeptide consisting of two L-tryptophan molecules
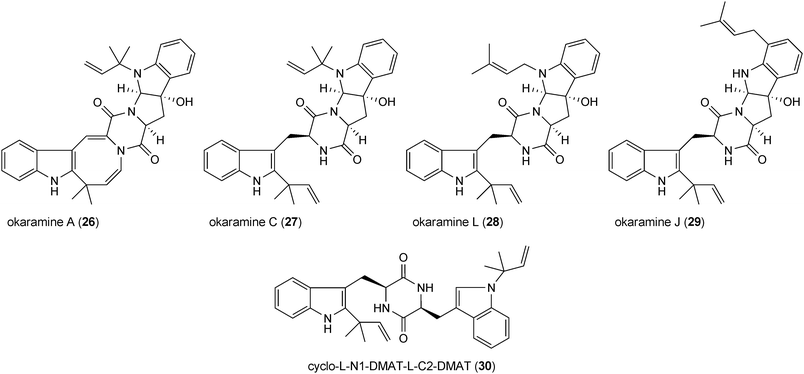
Fellutanines are reversely C2-prenylated derivatives of cyclo-L-Trp-L-Trp. Fellutanines B (20) and C (21) from Penicillium fellutanum are mono- and diprenylated derivatives with an intact diketopiperazine ring,41 while this ring system is fused with the indoline ring in fellutanine D (22) from Penicillium fellutanum and its diastereomer gypsetin (23) from Nannizzia gypsea.42 Amauromine (24) from Amauroascus sp.43 and epiamauromine (25) from Aspergillus ochraceus44 represent reversely C3-prenylated indolines. Okaramines (26–29) from Penicillium simplicissimum and Aspergillus aculeatus consist of at least 18 congeners.45–49 In the structures of the most okaramines, e.g. A (26) and C (27), one reverse prenyl moiety is found at position C2 and a second one at position N1 of another tryptophan unit. These okaramines very likely are derived from cyclo-L-N1-DMAT-L-C2-DMAT (30), which was also isolated from Penicillium simplicissimum.48 In the structure of okaramine L (28), position N1 is regularly prenylated instead. In some cases, the second prenyl residue can also be found at other positions than N1, e.g. in okaramine J (29) at position C7.48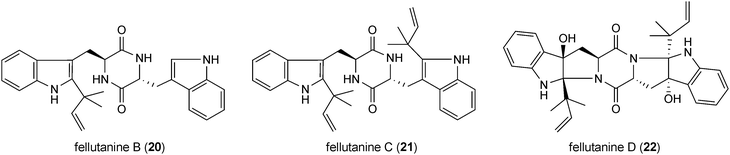
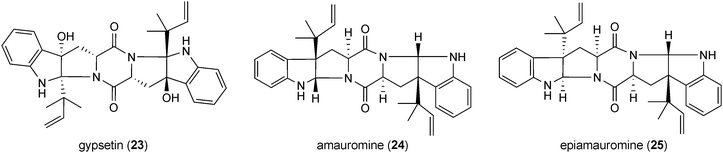
Fellutanine D (22) was reported to be cytotoxic against several cell lines,41 while its diastereomer gypsetin (23) showed cholesterol acyltransferase inhibiting activity.50 Amauromine (24) was reported to have a vasodilating activity.51 Its stereoisomer epiamauromine (25) showed, on the other hand, insecticidal activity.44 Insecticidal activity against silkworms has also been observed with several okaramines.52
2.3 Prenylated derivatives of cyclic dipeptides consisting of L-tryptophan and a second aromatic amino acid
All members of this group are derived from cyclic dipeptides containing an indoline ring, which is fused with a diketopiperazine or benzodiazepindinone ring. In addition, they carry reverse prenyl moieties at position C3 of the indoline ring. A prominent example is roquefortine C (31, also called roquefortine) derived from L-tryptophan and L-histidine. This compound was firstly isolated from Penicillium roqueforti53 and identified later in a number of Penicillium strains,54–57 including P. roqueforti strains used for cheese production.53,55 Roquefortine C (31) is also the precursor of several prenylated indole alkaloids such as roquefortine E (32), glandicoline B (33),58 meleagrin (34)58 and oxaline (35).58–60 Roquefortine E (32), containing an additional reverse prenyl moiety attached to the imidazole ring, was isolated from Gymnoascus reessii.61 A meleagrin derivative (36) carrying a modified prenyl moiety at the imidazole ring was recently isolated from a deep ocean sediment derived Penicillium sp.62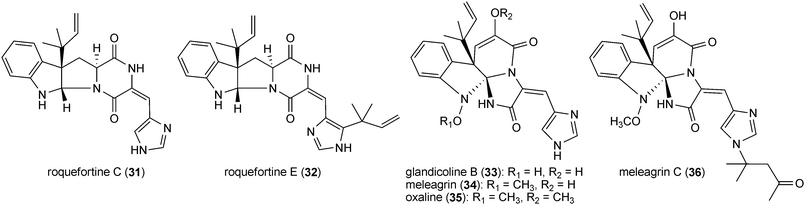
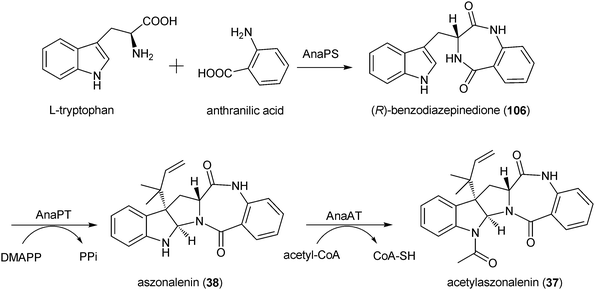
A second example from this group is the mycotoxin acetylaszonalenin (37) (called also LL-S490β) derived from the amino acids L-tryptophan and anthranilic acid. Acetylaszonalenin (37) was isolated initially from an unidentified Aspergillus species,63 Together with its non-acetylated form aszonalenin (38), it was also identified in various fungal strains, e.g. Aspergillus zonatus,64A. fumigatus,65A. carneus66 and Neosartorya fischeri.67,68 Their stereoisomers epi-aszonalenins A (39) and C (40) were isolated from Aspergillus novofumigatus.69
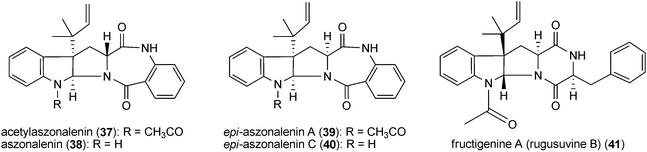
Fructigenine A (41) from Penicillium fructigenum,70 which was also identified as rugulosuvine B in Penicillium regulosum71 is a cyclic dipeptide derivative of L-tryptophan and L-phenylalanine. This compound was reported to be a plant growing inhibitor.70 Until now, no prenylated diketopiperazine derivatives containing tryptophan and tyrosine were isolated from the natural sources.
2.4 Prenylated peptide derivatives containing L-tryptophan and aliphatic amino acids
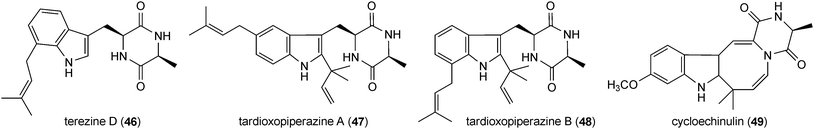
Prenylated indole alkaloids in this category are mainly derived from L-tryptophan and L-alanine or L-tryptophan and L-isoleucine. Prenylated cyclo-L-Trp-L-Ala derivatives including echinulin (42) or its analogues constitute a large group of fungal metabolites. They carry up to three prenyl moieties with a reverse one at position C2 of the indole ring. Echinulin (42), bearing prenyl moieties at positions C2, C5 and C7, was initially isolated from Aspergillus amstelodami72 and then from different Aspergillus strains.73–76 It should be mentioned that echinulin (42) was also identified in different plants of Anacardiaceae, Cucurbitaceae and Orchidaceae.77,78 Diverse echinulin analogues with different prenylation positions at the benzene ring and modifications, e.g. hydroxylation, oxidation and cyclisation of prenyl moieties were described in literature.74 Examples of such structures are variecolorin L (43) from Aspergillus variecolor with prenyl moieties at positions C2, C4 and C574 or arestrictin B (44) from Aspergillus penicilloides with prenyl moieties at positions C2, C6 and C7.79 A number of mono- and diprenylated cyclo-L-Trp-L-Ala derivatives were also isolated from various fungal strains,73–75e.g. preechinulin (45),73,74 terezine D (46),80 tardioxopiperazines A (47) and B (48).74,81 In the structure of cycloechinulin (49), the prenyl moiety at position C2 is connected to the diketopiperazine ring.82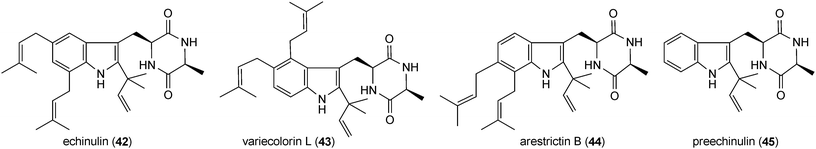
Astechrome (50) from Aspergillus terreus represents a rare iron-containing prenylated cyclo-Trp-Ala derivative.83 In the structure of 5-N-acetylardeemin (51), which was identified in Neosartorya fischeri var. brasiliensis,84 an anthranilic acid moiety is connected to the diketopiperazine ring consisting of tryptophan and alanine. Variecoloritide B (52) from Aspergillus variecolor contains a prenylated cyclo-Trp-Ala residue, which is condensed with an anthraquinone molecule.85 Fructigenine B (53) from Penicillium fructigenum,70 which was also identified as verrucofortine in Penicillium verucosum var. cyclopium,86 is a prenylated cyclic dipeptide of tryptophan and leucine. Together with fructigenine B (53), five new C3-prenylated derivatives including brevicompanine G (54) were isolated from a deep ocean sediment derived Penicillium sp.87 Brevicompanine G (54) is likely derived from tryptophan and valine. Both amino acids are also found in the structures of the prenylated linear peptides mellamide (55) and terpeptin (56), which were isolated from Aspergillus melleus88 and Aspergillus terreus,89 respectively.
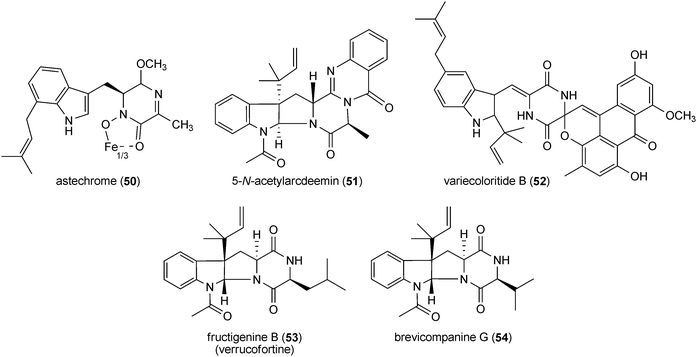
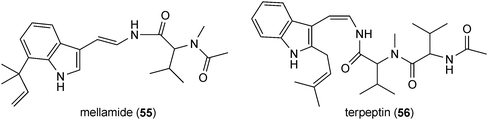
Paraherquamides (57 and 58) and related compounds marcfortines (59 and 60) are a structurally complex family of indole alkaloids. Many members of this group show potent anthelmintic and antinematodal activities and have been under intensive investigation for use in veterinary medicine to treat various intestinal parasites.3 The members of this group are derived from L-tryptophan and β-methylproline, which is in turn formed from L-isoleucine.3 These compounds were identified in early 1980s in Penicillium and Aspergillus strains, especially in Penicillium paraherquei.3,34
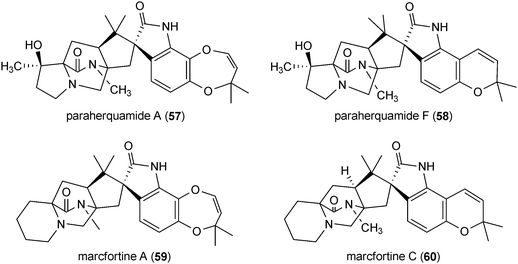
Most of the paraherquamides and analogues are diprenylated indole alkaloids. One prenyl moiety is found at position C3 or a structure derived thereof. The second one is connected to positions C6 and C7 of the indole ring via two oxygen atoms forming a seven-membered ring, as in the case of paraherquamide A (57) and macfortine A (59). This prenyl moiety exists also as a dimethyldihydropyran structure, as in the case of paraherquamide F (58) and macfortine C (60).
2.5 Prenylated bis(indolyl) benzoquinone derivatives
Asterriquinone (61), a symmetrical benzoquinone containing two reversely N1-penylated tryptophanyl moieties, was isolated from Aspergillus terreus90 and demonstrated to be active against tumour cells.91 Later on, a series of bis(indolyl) benzoquinones carrying one or two regular or reverse prenyl moieties was identified in A. terreus,15,92–94Chaetomium sp.95 and other fungi.96–100 Isoasterriquinone (62) from Aspergillus terreus contains reversely N1-prenylated and regularly C2-prenylated tryptophanyl moieties.94 Hinnuliquinone (63) from Nodulisporium hinnuleum101 is a symmetrical dimer of reversely C2-prenylated tryptophan components. Further examples of symmetrical dimers are asterriquinones CT5 (64) and CT3 (65), isocochliodinol (66) and asterriquinone CT4 (67), with regular prenylation at positions of C2, C5, C6 and C7, respectively. Asterriquinones CT3 (65), CT4 (67) and CT5 (64) were isolated from Humicola grisea, Humicola fuscoatra and Aspergillus terreus, respectively.94,99 Together with isocochliodinol (66), asterriquinones CT3 (65) and CT4 (67), termed cochliodinol and neocochliodinol, respectively, were also identified in a Chaetomium sp.95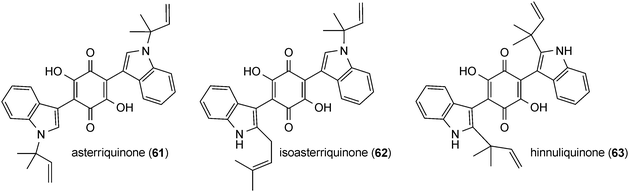
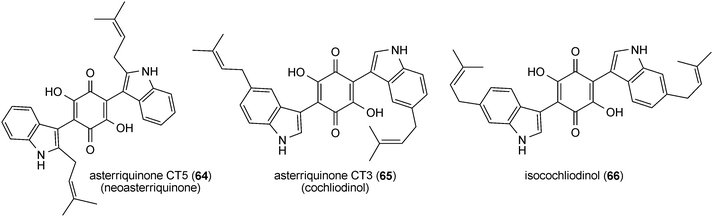
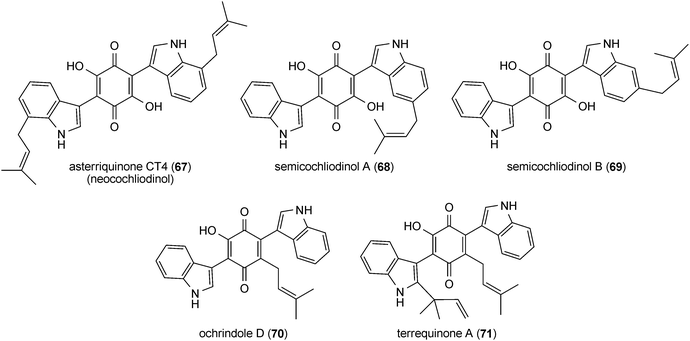
In addition to diprenylated derivatives, diverse monoprenylated indolyl benzoquinones were also reported, e.g. semicochliodinols A (68) and B (69) from Chrysophorium merdarium.100 In ochrindole D (70) from Aspergillus ochraceus, the prenyl moiety is not attached to the indole, but to the benzoquinone ring.102 Terrequinone A (71) from Aspergillus terreus and A. nidulans is a bis(indolyl) benzoquinone with a reverse prenyl moiety at position C2 of the indole and a regular one at the quinone ring.97,103 As in the cases of prenylated peptides, no C4-prenylated derivatives were found within the bis(indolyl) benzoquinones.
A number of asterrequinone derivatives showed cytotoxicity.104–107 Investigation on the structure–activity relationship showed that the indole ring is important for the effect.107 Hinnuliquinone (63), semicochliodinols A (68) and B (69) were reported to be active as inhibitors of HIV-1 protease.100,108
2.6 Ergot alkaloids and other C4-prenylated indole alkaloids
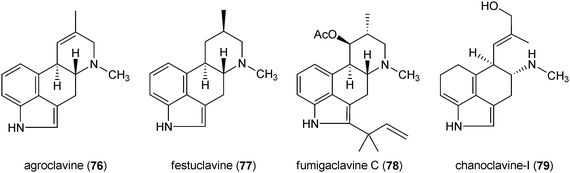
Ergot alkaloids are a complex family of toxins and important pharmaceuticals with diverse structures and biological activities.1,109 They are produced by fungi of two orders and plants of three families.110 The important producers are fungi of the genera Claviceps, Penicillium and Aspergillus.1,109 Both natural ergot alkaloids and their semi-synthetic derivatives are in widespread use in modern medicine.1,110 Chemically, ergot alkaloids are characterised by the presence of the tetracyclic ergoline ring (72) and can be divided into two classes according to their structural features, i.e. amide derivatives of D-lysergic acid (73) and the clavine alkaloids.109,111 The members of the first group are usually composed of lysergic acid (73) and an amino alcohol, e.g. ergometrine (74), or a peptide moiety, e.g. ergotamine (75). The clavines like agroclavine (76), festuclavine (77) and fumigaclavine C (78) or their precursors like chanoclavine-I (79) merely consist of a tetracyclic or even tricyclic ring system lacking an amide or a peptide moiety. Ergotamine (75) was described as the first ergot alkaloid in 1920. Since then about 50 different ergot alkaloids of both classes have been found in nature.109,111,112 Due to their significant importance as toxins and drugs, ergot alkaloids have been an important research field in secondary metabolism.111 Results on biology, chemistry, molecular biology and biotechnology have been reviewed extensively.1,109–113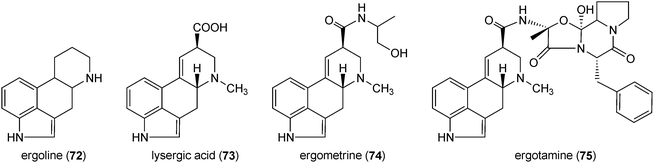
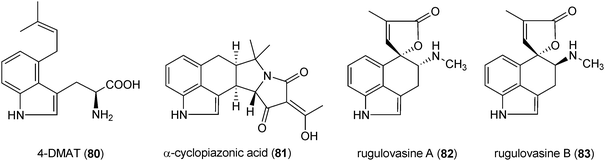
Biogenetically, the ergoline ring (72) is derived from the C4-prenylated tryptophan 4-DMAT (80). This building block was not found in tryptophan-containing diketopiperazine or bis(indolyl) benzoquinone alkaloids discussed above, but in the structures of α-cyclopiazonic acid (81) and rugulovasines (82 and 83). The fungal neurotoxin α-cyclopiazonic acid (CPA) (81), a nanomolar inhibitor of Ca2+-ATPase,114 has a pentacyclic indole tetramic acid scaffold and was firstly isolated from Penicillium cyclopium115 and followed by a large number of Penicillium and Aspergillus strains.57,116–119 α-Cyclopiazonic acid (81) was often found as a contaminant in food and feeds117,119,120 Rugulovasines A (82) and B (83) and their chlorinated derivatives were identified as a small group of C4-prenylated tryptophan derivatives in several Penicillium strains.121–123
2.7 Prenylated indole-diterpenes
Indole-diterpenes with a minimal structure (84) are a large, structurally and functionally diverse group of secondary metabolites produced by filamentous fungi with a common cyclic diterpen backbone and an indole residue.2 The first reported structure of this group, paspalitrem A (85), was isolated from Claviceps paspali in 1977.124 Two years later, the tremorgenic mycotoxin aflatrem (86) was identified in Aspergillus flavus.125,126 These mycotoxins are monoprenylated derivatives of paspalinine (87), which could also be isolated from various fungal strains.124,126 The structures of penitrem A (88) and analogues, a group of tremorgenic mycotoxins identified in several Penicillium strains, including P. crustosum,127–130 were determined in 1981.131 Lolilline (89) and the tremorgenic lolitrems A (90) and B (91) were identified in Neotyphodium-infected plants Lolium perenne and Festuca sp.11,132,133 In the structures of the mentioned compounds, a C19 skeleton derived from a geranylgeranyl moiety is fused to positions C2 and C3 of the indole ring.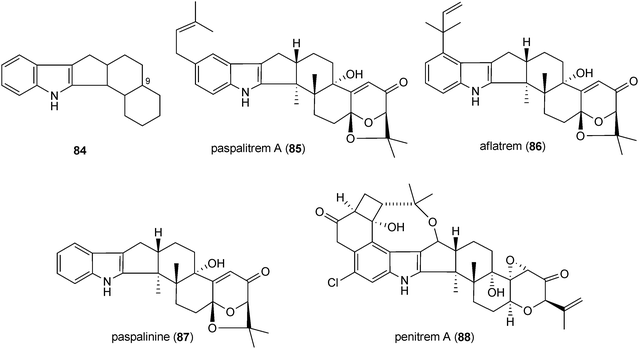
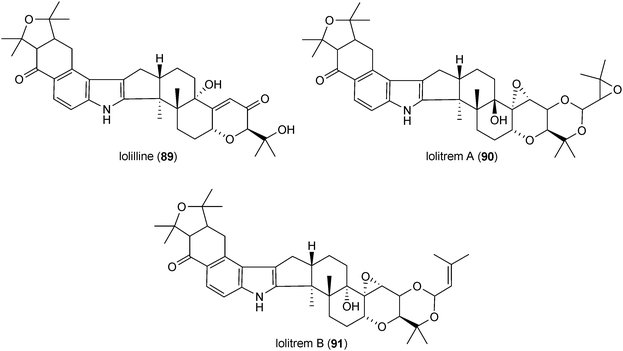
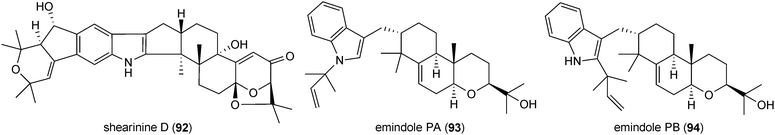
Similar structural features were also found in shearinines, e.g. shearinine D (92) from Penicillium strains.134,135 In comparison, indole-diterpenes with a C20 residue attached to indole moiety were also identified, e.g. emindoles PA (93) and PB (94).136 As paspalitrem A (85) and aflatrem (86), the other mentioned structures also carry additional intact or modified prenyl moieties at the indole rings. For example, emindoles PA (93) and PB (94) carry one prenyl moiety at N1 and C2, respectively, while two prenyl residues were found in lolilline (89), and lolitrems A (90) and B (91) at positions C4 and C5. Shearinine D (92) is diprenylated at C5 and C6. In the structures of lolitrem A (90) and B (91), an additional C5 unit is attached to the C19 residue.
In 1997, a potent insecticidal agent nodulisporic acid A (95) was identified in an endophytic fungus Nodulispororium sp. by bioassay-guided isolation.137 Its structure was determined as an indole-diterpene derivative carrying three modified dimethylallyl moieties at position C5, C6 and C7 of the indole ring.137,138 Since then, eighteen nontremorgenic nodulisporic acid derivatives have been isolated from Nodulispororium including the biosynthetic precursors of nodulisporic acid A such as nodulisporic acids B (96), C (97), D (98) and E (99).139–141 All of these compounds are potent anti-flea agents.141 Nodulisporic acids lack the tertiary hydroxyl group at C-9 of the structure (84) that is implicated in the tremorgenic properties of the related alkaloids.142
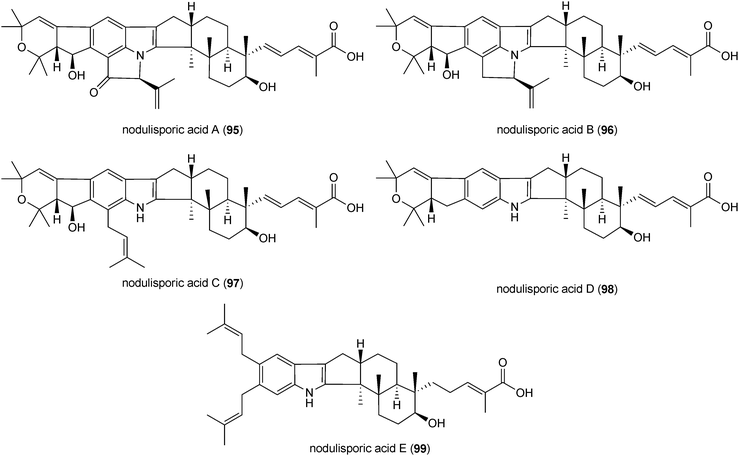
3 Biosynthesis
3.1 Feeding experiments and investigations with crude enzyme extracts
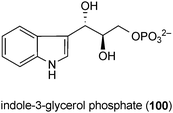
A large variety of feeding experiments with isotope-labelled precursors were carried out for ergot alkaloids in Claviceps,1,111 roquefortine C (31) in Penicillium roquerforti,143 aszonalenin (38) in Aspergillus zonatus,143 echinulin (42) in Aspergillus amstelodami,144 brevianamide A (10) in Penicillium brevicompactum, paraherquamide A (57) in Penicillium fellutanum3,34 and α-cyclopiazonic acid (81) in Penicillium cyclopium.145,146 The results of these experiments showed clearly that tryptophan and dimethylallyl diphosphate (DMAPP) serve as biosynthetic precursors of indole or indoline and prenyl moieties of these compounds, respectively.3 The prenyl moieties are derived from the acetate–mevalonate pathway. Incorporation of tryptophan and mevalonolactone into the alkaloid molecules has been clearly demonstrated. Furthermore, additional precursors such as S-adenosylmethionine for methyl groups or a second amino acid for diketopiperazine derivatives were also proven by feeding experiments. In addition, enzyme activities for a number of biosynthetic reactions were demonstrated with crude extracts or partially purified protein fractions. Several excellent reviews have summarized the results of feeding experiments on prenylated indole alkaloids and enzymatic characterisation of certain reactions.1,3,34,111,147 This work will therefore not be repeated in this review.It is worth mentioning the controversial discussion on the role of tryptophan as a biosynthetic precursor for indole-diterpenes.148 Feeding experiments in washed cells of a Nodulisporium mutant MF6244 showed clear incorporation of 2-13C-acetate and 2-13C-mevalonolactone into the indole-diterpene derivative nodulisporic acid A (95) and demonstrated the isoprene formation via the classical acetate–mevalonate pathway.142 Differing from the most prenylated indole alkaloids mentioned previously, the indole moiety of nodulisporic acid A (95) is most likely not derived from tryptophan, but from its precursor indole-3-glycerol phosphate (100). Incubations of Nodulisporium MF6244 with 14C- and 13C-tryptophan showed no incorporation of label into nodulisporic acid A (95). However, high levels of incorporation into nodulisporic acid A (95) were obtained with known tryptophan precursors such as 14C-, 13C-, and 15N-anthranilic acid as well as 14C- and 13C-ribose.142,148 These results are in contrast to those obtained for penitrem A (88) in Penicillium crustosum, which demonstrated a low incorporation of tryptophan into penitrem A (88).149
3.2 Identification of the biosynthetic gene clusters by genome mining and molecular biological and biochemical investigations on the biosynthesis of prenylated indole alkaloids
Significant progress has been achieved in the molecular biological and biochemical investigations on the biosynthesis of prenylated indole alkaloids in the last five years. In most cases, the biosynthetic genes were identified as a cluster in the genome sequences by bioinformatic approaches. Comparison of the orthologous gene clusters in different strains helps to identify a candidate gene for an enzymatic reaction, which can then be proven by molecular biological and biochemical characterisation.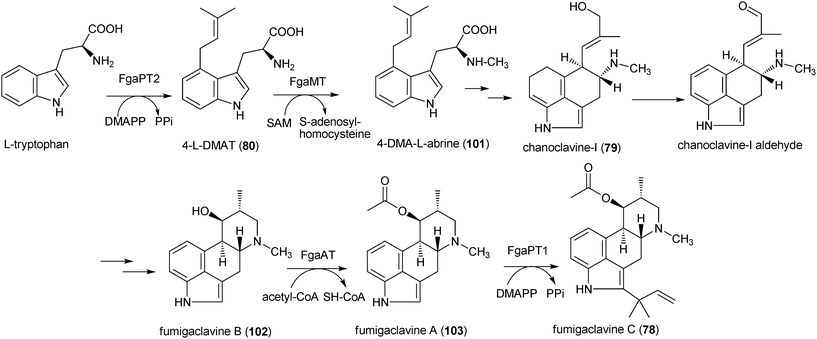 | ||
| Scheme 1 Proposed biosynthetic pathway for fumigaclavine C in Aspergillus fumigatus. | ||
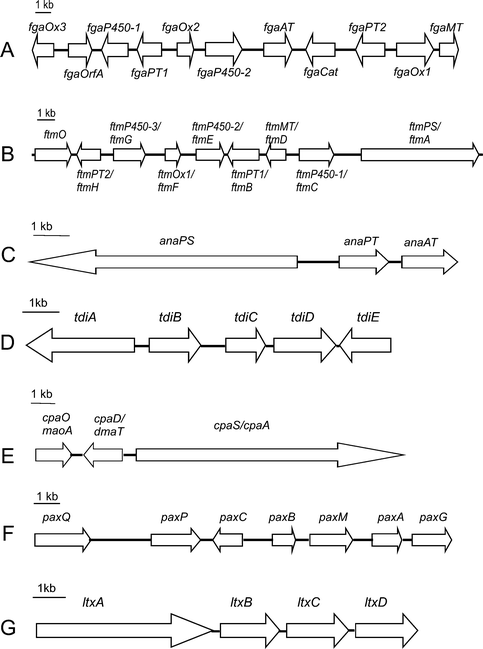 | ||
| Fig. 2 Biosynthetic gene clusters from different sources. A: fumigaclavine C cluster in Aspergillus fumigatus; B: fumitremorgin/verruculogen cluster in Aspergillus fumigatus; C: acetylaszonalenin cluster in Neosartorya fischeri; D: terrequinone A cluster in Aspergillus nidulans; E: α-cyclopiazonic acid cluster in Aspergillus flavus; F: paxilline cluster in Penicillium paxilli; G: lyngbyatoxin cluster in Lyngbya majuscula. | ||
 | ||
| Scheme 2 Biosynthetic pathway for verruculogen or fumitremorgin A in Aspergillus fumigatus. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 585–143349 of AAKE03000024.1 was identified in N. fischeri NRRL181 by homologous search and sequence analysis (Fig. 2C). An orthologous cluster was also identified in the genome sequence of Aspergillus terreus NIH2624.68 The putative genes anaPS, anaPT and anaAT in this cluster coding for the three putative enzymes mentioned above are likely involved in the biosynthesis of acetylaszonalenin (37).68AnaPS is predicted to encode a dimodular nonribosomal peptide synthetase (NRPS) with two adenylation domains (A), two peptidyl carrier domains (P) and two condensation domains (C), which is expected to be responsible for the condensation of the amino acids tryptophan and anthranilic acid (Scheme 3). AnaPT showed clear sequence similarity to indole prenyltransferases and was proven to be responsible for the reverse C3-prenylation of (R)-benzodiazopindinone (106) in the presence of DMAPP, resulting in formation of aszonalenin (38).68 These results provided evidence that AnaPT catalyses both the prenyl transfer reaction and the cyclisation between the indoline and the benzodiazepine ring system. By gene cloning and biochemical characterization of the recombinant gene product, it was shown that AnaAT catalyses the acetylation of aszonalenin (38), resulting in the formation of acetylaszonalenin (37) (Scheme 3).68 The biosynthetic pathway described for acetylaszonalenin (37) could also be plausible for other dipeptide derivatives with prenyl moieties at position C3 of the indoline rings, e.g. roquerfortine C (31), amauromines (24, 25), fructigenines (41, 53) and brevicompanine (54).62,68,87 This includes the formation of cyclic dipeptides with a diketopiperazine structure catalysed by NRPS, prenylation and cyclisation catalysed by a prenyltransferase, and acetylation (if necessary) by an acetyltransferase.
585–143349 of AAKE03000024.1 was identified in N. fischeri NRRL181 by homologous search and sequence analysis (Fig. 2C). An orthologous cluster was also identified in the genome sequence of Aspergillus terreus NIH2624.68 The putative genes anaPS, anaPT and anaAT in this cluster coding for the three putative enzymes mentioned above are likely involved in the biosynthesis of acetylaszonalenin (37).68AnaPS is predicted to encode a dimodular nonribosomal peptide synthetase (NRPS) with two adenylation domains (A), two peptidyl carrier domains (P) and two condensation domains (C), which is expected to be responsible for the condensation of the amino acids tryptophan and anthranilic acid (Scheme 3). AnaPT showed clear sequence similarity to indole prenyltransferases and was proven to be responsible for the reverse C3-prenylation of (R)-benzodiazopindinone (106) in the presence of DMAPP, resulting in formation of aszonalenin (38).68 These results provided evidence that AnaPT catalyses both the prenyl transfer reaction and the cyclisation between the indoline and the benzodiazepine ring system. By gene cloning and biochemical characterization of the recombinant gene product, it was shown that AnaAT catalyses the acetylation of aszonalenin (38), resulting in the formation of acetylaszonalenin (37) (Scheme 3).68 The biosynthetic pathway described for acetylaszonalenin (37) could also be plausible for other dipeptide derivatives with prenyl moieties at position C3 of the indoline rings, e.g. roquerfortine C (31), amauromines (24, 25), fructigenines (41, 53) and brevicompanine (54).62,68,87 This includes the formation of cyclic dipeptides with a diketopiperazine structure catalysed by NRPS, prenylation and cyclisation catalysed by a prenyltransferase, and acetylation (if necessary) by an acetyltransferase.
 | ||
| Scheme 3 Biosynthetic pathway for acetylaszonalenin in Neosartorya fischeri. | ||
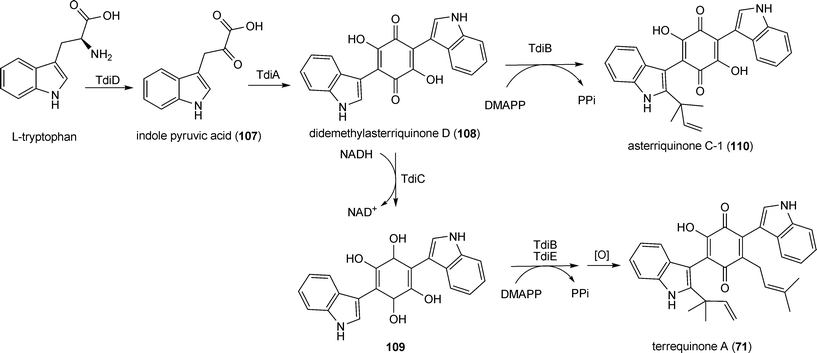 | ||
| Scheme 4 Biosynthetic pathway for terrequinone A in Aspergillus nidulans. | ||
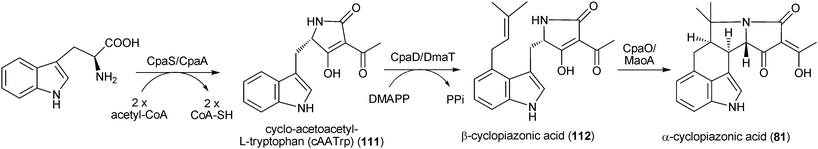 | ||
| Scheme 5 Proposed biosynthetic pathway for α-cyclopiazonic acid in Aspergillus flavus and Aspergillus oryzae. | ||
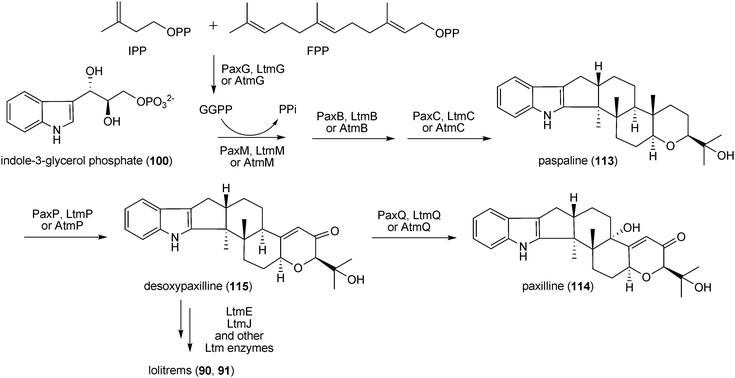 | ||
| Scheme 6 Proposed biosynthetic pathway for paxilline in Penicillium paxilli. | ||
A gene cluster containing homologues of paxG, paxM, and paxC, designated atmG, atmM, and atmC, respectively, was also identified in the aflatrem producer Aspergillus flavus NRRL 6541.175 Analysis of the genome sequences of A. flavus NRRL 3357 and Aspergillus oryzae RIB40 has revealed the presence of an aflatrem cluster similar to that of A. flavus NRRL 6541 mentioned above.11 These genomes also contain a second gene cluster at a separate chromosomal locus, which includes homologues of paxP, paxQ, paxA, paxB, and paxD.11 Thus, homologues of all seven genes necessary for paxilline (114) production in Penicillium paxilli are present in the genomes of A. flavus and A. oryzae, supporting the proposal that aflatrem production proceeds via paspaline (113) and paxilline (114) intermediates.11
4 Production of prenylated indole derivatives by chemoenzymatic synthesis
As discussed under section 2, many prenylated alkaloids are derived from the same amino acids. For example, tryprostatins (2, 4), fumitremorgins (6–8), brevianamides (10, 15), notoamides (12–14) and stephacidins (16, 19) are derivatives of cyclo-L-Trp-L-Pro. Cyclo-L-Trp-L-Trp is the common precursor of fellutanines (20–22), amauromines (24, 25) and okaramines (26–29). They differ from each other in prenylation position and manner (regular or reverse) as well as by further modifications. It can be expected that the respective cyclic dipeptide is formed by similar nonribosomal peptide synthetases in different producers, but diverse prenyltransferases contribute significantly to the large structure diversity within the prenylated indole alkaloids. The same conclusion is also applicable for asterriquinones and related compounds (61–71), which distinguish from each other by number and position of the prenyl moieties at the indole rings. Therefore, prenyl transfer reactions catalysed by prenyltransferases represent key steps in the biosynthesis of natural products. For this reason, prenyltransferases from different sources are important research topics in the field of secondary metabolism.9,176In recent years, ten indole prenyltransferase genes from various fungi were cloned and overexpressed in Escherichia coli or Saccharomyces cerevisiae.10,12 The overproduced proteins were proven to be soluble proteins and could be purified to near homogeneity by affinity chromatography in good yields.9 These prenyltransferases catalyse diverse prenyl transfer reactions onto different indole positions of various substrates.9,10 Investigations on substrate specificity of the characterised indole prenyltransferases revealed that these enzymes were specific for DMAPP as prenyl donor. Product formation was only observed with DMAPP, but not with geranyl diphosphate under the tested conditions.16,68,151,155,160,177,178 In contrast, they showed notable substrate flexibility towards their aromatic substrates.16,162,177–179 Diverse simple tryptophan derivatives (116) and tryptophan-containing cyclic dipeptides (117) were accepted by several prenyltransferases as substrates and converted to prenylated derivatives. For example, the two dimethylallyltryptophan synthases FgaPT2 and 7-DMATS, which catalyse the regular prenylation at positions C4 and C7 of the indole ring of L-tryptophan, respectively, accepted a series of simple tryptophan derivatives (116) with modifications at the side chain and at the indole nucleus as substrates.179,180 Notably, they also accepted tryptophan-containing cyclic dipeptides (117) as aromatic substrates.181 Conversely, the cyclic dipeptide prenyltransferases FtmPT1 and CdpNPT accepted not only tryptophan-containing cyclic dipeptides (117),16,162 but also simple indole derivatives (116) as substrates.182 More importantly, the prenyl transfer reactions catalysed by indole prenyltransferases are highly regiospecific.9,12 These features of substrate flexibility and reaction regiospecificity were successfully used for production of prenylated indole derivatives.12
By using chemoenzymatic synthesis with purified enzymes, it was possible to obtain three monoprenylated products from one simple indole derivative (116). The prenyltransferases FtmPT1 or CdpNPT, FgaPT2, and 7-DMATS converted the same substrate to derivatives with a prenyl moiety at position N1 (118), C4 (119) or C7 (120) of the indole ring, respectively (Scheme 7)179,180,182 By a tandem incubation with FgaPT2 and 7-DMATS, 4,7-diprenylated indole derivatives (121) have also been produced (Scheme 7).183
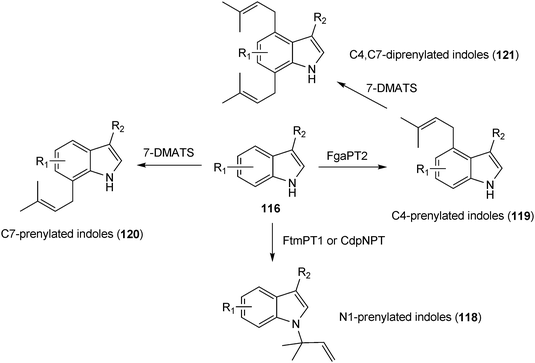 | ||
| Scheme 7 Chemoenzymatic synthesis of prenylated simple indole derivatives. | ||
By using the same strategy, at least five derivatives with prenyl moieties at N1 (122), C2 (123), C3 (124), C4 (125) and C7 (126) could be obtained from one tryptophan-containing cyclic dipeptide (Scheme 8). CdpNPT prenylates these compounds at position N1 of the indole ring, resulting in the formation of reversely prenylated derivatives (122).162 FtmPT1 is able to prenylate tryptophan-containing cyclic dipeptides at position C2 of the indole moiety.16,184 C3, C4 and C7-prenylated derivatives (124–126) could be obtained by using AnaPT, FgaPT2 or 7-DMATS, respectively.181 (Yin and Li, Kremer and Li, unpublished results)
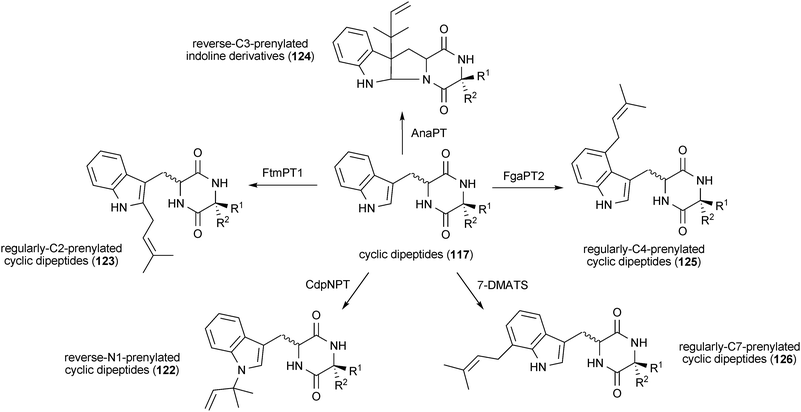 | ||
| Scheme 8 Chemoenzymatic synthesis of prenylated cyclic dipeptides. | ||
5 Prenylated indole alkaloids from other sources
Prenylated indole alkaloids are mainly, but not exclusively, found in fungi. They exist also in other domains of life such as bacteria, especially in actinomycetes and cyanobacteria, plants, as well as bryozoans.Lyngbyatoxin A (127) from the cyanobacterium Lyngbya majuscula is a derivative of indolactam-V (128), a cyclic dipeptide consisting of L-valine and L-tryptophan, which is connected via its C7 to a geranyl moiety.185 Derivatives of indolactam-V (128) such as the tumour promoters teleocidin B-4 (129), olivoretin A (130) and blastmycetin E (131) were identified in various actinomycetes such as Streptomyces mediocidicus, Streptoverticillium olivoreticuli and Streptoverticillium blastmycetium.186–190
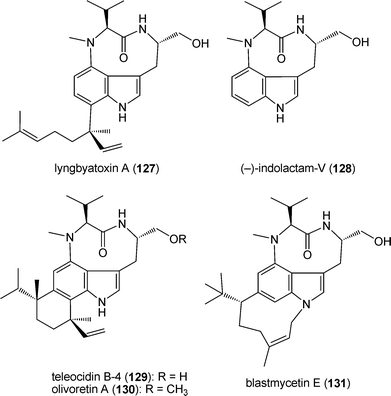
The biosynthesis of lyngbyatoxin A (127) was extensively studied by identification of a biosynthetic gene cluster191 and by biochemical investigation with an overproduced enzyme.192 The gene cluster (Fig. 2G) consists of four genes ltxABCD, which encode for a nonribosomal peptide synthetase, a cytochrome P450 monooxygenase, a prenyltransferase and an oxidoreductase, respectively.191 LtxA, containing additional N-methylation and reduction domains, catalyses the condensation of the amino acids L-valine and L-tryptophan, resulting in the formation of the linear dipeptide (132), which is then cyclised by LtxB to (−)-indolactam-V (128) (Scheme 9). Conversion of (−)-indolactam (128) to lyngbyatoxin A (127) would be catalysed by LtxC. LtxD would convert lyngbyatoxin A (127) to lyngbyatoxin B (133) (Scheme 9).191,192 In contrast, little is known about the biosynthesis of teleocidins, olivoretins or blastmycetins in actinomyces on the molecular biological and biochemical level.
 | ||
| Scheme 9 Proposed biosynthetic pathway for lynbyatoxins in Lynbya majuscula. | ||
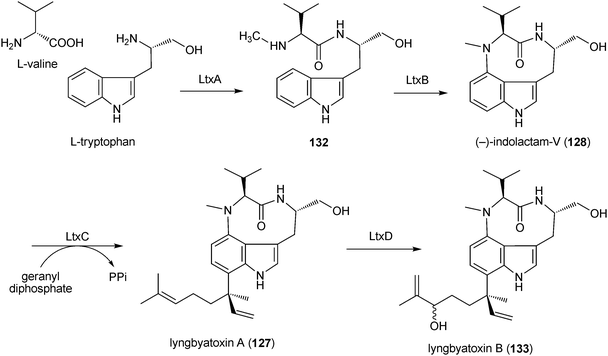
From the cyanobacterium Fischerella sp., nine antimicrobial isonitrile-containing indole alkaloids were isolated.193 These compounds, like ambiguine H isonitrile (134) and ambiguine F isonitrile (135), can be considered as tryptophan derivatives, which are connected to a reverse dimethylallyl and geranyl moiety at positions C2 and C4, respectively. Monoprenylated derivatives like 12-epi-hapalindole H (136) have also been reported.193
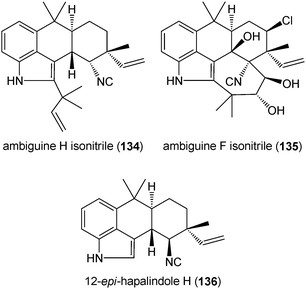
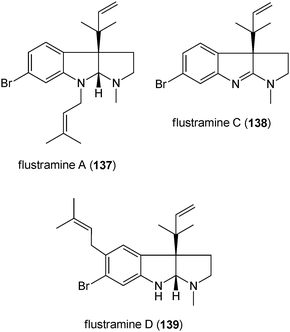
A number of simple prenylated indole derivatives were identified in the bryozoan Flustra foliacea.194–196 They are often brominated tryptamine derivatives with prenyl moieties at different positions of the indole ring like flustramines A (137), C (138) and D (139).197,198
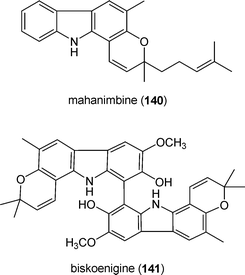
Prenylated indole derivatives occur also in higher plants. A large number of carbazole alkaloids have been isolated from higher plants of the genera Murraya, Glycosmis and Clausena, all belonging to the family Rutaceae.199 Two examples are mahanimbine (140) and biskoenigine (141) from Murraya koenigii.200,201
6 Conclusions
A large number of prenylated indole alkaloids with various structural features and important pharmacological activities have been identified in different sources, especially in filamentous fungi. Investigations on their biosynthesis in the last century have been mainly limited to feeding experiments with precursors and biochemical characterisation with crude extracts or partially purified enzymes from producers.3,111 Molecular biological and biochemical research with structural genes began ten years ago,202 but significant progress was achieved only after sequence release of genome sequencing projects for fungal strains, especially for Aspergillus strains in 2003/2004. The availability of more and more genome sequences provides an enormous chance for the study of secondary metabolites. Meanwhile, this is also a challenge for natural product research. Numerous genes and gene clusters with unknown functions have been identified in genome sequences,203 and functional proof of these genes and identification of the natural products coded by the gene clusters will be the main challenges for natural product chemists and biologists in the coming years or even decades. Therefore, it can be expected that more and more designed biologically-active substances will be produced by genetic manipulation.7 Acknowledgements
The work in the author's laboratory was supported by grants from the Deutsche Forschungsgemeinschaft.8 References
- C. L. Schardl, D. G. Panaccione and P. Tudzynski, Alkaloids: Chem. Biol., 2006, 63, 45–86 Search PubMed.
- H. Sings and S. Singh, Alkaloids: Chem. Biol., 2003, 60, 51–163 Search PubMed.
- R. M. Williams, E. M. Stocking and J. F. Sanz-Cervera, Top. Curr. Chem., 2000, 209, 97–173 CAS.
- M. G. Jones, Microbiology, 2007, 153, 1–6 CrossRef CAS.
- D. M. Raskin, R. Seshadri, S. U. Pukatzki and J. J. Mekalanos, Cell, 2006, 124, 703–714 CrossRef CAS.
- B. Wilkinson and J. Micklefield, Nat. Chem. Biol., 2007, 3, 379–386 CrossRef CAS.
- M. Zerikly and G. L. Challis, ChemBioChem, 2009, 10, 625–633 CrossRef CAS.
- C. Corre and G. L. Challis, Nat. Prod. Rep., 2009, 26, 977–986 RSC.
- N. Steffan, A. Grundmann, W.-B. Yin, A. Kremer and S.-M. Li, Curr. Med. Chem., 2009, 16, 218–231 CrossRef CAS.
- S.-M. Li, Phytochemistry, 2009, 70, 1746–1757 CrossRef CAS.
- S. Saikia, M. J. Nicholson, C. Young, E. J. Parker and B. Scott, Mycol. Res., 2008, 112, 184–199 Search PubMed.
- S.-M. Li, Appl. Microbiol. Biotechnol., 2009, 84, 631–639 CrossRef CAS.
- K. Higuchi and T. Kawasaki, Nat. Prod. Rep., 2007, 24, 843–868 RSC.
- M. Toyota and M. Ihara, Nat. Prod. Rep., 1998, 15, 327–340 RSC.
- K. Arai and Y. Yamamoto, Chem. Pharm. Bull., 1990, 38, 2929–2932 CAS.
- A. Grundmann and S.-M. Li, Microbiology, 2005, 151, 2199–2207 CrossRef CAS.
- H. Kato, T. Yoshida, T. Tokue, Y. Nojiri, H. Hirota, T. Ohta, R. M. Williams and S. Tsukamoto, Angew. Chem., Int. Ed., 2007, 46, 2254–2256 CrossRef CAS.
- S. Tsukamoto, H. Kato, M. Samizo, Y. Nojiri, H. Onuki, H. Hirota and T. Ohta, J. Nat. Prod., 2008, 71, 2064–2067 CrossRef CAS.
- M. Yamazaki, K. Sasago and K. Miyaki, J. Chem. Soc., Chem. Commun., 1974, 408–409 RSC.
- M. Yamazaki and S. Suzuki, Dev. Toxicol. Environ. Sci., 1986, 12, 273–282 Search PubMed.
- R. T. Gallagher and G. C. M. Latch, Appl. Environ. Microbiol., 1977, 33, 730–731 CAS.
- M. Sabater-Vilar, S. Nijmeijer and J. Fink-Gremmels, J. Food Prot., 2003, 66, 2123–2129 CAS.
- I. Kosalec, M. S. Klaric and S. Pepeljnjak, Acta Pharm., 2005, 55, 357–364 CAS.
- K. Khoufache, O. Puel, N. Loiseau, M. Delaforge, D. Rivollet, A. Coste, C. Cordonnier, E. Escudier, F. Botterel and S. Bretagne, BMC Microbiol., 2007, 7, 5 CrossRef.
- M. Yamazaki, H. Fujimoto and T. Kawasaki, Tetrahedron Lett., 1975, 16, 1241–1244 CrossRef.
- C. B. Cui, H. Kakeya, G. Okada, R. Onose, M. Ubukata, I. Takahashi, K. Isono and H. Osada, J. Antibiot., 1995, 48, 1382–1384 CAS.
- M. Kondoh, T. Usui, T. Mayumi and H. Osada, J. Antibiot., 1998, 51, 801–804 CAS.
- S. Zhao, K. S. Smith, A. M. Deveau, C. M. Dieckhaus, M. A. Johnson, T. L. Macdonald and J. M. Cook, J. Med. Chem., 2002, 45, 1559–1562 CrossRef CAS.
- S. K. Rabindran, D. D. Ross, L. A. Doyle, W. Yang and L. M. Greenberger, Cancer Res., 2000, 60, 47–50 CAS.
- J. D. Allen, A. van Loevezijn, J. M. Lakhai, d. van, V, O. van Tellingen, G. Reid, J. H. Schellens, G. J. Koomen and A. H. Schinkel, Mol. Cancer Ther., 2002, 1, 417–425 CAS.
- S. K. Rabindran, H. He, M. Singh, E. Brown, K. I. Collins, T. Annable and L. M. Greenberger, Cancer Res., 1998, 58, 5850–5858 CAS.
- H. D. Jain, C. Zhang, S. Zhou, H. Zhou, J. Ma, X. Liu, X. Liao, A. M. Deveau, C. M. Dieckhaus, M. A. Johnson, K. S. Smith, T. L. Macdonald, H. Kakeya, H. Osada and J. M. Cook, Bioorg. Med. Chem., 2008, 16, 4626–4651 CrossRef CAS.
- H. Woehlecke, H. Osada, A. Herrmann and H. Lage, Int. J. Cancer, 2003, 107, 721–728 CrossRef CAS.
- R. M. Williams and R. J. Cox, Acc. Chem. Res., 2003, 36, 127–139 CrossRef CAS.
- A. J. Birch and J. J. Wright, J. Chem. Soc. D, 1969, 644b–645 RSC.
- P. S. Steyn, Tetrahedron, 1973, 29, 107–120 CrossRef CAS.
- G.-Y. Li, T. Yang, Y.-G. Luo, X.-Z. Chen, D. M. Fang and G.-L. Zhang, Org. Lett., 2009, 11, 3714–3717 CrossRef CAS.
- S. Tsukamoto, T. Kawabata, H. Kato, T. J. Greshock, H. Hirota, T. Ohta and R. M. Williams, Org. Lett., 2009, 11, 1297–1300 CrossRef CAS.
- J. Qian-Cutrone, S. Huang, Y. Z. Shu, D. Vyas, C. Fairchild, A. Menendez, K. Krampitz, R. Dalterio, S. E. Klohr and Q. Gao, J. Am. Chem. Soc., 2002, 124, 14556–14557 CrossRef CAS.
- T. J. Greshock, A. W. Grubbs, S. Tsukamoto and R. M. Williams, Angew. Chem., Int. Ed., 2007, 46, 2262–2265 CrossRef.
- A. G. Kozlovsky, N. G. Vinokurova, V. M. Adanin, G. Burkhardt, H. M. Dahse and U. Gräfe, J. Nat. Prod., 2000, 63, 698–700 CrossRef CAS.
- B. Nuber, F. Hansske, C. Shinohara, S. Miura, K. Hasumi and A. Endo, J. Antibiot., 1994, 47, 168–172 CAS.
- S. Takase, Y. Kawai, I. Uchida, H. Tanaka and H. Aoki, Tetrahedron, 1985, 41, 3037–3048 CrossRef CAS.
- F. S. De Guzman and J. B. Glober, J. Nat. Prod., 1992, 55, 931–939 CrossRef CAS.
- H. Hayashi, K. Takiuchi, S. Murao and M. Arai, Agric. Biol. Chem., 1989, 53, 461–469 CAS.
- H. Hayashi, Y. Asabu, S. Murao and M. Arai, Biosci., Biotechnol., Biochem., 1995, 59, 246–250 CrossRef CAS.
- H. Hayashi, K. Furutsuka and Y. Shiono, J. Nat. Prod., 1999, 62, 315–317 CrossRef CAS.
- Y. Shiono, K. Akiyama and H. Hayashi, Biosci., Biotechnol., Biochem., 1999, 63, 1910–1920 CAS.
- Y. Shiono, K. Akiyama and H. Hayashi, Biosci., Biotechnol., Biochem., 2000, 64, 103–110 CAS.
- C. Shinohara, K. Hasumi, Y. Takei and A. Endo, J. Antibiot., 1994, 47, 163–167 CAS.
- S. Takase, Y. Kawai, I. Uchida, H. Tanaka and H. Aoki, Tetrahedron Lett., 1984, 25, 4673–4676 CrossRef CAS.
- H. Hayashi, Foods Food Ingredients J. Jpn., 2008, 213, 1079–1085 Search PubMed.
- P. M. Scott and B. P. C. Kennedy, J. Agric. Food Chem., 1976, 24, 865–868 CrossRef CAS.
- T. Rundberget, I. Skaar and A. Flaoyen, Int. J. Food Microbiol., 2004, 90, 181–188 CrossRef CAS.
- C. Finoli, A. Vecchio, A. Galli and I. Dragoni, J. Food Prot., 2001, 64, 246–251 CAS.
- M. O'Brien, K. F. Nielsen, P. O'Kiely, P. D. Forristal, H. T. Fuller and J. C. Frisvad, J. Agric. Food Chem., 2006, 54, 9268–9276 CrossRef CAS.
- J. C. Frisvad, J. Smedsgaard, T. O. Larsen and R. A. Samson, Stud. Mycol., 2004, 49, 201–241 Search PubMed.
- T. A. Reshetilova, N. G. Vinokurova, V. N. Khmelenina and A. G. Kozlovskii, Microbiology (Moscow), 1995, 64, 27–29 Search PubMed.
- P. S. Steyn and R. Vleggaar, J. Chem. Soc., Chem. Commun., 1983, 560–561 RSC.
- D. P. Overy, K. F. Nielsen and J. Smedsgaard, J. Chem. Ecol., 2005, 31, 2373–2390 CrossRef CAS.
- B. Clark, R. J. Capon, E. Lacey, S. Tennant and J. H. Gill, J. Nat. Prod., 2005, 68, 1661–1664 CrossRef CAS.
- L. Du, D. Li, T. Zhu, S. Cai, F. Wang, X. Xiao and Q. Gu, Tetrahedron, 2009, 65, 1033–1039 CrossRef CAS.
- G. A. Ellestad, P. Mirando and M. P. Kunstmann, J. Org. Chem., 1973, 38, 4204–4205 CrossRef CAS.
- Y. Kimura, T. Hamasaki, H. Nakajima and A. Isogai, Tetrahedron Lett., 1982, 23, 225–228 CrossRef.
- A. Hayashi, S. Fujioka, M. Nukina, T. Kawano, A. Shimada and Y. Kimura, Biosci., Biotechnol., Biochem., 2007, 71, 1697–1702 CrossRef CAS.
- R. J. Capon, C. Skene, M. Stewart, J. Ford, R. A. J. O'Hair, L. Williams, E. Lacey, J. H. Gill, K. Heiland and T. Friedel, Org. Biomol. Chem., 2003, 1, 1856–1862 RSC.
- D. Wakana, T. Hosoe, T. Itabashi, K. Nozawa, K. Okada, G. M. d. C. Takaki, T. Yaguchi, K. Fukushima and K. I. Kawai, Mycotoxins, 2006, 56, 3–6 CrossRef CAS.
- W.-B. Yin, A. Grundmann, J. Cheng and S.-M. Li, J. Biol. Chem., 2009, 284, 100–109 CAS.
- C. Rank, R. K. Phipps, P. Harris, J. C. Frisvad, C. H. Gotfredsen and T. O. Larsen, Tetrahedron Lett., 2006, 47, 6099–6102 CrossRef CAS.
- K. Arai, K. Kimura, T. Mushiroda and Y. Yamamoto, Chem. Pharm. Bull., 1989, 37, 2937–2939 CAS.
- A. G. Kozlovsky, V. M. Adanin, H. M. Dahse and U. Gräfe, Appl. Biochem. Microbiol., 2001, 37, 253–256 Search PubMed.
- A. J. Birch, G. E. Blance, S. David and H. Smith, J. Chem. Soc., 1961, 3128–3131 RSC.
- R. D. Stipanovic and H. W. Schroeder, Trans. Br. Mycol. Soc., 1976, 66, 178–179 Search PubMed.
- W. L. Wang, Z. Y. Lu, H. W. Tao, T. J. Zhu, Y. C. Fang, Q. Q. Gu and W. M. Zhu, J. Nat. Prod., 2007, 70, 1558–1564 CrossRef CAS.
- Y. Li, X. Li, J. S. Kang, H. D. Choi and B. W. Son, J. Antibiot., 2004, 57, 337–340 CAS.
- G. J. Slack, E. Puniani, J. C. Frisvad, R. A. Samson and J. D. Miller, Mycol. Res., 2009, 113, 480–490 Search PubMed.
- W. Zhao, Y. Guo, Y. Tezuka and T. Kikuchi, Zhongguo Zhongyao Zazhi, 1991, 16, 425–426 Search PubMed.
- S. K. Talapatra, S. K. Mandal, A. Bhaumik, S. Mukhopadhyay, P. Kar, A. Patra and B. Talapatra, J. Indian Chem. Soc., 2001, 78, 773–777 CAS.
- T. Itabashi, N. Matsuishi, T. Hosoe, N. Toyazaki, S. Udagawa, T. Imai, M. Adachi and K. Kawai, Chem. Pharm. Bull., 2006, 54, 1639–1641 CrossRef CAS.
- Y. Wang, J. B. Gloer, J. A. Scott and D. Malloch, J. Nat. Prod., 1995, 58, 93–99 CrossRef CAS.
- H. Fujimoto, T. Fujimaki, E. Okuyama and M. Yamazaki, Chem. Pharm. Bull., 1999, 47, 1426–1432 CAS.
- L. K. Hansen, F. C. Størmer, D. Petersen and A. J. Aasen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2001, 57, o909–o912 CrossRef.
- K. Arai, S. Sato, S. Shimizu, K. Nitta and Y. Yamamoto, Chem. Pharm. Bull., 1981, 29, 1510–1517 CAS.
- J. E. Hochlowski, M. M. Mullally, S. G. Spanton, D. N. Whittern, P. Hill and J. B. McAlpine, J. Antibiot., 1993, 46, 380–386 CAS.
- W. L. Wang, T. J. Zhu, H. W. Tao, Z. Y. Lu, Y. C. Fang, Q. Q. Gu and W. M. Zhu, Chem. Biodiversity, 2007, 4, 2913–2919 CrossRef CAS.
- R. P. Hodge, C. M. Harris and T. M. Harris, J. Nat. Prod., 1988, 51, 66–73 CrossRef CAS.
- L. Du, X. Yang, T. Zhu, F. Wang, X. Xiao, H. Park and Q. Gu, Chem. Pharm. Bull., 2009, 57, 873–876 CrossRef CAS.
- J. G. Ondeyka, A. W. Dombrowski, J. P. Polishook, T. Felcetto, W. L. Shoop, Z. Guan and S. B. Singh, J. Ind. Microbiol. Biotechnol., 2003, 30, 220–224 CAS.
- T. Kagamizono, N. Sakai, K. Arai, K. Kobinata and H. Osada, Tetrahedron Lett., 1997, 38, 1223–1226 CrossRef CAS.
- Y. Yamamoto, K. Nishimura and N. Kiriyama, Chem. Pharm. Bull., 1976, 24, 1853–1859 CAS.
- Y. Yamamoto, N. Kiriyama, S. Shimizu and S. Koshimura, Gann, 1976, 67, 623–624 CAS.
- K. Arai, K. Masuda, N. Kiriyama, K. Nitta, Y. Yamamoto and S. Shimizu, Chem. Pharm. Bull., 1981, 29, 961–969.
- K. Arai, S. Shimizu and Y. Yamamoto, Chem. Pharm. Bull., 1981, 29, 1005–1012 CAS.
- A. Kaji, T. Iwata, N. Kiriyama, S. Wakusawa and K. Miyamoto, Chem. Pharm. Bull., 1994, 42, 1682–1684 CAS.
- S. Sekita, Chem. Pharm. Bull., 1983, 31, 2998–3001 CAS.
- M. A. O'Leary and J. R. Hansen, Tetrahedron Lett., 1982, 23, 1855–1856 CrossRef CAS.
- J. He, E. M. K. Wijeratne, B. P. Bashyal, J. Zhan, C. J. Seliga, M. X. Liu, E. E. Pierson, L. S. Pierson, H. D. VanEtten and A. A. L. Gunatilaka, J. Nat. Prod., 2004, 67, 1985–1991 CrossRef CAS.
- M. Ooike, K. Nozawa, S. Udagawa and K. Kawai, Chem. Pharm. Bull., 1997, 45, 1694–1696 CAS.
- U. Mocek, L. Schultz, T. Buchan, C. Baek, L. Fretto, J. Nzerem, L. Sehl and U. Sinha, J. Antibiot., 1996, 49, 854–859 CAS.
- A. Fredenhagen, F. Petersen, M. Tintelnot-Blomley, J. Rosel, H. Mett and P. Hug, J. Antibiot., 1997, 50, 395–401 CAS.
- M. A. O'Leary, J. R. Hanson and B. L. Yeoh, J. Chem. Soc., Perkin Trans. 1, 1984, 567–569 RSC.
- F. S. De Guzman, D. R. Bruss, J. M. Rippentrop, K. B. Gloer and J. B. Gloer, J. Nat. Prod., 1994, 57, 634–639 CrossRef CAS.
- J. W. Bok, D. Hoffmeister, L. A. Maggio-Hall, R. Murillo, J. D. Glasner and N. P. Keller, Chem. Biol., 2006, 13, 31–37 CrossRef CAS.
- A. Kaji, R. Saito, M. Nomura, K. Miyamoto and N. Kiriyama, Anticancer Res., 1997, 17, 3675–3679 CAS.
- A. Kaji, R. Saito, M. Nomura, K. Miyamoto and N. Kiriyama, Biol. Pharm. Bull., 1998, 21, 945–949 CAS.
- A. Kaji, T. Iwata, N. Kiriyama, M. Nomura and K. Miyamoto, J. Antibiot., 1998, 51, 235–238 CAS.
- A. Kaji, K. Kimura, M. Teranishi, N. Kiriyama, M. Nomura and K. Miyamoto, Chem. Pharm. Bull., 1998, 46, 1325–1329 CAS.
- S. B. Singh, J. G. Ondeyka, N. Tsipouras, C. Ruby, V. Sardana, M. Schulman, M. Sanchez, F. Pelaez, M. W. Stahlhut, S. Munshi, D. B. Olsen and R. B. Lingham, Biochem. Biophys. Res. Commun., 2004, 324, 108–113 CrossRef CAS.
- M. Flieger, M. Wurst and R. Shelby, Folia Microbiol., 1997, 42, 3–30 Search PubMed.
- T. Haarmann, Y. Rolke, S. Giesbert and P. Tudzynski, Mol. Plant Pathol., 2009, 10, 563–577 CrossRef CAS.
- D. Gröger and H. G. Floss, Alkaloids: Chem. Biol., 1998, 50, 171–218 Search PubMed.
- R. Krska and C. Crews, Food Addit. Contam., Part A, 2008, 25, 722–731 Search PubMed.
- P. Tudzynski, T. Correia and U. Keller, Appl. Microbiol. Biotechnol., 2001, 57, 593–605 CrossRef CAS.
- K. Moncoq, C. A. Trieber and H. S. Young, J. Biol. Chem., 2007, 282, 9748–9757 CrossRef CAS.
- C. W. Holzapfel, R. D. Hutchison and D. C. Wilkins, Tetrahedron, 1970, 26, 5239–5246 CrossRef CAS.
- N. G. Vinokurova, N. E. Ivanushkina, I. I. Khmel'nitskaia and M. U. Arinbasarov, Appl. Biochem. Microbiol., 2007, 43, 435–438 Search PubMed.
- C. Finoli, A. Vecchio, A. Galli and L. Franzetti, J. Food Prot., 1999, 62, 1198–1202 CAS.
- K. Hermansen, J. C. Frisvad, C. Emborg and J. Hansen, FEMS Microbiol. Lett., 1984, 21, 253–261 CrossRef CAS.
- F. Nunez, C. D. Westphal, E. Bermudez and M. A. Asensio, J. Food Prot., 2007, 70, 2829–2836 CAS.
- A. S. Moldes-Anaya, T. N. Asp, G. S. Eriksen, I. Skaar and T. Rundberget, J. Chromatogr., A, 2009, 1216, 3812–3818 CrossRef CAS.
- M. Abe, S. Ohmomo, T. Ohashi and T. Tabuchi, Agric. Biol. Chem., 1969, 33, 469–471 CAS.
- R. J. Cole, J. W. Kirksey, S. M. Weinreb, P. Singh, D. Kim, J. Clardy and N. Eickman, Tetrahedron Lett., 1976, 17, 3849–3852 CrossRef.
- J. W. Dorner, R. J. Cole, R. Hill, D. Wicklow and R. H. Cox, Appl. Environ. Microbiol., 1980, 40, 685–687 CAS.
- R. J. Cole, J. W. Dorner, J. A. Lansden, R. H. Cox, C. Pape, B. Cunfer, S. S. Nicholson and D. M. Bedell, J. Agric. Food Chem., 1977, 25, 1197–1201 CrossRef CAS.
- R. J. Cole, J. W. Dorner, J. P. Springer and R. H. Cox, J. Agric. Food Chem., 1981, 29, 293–295 CrossRef CAS.
- R. T. Gallagher and B. J. Wilson, Mycopathologia, 1979, 66, 183–185 CrossRef CAS.
- R. E. Wagener, N. D. Davis and U. L. Diener, Appl. Environ. Microbiol., 1980, 39, 882–887 CAS.
- J. L. Richard and L. H. Arp, Mycopathologia, 1979, 67, 107–109 CAS.
- A. Ciegler, Mycopathologia, 1978, 65, 5–11 CrossRef CAS.
- T. Rundberget, I. Skaar, O. O'Brien and A. Flåoyen, Mycopathologia, 2004, 157, 349–357 CrossRef.
- A. E. De Jesus, P. S. Steyn, F. R. van Heerden, R. Vleggaar, P. L. Wessels and W. E. Hull, J. Chem. Soc., Chem. Commun., 1981, 289–291 RSC.
- R. T. Gallagher, E. P. White and P. H. Mortimer, N. Z. Vet. J., 1981, 29, 189–190 Search PubMed.
- R. X. Tan and W. X. Zou, Nat. Prod. Rep., 2001, 18, 448–459 RSC.
- O. F. Smetanina, A. I. Kalinovsky, Y. V. Khudyakova, M. V. Pivkin, P. S. Dmitrenok, S. N. Fedorov, H. Ji, J. Y. Kwak and T. A. Kuznetsova, J. Nat. Prod., 2007, 70, 906–909 CrossRef CAS.
- M. Xu, G. Gessner, I. Groth, C. Lange, A. Christner, T. Bruhn, Z. Deng, X. Li, S. H. Heinemann, S. Grabley, G. Bringmann, I. Sattler and W. Lin, Tetrahedron, 2007, 63, 435–444 CrossRef CAS.
- T. Hosoe, T. Itabashi, N. Kobayashi, S. I. Udagawa and K. I. Kawai, Chem. Pharm. Bull., 2006, 54, 185–187 CrossRef CAS.
- J. G. Ondeyka, G. L. Helms, O. D. Hensens, M. A. Goetz, D. L. Zink, A. Tsipouras, W. L. Shoop, L. Slayton, A. W. Dombrowski, J. D. Polishook, D. A. Ostlind, N. N. Tsou, R. G. Ball and S. B. Singh, J. Am. Chem. Soc., 1997, 119, 8809–8816 CrossRef CAS.
- D. A. Ostlind, T. Felcetto, A. Misura, J. Ondeyka, S. Smith, M. Goetz, W. Shoop and W. Mickle, Med. Vet. Entomol., 1997, 11, 407–408 CrossRef CAS.
- J. G. Ondeyka, A. M. Dahl-Roshak, J. S. Tkacz, D. L. Zink, M. Zakson-Aiken, W. L. Shoop, M. A. Goetz and S. B. Singh, Bioorg. Med. Chem. Lett., 2002, 12, 2941–2944 CrossRef CAS.
- J. G. Ondeyka, K. Byrne, D. Vesey, D. L. Zink, W. L. Shoop, M. A. Goetz and S. B. Singh, J. Nat. Prod., 2003, 66, 121–124 CrossRef CAS.
- S. B. Singh, J. G. Ondeyka, H. Jayasuriya, D. L. Zink, S. N. Ha, A. Dahl-Roshak, J. Greene, J. A. Kim, M. M. Smith, W. Shoop and J. S. Tkacz, J. Nat. Prod., 2004, 67, 1496–1506 CrossRef CAS.
- K. M. Byrne, S. K. Smith and J. G. Ondeyka, J. Am. Chem. Soc., 2002, 124, 7055–7060 CrossRef CAS.
- B. Bhat, D. M. Harrison and H. M. Lamont, Tetrahedron, 1993, 49, 10663–10668 CrossRef CAS.
- R. Marchelli, A. Dossena and G. Casnati, J. Chem. Soc., Chem. Commun., 1975, 779–780 RSC.
- C. W. Holzapfel and D. C. Wilkins, Phytochemistry, 1971, 10, 351–358 CrossRef CAS.
- R. M. McGrath, P. S. Steyn, N. P. Ferreira and D. C. Neethling, Bioorg. Chem., 1976, 5, 11–23 CrossRef CAS.
- E. M. Stocking and R. M. Williams, Angew. Chem., Int. Ed., 2003, 42, 3078–3115 CrossRef CAS.
- P. G. Mantle, Phytochemistry, 2009, 70, 7–10 CrossRef CAS.
- A. E. De Jesus, C. P. Gorst-Allman, P. S. Steyn, F. R. van Heerden, R. Vleggaar, P. L. Wessels and W. E. Hull, J. Chem. Soc., Perkin Trans. 1, 1983, 1863–1868 RSC.
- O. Rigbers and S.-M. Li, J. Biol. Chem., 2008, 283, 26859–26868 CrossRef CAS.
- I. A. Unsöld and S.-M. Li, Microbiology, 2005, 151, 1499–1505 CrossRef.
- C. M. Coyle, S. C. Kenaley, W. R. Rittenour and D. G. Panaccione, Mycologia, 2007, 99, 804–811 Search PubMed.
- U. Metzger, C. Schall, G. Zocher, I. Unsold, E. Stec, S.-M. Li, L. Heide and T. Stehle, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14309–14314 CrossRef CAS.
- X. Liu, L. Wang, N. Steffan, W.-B. Yin and S.-M. Li, ChemBioChem, 2009, 10, 2325–2328 CrossRef CAS.
- I. A. Unsöld and S.-M. Li, ChemBioChem, 2006, 7, 158–164 CrossRef.
- N. Steffan, A. Grundmann, A. Afiyatullov, H. Ruan and S.-M. Li, Org. Biomol. Chem., 2009, 7, 4082–4087 RSC.
- S. Maiya, A. Grundmann, S.-M. Li and G. Turner, ChemBioChem, 2006, 7, 1062–1069 CrossRef CAS.
- C. B. Cui, H. Kakeya and H. Osada, J. Antibiot., 1996, 49, 534–540 CAS.
- N. Kato, H. Suzuki, H. Takagi, Y. Asami, H. Kakeya, M. Uramoto, T. Usui, S. Takahashi, Y. Sugimoto and H. Osada, ChemBioChem, 2009, 10, 920–928 CrossRef CAS.
- A. Grundmann, T. Kuznetsova, S. S. Afiyatullov and S.-M. Li, ChemBioChem, 2008, 9, 2059–2063 CrossRef CAS.
- C. J. Balibar and C. T. Walsh, Biochemistry, 2006, 45, 15029–15038 CrossRef CAS.
- H.-L. Ruan, W.-B. Yin, J.-Z. Wu and S.-M. Li, ChemBioChem, 2008, 9, 1044–1047 CrossRef CAS.
- C. J. Balibar, A. R. Howard-Jones and C. T. Walsh, Nat. Chem. Biol., 2007, 3, 584–592 CrossRef CAS.
- P. Schneider, M. Weber and D. Hoffmeister, Fungal Genet. Biol., 2008, 45, 302–309 CrossRef CAS.
- M. Tokuoka, Y. Seshime, I. Fujii, K. Kitamoto, T. Takahashi and Y. Koyama, Fungal Genet. Biol., 2008, 45, 1608–1615 CrossRef CAS.
- P. K. Chang, B. W. Horn and J. W. Dorner, Fungal Genet. Biol., 2009, 46, 176–182 CrossRef CAS.
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS.
- Y. Seshime, P. R. Juvvadi, M. Tokuoka, Y. Koyama, K. Kitamoto, Y. Ebizuka and I. Fujii, Bioorg. Med. Chem. Lett., 2009, 19, 3288–3292 CrossRef CAS.
- C. Young, L. McMillan, E. Telfer and B. Scott, Mol. Microbiol., 2001, 39, 754–764 CrossRef CAS.
- S. Saikia, E. J. Parker, A. Koulman and B. Scott, FEBS Lett., 2006, 580, 1625–1630 CrossRef CAS.
- S. Saikia, E. J. Parker, A. Koulman and B. Scott, J. Biol. Chem., 2007, 282, 16829–16837 CrossRef CAS.
- C. A. Young, S. Felitti, K. Shields, G. Spangenberg, R. D. Johnson, G. T. Bryan, S. Saikia and B. Scott, Fungal Genet. Biol., 2006, 43, 679–693 CrossRef CAS.
- C. A. Young, M. K. Bryant, M. J. Christensen, B. A. Tapper, G. T. Bryan and B. Scott, Mol. Genet. Genomics, 2005, 274, 13–29 CrossRef.
- C. A. Young, B. A. Tapper, K. May, C. D. Moon, C. L. Schardl and B. Scott, Appl. Environ. Microbiol., 2009, 75, 2200–2211 CrossRef CAS.
- S. Zhang, B. J. Monahan, J. S. Tkacz and B. Scott, Appl. Environ. Microbiol., 2004, 70, 6875–6883 CrossRef CAS.
- L. Heide, Curr. Opin. Chem. Biol., 2009, 13, 171–179 CrossRef CAS.
- W.-B. Yin, H.-L. Ruan, L. Westrich, A. Grundmann and S.-M. Li, ChemBioChem, 2007, 8, 1154–1161 CrossRef CAS.
- A. Kremer, L. Westrich and S.-M. Li, Microbiology, 2007, 153, 3409–3416 CrossRef CAS.
- N. Steffan, I. A. Unsöld and S.-M. Li, ChemBioChem, 2007, 8, 1298–1307 CrossRef CAS.
- A. Kremer and S.-M. Li, Appl. Microbiol. Biotechnol., 2008, 79, 951–961 CrossRef CAS.
- N. Steffan and S.-M. Li, Arch. Microbiol., 2009, 191, 461–466 CrossRef CAS.
- H. Zou, X. Zheng and S.-M. Li, J. Nat. Prod., 2009, 72, 44–52 CrossRef CAS.
- H.-L. Ruan, E. Stec and S.-M. Li, Arch. Microbiol., 2009, 191, 791–795 CrossRef CAS.
- L. Wang, W.-B. Yin, S.-M. Li and X.-Q. Liu, Chin. J. Biochem. Mol. Biol., 2009, 25, 580–584 Search PubMed.
- M. E. van Apeldoorn, H. P. van Egmond, G. J. Speijers and G. J. Bakker, Mol. Nutr. Food Res., 2007, 51, 7–60 CrossRef CAS.
- Y. Hitotsuyanagi, H. Fujiki, M. Suganuma, N. Aimi, S. Sakai, Y. Endo, K. Shudo and T. Sugimura, Chem. Pharm. Bull., 1984, 32, 4233–4236 CAS.
- S. Sakai, N. Aimi, K. Yamaguchi, Y. Hitotsuyanagi, C. Watanabe, K. Yokose, Y. Koyama, K. Shudo and A. Itai, Chem. Pharm. Bull., 1984, 32, 354–357 CAS.
- K. Irie, A. Funaki, K. Koshimizu, H. Hayashi and M. Arai, Tetrahedron Lett., 1989, 30, 2113–2116 CrossRef CAS.
- K. Irie and K. Koshimizu, Comments Agric. Food Chem., 1993, 3, 1–25 Search PubMed.
- K. Irie, S. Kajiyama, A. Funaki, K. Koshimizu, H. Hayashi and M. Arai, Tetrahedron, 1990, 46, 2773–2788 CrossRef CAS.
- D. J. Edwards and W. H. Gerwick, J. Am. Chem. Soc., 2004, 126, 11432–11433 CrossRef CAS.
- J. A. Read and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 15762–15763 CrossRef CAS.
- A. Raveh and S. Carmeli, J. Nat. Prod., 2007, 70, 196–201 CrossRef CAS.
- J. S. Carlé and C. Christophersen, J. Am. Chem. Soc., 1979, 101, 4012–4013 CrossRef CAS.
- L. Peters, G. M. König, A. D. Wright, R. Pukall, E. Stackebrandt, L. Eberl and K. Riedel, Appl. Environ. Microbiol., 2003, 69, 3469–3475 CrossRef CAS.
- L. Peters, A. D. Wright, S. Kehraus, D. Gundisch, M. C. Tilotta and G. M. König, Planta Med., 2004, 70, 883–886 CrossRef CAS.
- L. Peters, G. M. Konig, H. Terlau and A. D. Wright, J. Nat. Prod., 2002, 65, 1633–1637 CrossRef CAS.
- N. Lysek, E. Rachor and T. Lindel, Z. Naturforsch., C: J. Biosci., 2002, 57, 1056–1061 Search PubMed.
- H. J. Knölker and K. R. Reddy, Alkaloids: Chem. Biol., 2008, 65, 1–410 Search PubMed.
- Y. S. Wang, H. P. He, Y. M. Shen, X. Hong and X. J. Hao, J. Nat. Prod., 2003, 66, 416–418 CrossRef CAS.
- R. S. Ramsewak, M. G. Nair, G. M. Strasburg, D. L. DeWitt and J. L. Nitiss, J. Agric. Food Chem., 1999, 47, 444–447 CrossRef CAS.
- P. Tudzynski, K. Holter, T. Correia, C. Arntz, N. Grammel and U. Keller, Mol. Gen. Genet., 1999, 261, 133–141 CrossRef CAS.
- T. E. Cleveland, J. Yu, N. Fedorova, D. Bhatnagar, G. A. Payne, W. C. Nierman and J. W. Bennett, Trends Biotechnol., 2009, 27, 151–157 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |