Mechanically induced reactions in organic solids: liquid eutectics or solid-state processes?†
Oleksandr
Dolotko
a,
Jerzy W.
Wiench
a,
Kevin W.
Dennis
a,
Vitalij K.
Pecharsky
*a and
Viktor P.
Balema
*b
aThe Ames Laboratory, U.S. Department of Energy (OD JWW VKP) and Department of Materials Science and Engineering (VKP), Iowa State University, Ames, Iowa 50011-3020, USA. E-mail: vitkp@ameslab.gov; Fax: +1 (515) 294-9579
bAldrich Materials Science, Sigma-Aldrich Corporation, 6000 N. Teutonia Avenue, Milwaukee, WI 53209, USA. E-mail: vbalema@sial.com; Fax: +1 (414) 438-4224
First published on 24th November 2009
Abstract
The solvent-free reaction between o-vanillin and p-toluidine was investigated using NMR, DSC and XRD analyses. At room temperature, o-vanillin and p-toluidine react in a liquid eutectic formed upon grinding, while below 10 °C the same materials appear to react without the formation of a liquid phase, which most likely remains hidden behind solid reactants and reaction products.
Solvent-free chemical transformations are commonly known as mechanochemical reactions, or mechanochemistry, which reflects a crucial role of the mechanical processing in the process.1 Usually, mechanochemical reactions are carried out by grinding solid reactants using a mortar and a pestle,1 or by milling them in commercially available ball-mills.2 Products of such transformations often form in high yields, and require little or no purification.3,4 The mechanochemical approach has been successfully used to perform both stoichiometric and catalytic solvent-free reactions, as well as to carry out multi-step transformations as one-pot processes.5–10 As a rule, mechanochemistry significantly reduces the waste stream of hazardous materials and demonstrates an excellent atom economy,5 thus, satisfying major requirements of green chemistry.11
Despite recent advancements of the preparative mechanochemistry, understanding of physical and chemical events that take place in organic solids during mechanical processing remains limited. Often, it is even not apparent in which phase (liquid or solid) mechanochemical reactions really occur. Initially, mechanically induced transformations of solid reactants into solid reaction products were described as solid-state processes.1,3 However, Rothenberg et al. discovered that in some cases mechanically facilitated chemical transformations may proceed through an intermediate formation of short-lived liquid phases.12 The same group even hypothesized that all mechanochemical organic reactions might take place in low melting eutectics formed upon mechanical treatment of starting materials. However, later they admitted that similar mechanochemical reactions may occur in both liquid and solid phases,13 thus leaving the question about the true mechanism of mechanochemical transformations open. Although the formation of low melting eutectics during mechanochemical processing of organic solids was shown in model experiments,9,12 no direct evidence that an eutectic might be an essential element of a mechanochemical reaction has been provided to date. Furthermore, the vast majority of the known mechanochemical organic reactions are multi-step processes, which may generate liquid intermediates. An example is the condensation of o-vanillin (further abbreviated as oV) and p-toluidine (further abbreviated as pT); a two-step process that proceeds through a potentially liquid, unstable alcohol 1 (Fig. 1). When performed under solvent-free mechanochemical conditions at room temperature, this reaction produces an orange liquid that quickly crystallizes yielding the solid azomethine 2.12 The reaction had been originally reported by Toda et al. as a solid-state process,1 and the intermediate melting during the mechanical processing was noticed only a few years later.12
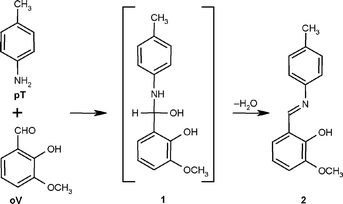 | ||
| Fig. 1 The reaction between o-vanillin (oV) and p-toluidine (pT). | ||
According to the literature,12 the life time of the liquid formed during grinding oV and pT might be long enough to analyze it using direct analytical techniques. Therefore, we revisited the reaction between o-vanillin and p-toluidine. Below, we report the results of our studies.
Our experiments have confirmed the formation of an orange liquid immediately after oV and pT are ground together at room temperature. The liquid also forms if both compounds are ground separately using a mortar and a pestle, and then thoroughly mixed together at room temperature. On the other hand, no phase or color changes occur if the materials are ground at 0 °C for 5 min. The prepared powder, however, turns orange and melts once it is quickly warmed up to room temperature. The melt that forms remains liquid for several minutes, i.e. long enough to be studied by liquid-state 1H and 13C NMR techniques.
For NMR experiments, the mixture of oV and pT (refrigerated in liquid nitrogen) was quickly placed into the NMR probe (pre-heated to 25 °C) and a series of 1H NMR spectra were recorded every 4 s. (Fig. 2, see also ESI†).
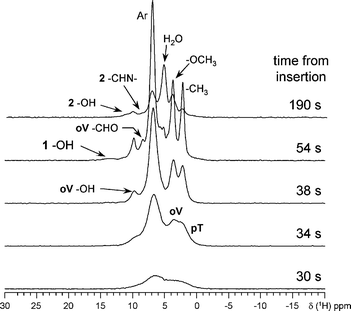 | ||
| Fig. 2 Selected 1H spectra of the mixture of oV and pT ground at 0 °C for 5 min and quickly warmed up to room temperature in the probe. The melting of the sample becomes apparent after 34 s at room temperature. | ||
Full width at half maximum (FWHM) of the broadest resonances in these spectra does not exceed 2 kHz (∼5 ppm), which is one order of magnitude lower than the line width expected for rigid solid samples.14 This indicates that the spectra represent a predominantly liquid material. In the first spectrum, recorded 30 s after the insertion of the sample into the probe, broad resonances are observed representing partially mobile species. As time progresses, the spectral resolution increases (i.e. FWHMs decrease) reaching its maximum at 54 s and then decreases again in subsequent spectra. This clearly indicates liquefying of the mixture followed by the condensation reaction and precipitation of the product 2. The resonances corresponding to –CH3 (2.2 ppm), –OCH3 (3.7 ppm), water (4.8 ppm), aromatic (Ar, ∼7 ppm), –CHO (8.6 ppm), –CHN– (8.5 ppm), and –OH (10.0 ppm in oV, ∼11.0 in 2, and ∼13.8 ppm in 1) groups in oV, pT, 1, and 2, are observed at the expected locations.
As time progresses, integrated intensities of peaks corresponding to the protons attached to carbon nuclei (–CH3, Ar, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) quickly increase up to ∼80% of the expected intensity at 54 s and then decrease down to ∼20% at ∼170 s. The resonance signal from the water is first observed in the spectrum taken after 46 s and continues to grow until the reaction comes to an end (for details see ESI†). At the same time, the resonance at ∼8.6 ppm shifts to 8.5 ppm as –CHO transforms into –CHN–. Finally, the signal of the –OH group shifts to the lower field by ∼1 ppm, when the adjacent aldehyde group transforms into an imine group. In addition, a broad, short lived peak at ∼13.8 ppm is present in the spectra recorded between 50 and 64 s from the insertion. This resonance most likely corresponds to the hydroxyl group at the C7 atom of the intermediate 1, which is present in the reaction mixture in minor quantities due to its instability. At the same time, this peak may also represent the overlapped signals from the –OH group at the C2 atoms in 1 and 2.15 For 13C direct polarization (DP) spectrum, a fresh cold ground mixture of oV and pT was quickly loaded into the probe. The spectrum was acquired for 6 min beginning at 40 s after the insertion (see Fig. 3a). 13C DP spectra for 2, pT, and oV samples in acetone solution were also recorded, compared with literature data (oV,16pT,17 and 215), and used as references (Fig. 3b–d). The spectrum (a) clearly demonstrates the presence of starting materials (oV and pT) and a small quantity of the final product 2 in the reaction mixture (evident from the resonances positions corresponding to C7 in oV and 2, C3 and C12 in 2, and C10 in pT). oV, pT and the azomethine 2 are present as major components in the reaction mixture throughout the entire reaction. The line widths for the resonances representing these materials gradually decrease as oV and pT melt and increase due to precipitation of the azomethine 2. Although trace amounts of the intermediate amido-alcohol 1 could also be detected, its quantities present in the reaction mixture were close to detection limits. No other substances or phases could be detected.
quickly increase up to ∼80% of the expected intensity at 54 s and then decrease down to ∼20% at ∼170 s. The resonance signal from the water is first observed in the spectrum taken after 46 s and continues to grow until the reaction comes to an end (for details see ESI†). At the same time, the resonance at ∼8.6 ppm shifts to 8.5 ppm as –CHO transforms into –CHN–. Finally, the signal of the –OH group shifts to the lower field by ∼1 ppm, when the adjacent aldehyde group transforms into an imine group. In addition, a broad, short lived peak at ∼13.8 ppm is present in the spectra recorded between 50 and 64 s from the insertion. This resonance most likely corresponds to the hydroxyl group at the C7 atom of the intermediate 1, which is present in the reaction mixture in minor quantities due to its instability. At the same time, this peak may also represent the overlapped signals from the –OH group at the C2 atoms in 1 and 2.15 For 13C direct polarization (DP) spectrum, a fresh cold ground mixture of oV and pT was quickly loaded into the probe. The spectrum was acquired for 6 min beginning at 40 s after the insertion (see Fig. 3a). 13C DP spectra for 2, pT, and oV samples in acetone solution were also recorded, compared with literature data (oV,16pT,17 and 215), and used as references (Fig. 3b–d). The spectrum (a) clearly demonstrates the presence of starting materials (oV and pT) and a small quantity of the final product 2 in the reaction mixture (evident from the resonances positions corresponding to C7 in oV and 2, C3 and C12 in 2, and C10 in pT). oV, pT and the azomethine 2 are present as major components in the reaction mixture throughout the entire reaction. The line widths for the resonances representing these materials gradually decrease as oV and pT melt and increase due to precipitation of the azomethine 2. Although trace amounts of the intermediate amido-alcohol 1 could also be detected, its quantities present in the reaction mixture were close to detection limits. No other substances or phases could be detected.
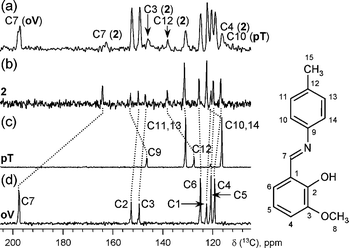 | ||
| Fig. 3 13C spectra recorded for (a) reaction mixture, (b) product 2, (c) pT, and (d) oV. For simplicity, only the aromatic region is shown. Signal assignment is indicated in the right hand inset. | ||
DSC experiments (Fig. 4a) have also confirmed the melting of the oV–pT mixture (1 : 1) just below room temperature. The onset of the endothermic event, corresponding to the melting of the material, is observed at 11 °C. The endothermic event reaches its maximum at ∼20 °C and is followed by/overlaps with an exothermic event, which peaks at ∼27 °C and slowly diminishes by ∼45 °C. Since thermal events corresponding to melting of free oV and pT are not present in the DSC trace, it is logical to conclude that this exothermic event is caused by the reaction leading to 2.
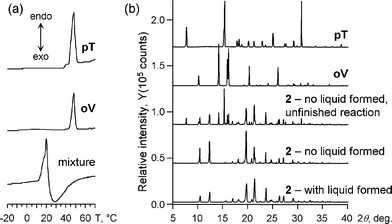 | ||
| Fig. 4 (a) DSC spectra of pT, oV, and physical mixture. (b) X-Ray powder diffraction patterns of pT, oV, and the product 2 (see text for details). | ||
Thus, our experiments confirm the hypothesis12 that the liquid formed upon grinding of oV and pT at room temperature is indeed a low melting eutectic, and these compounds react with one another in the liquid phase. So, was the earlier report by Toda et al.,1 who described their reaction as a solid-state process, a result of an unfortunate error? Not quite. Our studies also revealed facts that may shed light on this and some other contradictions12,13 concerning the nature of the mechanically induced reactions of organic solids.
It turned out that if the mixture of oV and pT is ground at ∼0 °C and kept between 5 and 10 °C for at least 2 min, a gradual color change occurs without noticeable formation of a liquid. Also, no melting of the material is observed when the sample temperature is further increased to room temperature. The X-ray powder diffraction analysis (XRD) of the obtained orange solid, revealed the formation of 2 in the reaction that appeared to be a solid-state process (Fig. 4b). Furthermore, the grinding of oV and pT at the temperature between 0° and 10 °C for one hour has a similar effect. The material turns orange upon grinding, and warming up to room temperature does not cause melting of the sample. According to XRD, the solid formed is identical to the azomethine 2. Remarkably, once at room temperature, the powder still contains some residual amount of the starting materials, which disappear after one hour at room temperature.
As a result of our experiments, we obtained the direct experimental evidence that mechanical processing of organic solids can produce liquid eutectics and create conditions for liquid-state reactions even if the reactants and the reaction products are solids. Under certain circumstances, for example at low temperatures, same reaction can appear to proceed as a solid-state process with no visible formation of a liquid phase. It can also take place in a self-propagating mode after the mechanical processing is terminated. Most likely, the heat generated during the reaction is a major factor promoting such self-propagating reactions.1,3,18
We also found that both the liquid-state and the liquid-less reactions between oV and pT produce the same reaction product—the azomethine 2. They are accompanied by similar color changes and the “solid-state” reaction is as efficient as the liquid-state process. In other words, appearance, i.e. the presence or the absence of a visible liquid phase, constitutes the major difference between the two processes. The fact that the liquid is not observed during mechanical processing of oV and pT below 10 °C does not necessarily mean that the liquid eutectic is not forming during the process. It may initially form in limited quantities in “hot-spot” areas, where materials particles interact with each other, and remain hidden behind the solid reactants and the reaction product throughout the entire process. The crystallization rate of the reaction product 2 may also affect the appearance of the reaction mixture. The fast crystallization of 2 would create an additional surface capable of absorbing the liquid eutectic and hiding its presence in the material.
Our results have revealed a distinct possibility that mechanochemical reactions, which appear to be true solid-state processes,1,3 in fact occur in a liquid phase. The major condition for such reactions is the formation of low-melting eutectics from starting materials upon mechanical processing. The mechanical processing is an essential element of mechanochemical reactions since it enhances surface interactions between reacting materials, supports their intimate mixing, provides mass transfer and initial energy required for the formation of low-melting eutectics.19,20
Work at the Ames Laboratory is supported by the Office of Basic Energy Sciences of the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-07CH11358 with Iowa State University.
Experimental
The starting materials o-vanillin (oV, 99 wt.% purity) and p-toluidine (pT, 99 wt.% purity) were purchased from Sigma-Aldrich Co. and used without a further purification. A 1 : 1 mixture of oV and pT was ground in an agate mortar at ambient or low temperature. The reaction products have been identified by comparison with the literature data12, see ESI† for further details.NMR: Varian Infinity 400 (400.0 MHz for 1H and 100.6 MHz for 13C, TMS as a reference). Sample preparation: oV and pT, a spatula, 5 mm glass NMR tubes, an agate mortar and pestle were cooled down to ∼0 °C. The cold mixture of oV and pT was ground in the mortar, than quickly transferred into a NMR tube, sealed, and placed in liquid nitrogen.†
XRD: PANalytical powder diffractometer (Cu-Kα1, 2θ range 5–70°, 0.02° step).†
DSC: Perkin Elmer “Pyris 1” (heating rate of 10 °C min−1).†
References
- K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS.
- V. P. Balema, Mater. Matters, 2007, 2, 16 Search PubMed.
- G. Kaupp, Top. Curr. Chem., 2005, 254, 95 CAS.
- V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 724 RSC.
- V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, J. Am. Chem. Soc., 2002, 124, 6244 CrossRef CAS.
- Z. Zhang, Y.-W. Dong, G.-W. Wang and K. Komatsu, Synlett, 2004, 61; Z. Zhang, Y.-W. Dong, G.-W. Wang and K. Komatsu, Chem. Lett., 2004, 33, 168 CrossRef CAS; L. Li, Z.-J. Ren, W.-G. Cao, P.-G. Huang and H. Zhu, Chin. J. Org. Chem., 2007, 27, 120 CAS; F. Toda, T. Suzuki and S. Higa, J. Chem. Soc., Perkin Trans. 1, 1998, 3521 RSC; F. Toda, K. Tanaka and S. Iwata, J. Org. Chem., 1989, 54, 3007 CrossRef CAS; X. Yang, C. Xi and Y. Jiang, Synth. Commun., 2006, 36, 2413 CrossRef CAS; E. Tullberg, D. Peters and T. Frejd, J. Organomet. Chem., 2004, 689, 3778 CrossRef CAS; E. Tullberg, F. Schacher, D. Peters and T. Frejd, Synthesis, 2006, 1183 CAS; Z. Zhang, X. Wang and Z. Li, Synth. Commun., 2004, 34, 1407 CrossRef CAS; M. Nüchter, B. Ondruschka and R. Trotzki, J. Prakt. Chem., 2000, 342, 720 CrossRef CAS; X. Bu, H. Jing, L. Wang, T. Chang, L. Jin and Y. Liang, J. Mol. Catal. A: Chem., 2006, 259, 121 CrossRef CAS; F. Toda and K. Okuda, J. Chem. Soc., Chem. Commun., 1991, 1212 RSC; A. L. Garay, A. Pichon and S. L. James, Chem. Soc. Rev., 2007, 36, 846 RSC.
- F. Toda, H. Takumi and M. Akehi, J. Chem. Soc., Chem. Commun., 1990, 1270 RSC.
- D. Braga, D. D'Addario, S. L. Giaffreda, L. Maini, M. Polito and F. Grepioni, Top. Curr. Chem., 2005, 254, 71 CAS.
- V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 1606 RSC.
- K. Komatsu, Top. Curr. Chem., 2005, 254, 185 CAS.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- G. Rothenberg, A. P. Downie, C. L. Raston and J. L. Scott, J. Am. Chem. Soc., 2001, 123, 8701 CrossRef CAS.
- W. H. Correa, S. Papadopoulos, P. Radnidge, B. A. Roberts and J. L. Scott, Green Chem., 2002, 4, 245 RSC.
- A.-R. Grimmer and B. Blumich, in NMR Basic Principles and Progress, ed. P. Diehl, E. Fluck, H. Gunther, R. Kosfeld and J. Seelig, Springer-Verlag, Berlin, 1994, vol. 30, pp. 1 Search PubMed.
- G.-Y. Yeap, S.-T. Ha, N. Ishizawa, K. Suda, P.-L. Boey and W. A. Kamil Mahmood, J. Mol. Struct., 2003, 658, 87 CrossRef CAS.
- A. Pelter, R. S. Ward and T. I. Gray, J. Chem. Soc., Perkin Trans. 1, 1976, 2475 RSC; F. Imasihro, S. Maeda, K. Takegoshi, T. Terao and A. Saika, Chem. Phys. Lett., 1983, 99, 189 CrossRef.
- P. C. Lauterbur, J. Chem. Phys., 1963, 38, 1415 CAS; G. Miyajima, Y. Sasaki and M. Suzuki, Chem. Pharm. Bull., 1971, 19, 2301 CAS.
- F. Toda, J. Okada and K. Mori, Angew. Chem., 1988, 100, 872 CrossRef CAS; F. Toda and Y. Tokumaru, Chem. Lett., 1990, 987; B. R. Madje, S. S. Shindalkar, M. N. Ware and M. S. Shingare, ARKIVOC, 2005, 82 CAS.
- At the same time, mechanochemical transformations of molecular and ionic solids, which do not form low-melting eutectics,5,9,10 should be qualified as true solid-state processes unless new experimental data prove the opposite.
- We found no evidence that mechanochemical transformations described in this and other our publications2,4,5,9 may proceed through an intermediate formation of mixed crystalline phases (co-crystals21).
- M. L. Cheney, M. J. Zaworotko, S. Beaton and R. D. Singer, J. Chem. Educ., 2008, 85, 1649 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b9nj00588a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |