Modulating the luminescence of an iridium(III) complex incorporating a di(2-picolyl)anilino-appended bipyridine ligand with Zn2+ cations†
Juan C.
Araya
a,
Juana
Gajardo
a,
Sergio A.
Moya
a,
Pedro
Aguirre
b,
Loïc
Toupet
c,
J. A. Gareth
Williams
d,
Muriel
Escadeillas
e,
Hubert
Le Bozec
e and
Véronique
Guerchais
*e
aUniversidad de Santiago de Chile, Facultad de Química y Biología, Avda. Libertador Bernardo O’Higgins 3363, Santiago de Chile, Chile
bUniversidad de Chile, Facultad de Ciencias Química y Farmacéuticas, Casilla 233, Santiago de Chile, Chile
cUMR 6626 (Institut de Physique de Rennes) CNRS-Université de Rennes 1, Campus de Beaulieu, 35042 Rennes, France
dDepartment of Chemistry, University of Durham, Durham, UK DH1 3LE
eUMR 6226 (Sciences Chimiques de Rennes) CNRS-Université de Rennes 1, Campus de Beaulieu, 35042 Rennes, France. E-mail: veronique.guerchais@univ-rennes1.fr; Fax: +33 (0)2 23 23 69 39
First published on 1st December 2009
Abstract
A novel iridium complex incorporating a di(2-picolyl)anilino-appended bipyridine ligand was synthesized and its optical properties studied. The presence of Zn2+ ions specifically perturbs the excited state, giving rise to a blue-shifted absorption and emission, and a shorter luminescence lifetime.
Cyclometallated iridium(III) complexes are well known for their rich photochemical and photophysical properties.1 These complexes, with low-lying triplet excited states and lifetimes of around a microsecond, behave as a promising class of phosphorescent dyes in optoelectronic devices2 and as new luminescent sensors for analytes,3 including protons,4 ions5 and biomolecules.6 Among them, cationic N⁁N-diimine complexes [Ir(N⁁C-ppy)2(N⁁N)]+ (where Hppy is 2-phenylpyridine and N⁁N represents a diimine, such as bpy or phen) are particularly attractive.7,8 They are readily synthesized, and the facile functionalization of diimine ligands provides access to diverse emissive states, allowing fine tuning of their optical properties.
We have previously shown that 2-phenylpyridines incorporating π-conjugated substituents are good building blocks for the construction of luminophores such as neutral, bis-9a and tris-cyclometalated9b 4-(4-(donor)styryl-2-phenylpyridine) iridium(III) and platinum(II)9c complexes, the interesting phosphorescent properties of which are governed by strong triplet intraligand transitions, promoted by an admixture of MLCT character {d/π → π*}.
With our ongoing interest in the design of Ir(III) complexes, we have turned our attention to cationic bipyridine complexes incorporating a styryl group substituted with a di(2-picolyl)amino (DPA) group (Scheme 1). The affinity of the nitrogen-based DPA group towards metal ions, such as Ni2+, Zn2+ and Cd2+ cations, has previously been demonstrated.10,11 We anticipated that attachment of this receptor to an Ir-coordinated bipyridine ligand would allow modulation of the electronic structure upon coordination of metal cations at the DPA site by attenuating the electron donating ability of the amino group; the emission properties should be perturbed as a result.
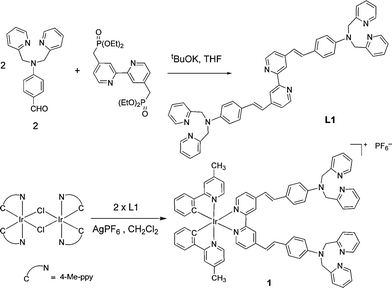 | ||
| Scheme 1 Preparation of L1 and 1. | ||
We report herein the synthesis, characterization and photophysical properties of the Ir complex [Ir(N⁁C-4-Me-ppy)2(N⁁N-bpy-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–C6H4-DPA)][PF6] (1) and the changes to its optical properties upon binding Ni2+, Zn2+ and Cd2+. The addition of Zn2+ gives a unique response, including a significant emission wavelength shift and a change in luminescence lifetime.
CH–C6H4-DPA)][PF6] (1) and the changes to its optical properties upon binding Ni2+, Zn2+ and Cd2+. The addition of Zn2+ gives a unique response, including a significant emission wavelength shift and a change in luminescence lifetime.
Target bipyridine L1 was readily prepared in 88% yield by means of a Horner–Wadsworth–Emmons condensation between a bisphosphonate-bipyridine derivative12 and the appropriate benzaldehyde amine, 2 (Scheme 1). The latter compound was obtained by following a reported procedure for the substitution of aniline by two equivalents of 2-chloromethylpyridine, followed by oxidation with POCl3 in DMF to introduce the aldehyde functionality.10e The 1H and 13C NMR spectra of L1 were in agreement with the proposed structure. Complex 1 was subsequently prepared by the reaction of L1 with Ir dimer [Ir(N⁁C-4-Me-ppy)2(μ-Cl)]2 in CH2Cl2 in the presence of AgPF6 (Scheme 1). It was isolated as orange-red microcrystals in 70% yield after recrystallization from CH2Cl2–pentane; the structure determined by X-ray diffraction is shown in Fig. 1. The initial E configuration of the two CC double bonds was retained upon metal complexation. However, upon UV irradiation, the CC bonds of 1 underwent a slow E–Z isomerization process, as revealed by 1H NMR spectroscopy.
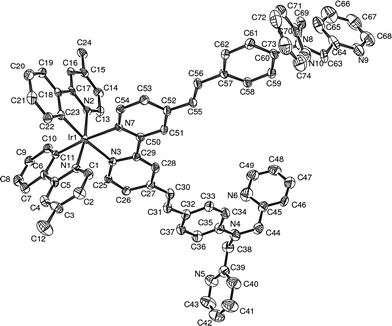 | ||
| Fig. 1 An ORTEP plot of 1 with thermal ellipsoids at the 50% probability level; T = 120 K. Hydrogen atoms are omitted for clarity. | ||
The electronic absorption and luminescence data for 1 and L1 are summarized in Table S1.† The absorption spectrum of 1 displays high energy bands in the UV region (250–340 nm) assigned to intraligand IL (π → π*) (bpy and ppy) transitions (Fig. 2). An intense low energy absorption band centered at 449 nm (ε = 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 dm3 mol−1 cm−1) probably arises from the influence of the electron donating nature of the amino group. The effect of the amino substituent is also seen in the behavior of the corresponding uncoordinated ligand L1, which displays a low energy absorption band (λmax = 385 nm). The 64 nm red shift in the IL band of L1 upon complexation to iridium(III) (highlighted in Figure S1†) can be attributed to the enhanced π-acceptor character of the bpy moiety upon coordination, lowering the LUMO energy level.13 Based on the spectra of complexes such as [Ir(N⁁C-ppy)2(N⁁N-bpy)]+,7,8 one or more bands due to MLCT transitions [dπ(Ir) → π*(diimine and ppy)] would be anticipated over the range 330–400 nm; in the present complex, they are superimposed by the tail of the low energy band associated with the aminostyryl unit.
500 dm3 mol−1 cm−1) probably arises from the influence of the electron donating nature of the amino group. The effect of the amino substituent is also seen in the behavior of the corresponding uncoordinated ligand L1, which displays a low energy absorption band (λmax = 385 nm). The 64 nm red shift in the IL band of L1 upon complexation to iridium(III) (highlighted in Figure S1†) can be attributed to the enhanced π-acceptor character of the bpy moiety upon coordination, lowering the LUMO energy level.13 Based on the spectra of complexes such as [Ir(N⁁C-ppy)2(N⁁N-bpy)]+,7,8 one or more bands due to MLCT transitions [dπ(Ir) → π*(diimine and ppy)] would be anticipated over the range 330–400 nm; in the present complex, they are superimposed by the tail of the low energy band associated with the aminostyryl unit.
![Top: UV-vis absorption spectral changes of 1 (conc. = 1 × 10−5 M in CH3CN) after the addition of 50 equivalents of Ba2+, Ca2+, Mg2+, Cd2+, Zn2+ and Ni2+ ions. Bottom: UV-vis absorption spectral changes of 1 upon addition of incremental (0.5 equivalent) amounts of Zn(ClO4)2. The inset shows a plot of absorbance against [Zn2+] monitored at 430 nm.](/image/article/2010/NJ/b9nj00515c/b9nj00515c-f2.gif) | ||
| Fig. 2 Top: UV-vis absorption spectral changes of 1 (conc. = 1 × 10−5 M in CH3CN) after the addition of 50 equivalents of Ba2+, Ca2+, Mg2+, Cd2+, Zn2+ and Ni2+ ions. Bottom: UV-vis absorption spectral changes of 1 upon addition of incremental (0.5 equivalent) amounts of Zn(ClO4)2. The inset shows a plot of absorbance against [Zn2+] monitored at 430 nm. | ||
Uncoordinated ligand L1 is fluorescent in solution at room temperature (RT) (λmax = 486 nm in CH2Cl2). In contrast, complex 1 is non-emissive in solution at RT. Simpler iridium(III) complexes, such as [Ir(N⁁C-ppy)2(N⁁N-bpy)]+, display luminescence from the lowest-lying triplet state under these conditions, promoted by spin–orbit coupling. The absence of RT phosphorescence in the present case suggests the introduction of a non-radiative decay pathway that is not open to simpler derivatives and/or a radiative rate constant, kr, that is unusually low; this is confirmed by its behavior at 77 K. Under these conditions, the complex emits brightly, displaying a highly structured spectrum deep into the red region (λmax0–0 = 640 nm, Δν = 1400 cm−1) (Fig. 3). The lifetime of 67 μs is exceptionally long, confirming that kr is indeed low. The values are in stark contrast to those of, for example, [Ir(N⁁C-ppy)2(N⁁N-4,4′-Me2-bpy)]+ (λem = 506 nm at 77 K, τ = 4.83 μs),7d where the emission is attributed to an MLCT state. Evidently, in complex 1, the emission emanates from a state of 3IL character localized on the styryl-substituted bpy ligand, with relatively low metal participation; triplet excited states, in which the metal plays only a minor role, are expected to have longer radiative lifetimes owing to less efficient spin–orbit coupling pathways. The triplet radiative rate constant is probably so low that, at RT, the excited state is essentially fully deactivated by the faster E–Z isomerization process, accounting for the lack of emission. In free ligand L1, in contrast, the fluorescence is fast enough to be competitive with this process.
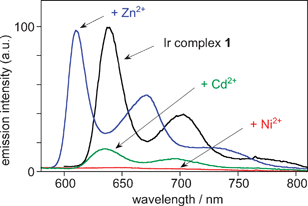 | ||
| Fig. 3 Luminescence spectral changes (77 K) of 1 (conc. = 3 × 10−6 M) in ethanol–methanol (4:1, v/v) upon the addition of M(ClO4)2 (M = Ni, Zn, Cd) (conc. = 6 × 10−5 M); λex = 385 nm (isosbestic point). | ||
The behavior of 1 in the presence of various metal cations was studied (Ba2+, Ca2+, Mg2+, Ni2+, Zn2+ and Cd2+ as their perchlorate salts in CH3CN). The absorption spectrum was dramatically modified upon adding Ni2+, Zn2+ and Cd2+ (Fig. 2). The solution color visibly changed from orange to colorless, the low energy absorption band (λmax 430 nm in CH3CN) decreased and a new band at ∼330 nm concomitantly evolved (Fig. 2 and Fig. S3†). In contrast, no significant changes were observed with Ba2+, Ca2+ and Mg2+.
The blue shift of the absorption band with Ni2+, Zn2+ and Cd2+ is intuitively consistent with the binding of the metal ion at the DPA amine nitrogen, which would reduce its donating ability, lowering the energy of the highest-occupied molecular orbital. The stoichiometric ratio of the species formed between complex 1 and M2+ was estimated to be 1:2 in accordance with the presence of two DPA receptors. An isosbestic point at 385 nm is consistent with a two-species equilibrium (1 + 2M2+ → 1·(M2+)2). The following binding constants, log Ks, were determined: Ni2+ = 7.01 ± 0.03, Cd2+ = 6.56 ± 0.61 and Zn2+ = 5.46 ± 0.12.
The effect of the same set of metal ions on the luminescence of 1 at 77 K was also investigated. The emission (λmax = 640 nm) was completely quenched upon adding Ni2+ ions, whereas Cd2+ lead only to partial quenching and a scarcely perceptible blue shift (3 nm) (Fig. 3). Most strikingly, the coordination of Zn2+ ions induced a unique response: a blue-shifted emission (λmax0–0 = 610 nm) of the intensity comparable to 1 (for excitation at the isosbestic point at 385 nm) with a lifetime of 34 μs was observed, indicative of the formation of a new emissive species. In the presence of a sub-stoichiometric quantity of Zn2+, the emission profiles of the two species—the ion-free complex 1 and the Zn2+-coordinated complex—were independently observable at 77 K (Fig. S5†). Two distinct sets of vibrational bands were clear, and the distinctly different decay curves registered at 610 and 637 nm, respectively, are in line with those of the fully bound and unbound species. Interestingly, the addition of H+ ions (from trifluoroacetic acid) lead to related but much smaller effects on the emission: a blue shift of 12 nm and a small decrease in the lifetime to 50 μs.
As for the absorption, the emission blue shift can be interpreted in terms of a stabilisation of the highest occupied molecular orbitals as the N lone pair becomes bound to the Zn2+ ion. That selectivity for Cd2+ is observed only in emission and not in absorption presumably reflects a subtle difference in the electronic distribution in the triplet and singlet excited states. The selective blue shift induced by Zn2+ implies that, in the excited state, the more charge-dense Zn2+ ion interacts more strongly with the amino group of the DPA unit than does Cd2+. The softer nature of the Cd2+ ion probably means that its predominant mode of binding to the DPA in the excited state is via the picolyl units.
In summary, complex 1 displays an unusual phenomenon of selective zinc-dependent wavelength- and lifetime-modulation of its emission.
This work was supported by ECOS-CONICYT (Action CO7E02), COST D35 and Region Bretagne (SIE 211-B3-11). This research was performed as part of the Chilean–French “Joint Laboratory for Inorganic Functional Materials “(LIAMIF_836_). We thank J.-L. Fillaut and P.-H. Lanoë for assistance. J. C. A., J. G., P. A. and S. A. M. thank Fondecyt 1085135.
Experimental
Preparation of 1
Chloride-bridged dimer [Ir(N⁁C-4-Me-ppy)2(μ-Cl)]2 (100 mg, 0.09 mmol), bipyridine derivative L1 (140 mg, 0.19 mmol) and AgPF6 (60 mg, 0.24 mmol) were mixed in CH2Cl2 (10 mL). The reaction mixture was stirred under Ar for 2 h. The solution was then concentrated to dryness and the product extracted with CH2Cl2 (3 × 5 mL). Crystallization from a CH2Cl2–diethyl ether mixture gave an orange-red powder (186 mg, 70%). 1H NMR (500 MHz, CDCl3): 8.68 (s, 2H, H3-bpy), 8.61 (d, 4H, H6-Py*, 3J = 4.4 Hz), 7.70 (s, 2H, H6-bpy), 7.65 (m, 2H, H3-Ph, 4H, H3-Py* and 2H, H3-Py), 7.48 (d, 4H, C6H4, 3J = 8.9 Hz), 7.44 (d, 2H, H6-Py, 3J = 6.0 Hz), 7.41 (d, 2H,CH8, 3J = 16.2 Hz), 7.27 (m, 2H, H5-bpy), 7.25 (d, 4H, H4-Py*, 3J = 7.9 Hz), 7.20 (dd, 4H, H5-Py*, 3J = 6.8 Hz, 3J = 5.03 Hz), 7.11 (d, 2H, CH7, 3J = 16.2 Hz), 7.01 (td, 2H, H4-Ph, 3J = 7.5 Hz, 4J = 1.0 Hz), 6.90 (td, 2H, H5-Ph, 3J = 7.5 Hz, 4J = 1.2 Hz), 6.84 (dd, 2H, H5-Py, 3J = 6.0 Hz, 4J = 1.4 Hz), 6.69 (d, 4H, C6H4, 3J = 8.8 Hz), 6.34 (dd, 2H, H6-Ph, 3J = 7.5 Hz, 4J = 0.8 Hz), 4.86 (s, 8H, CH2) and 2.49 (s, 6H, CH3). 13C[1H] NMR (75 MHz, CDCl3): 167.23 (C2-py), 158.13 (C2-py*), 156.05 (C2-bpy), 151.67 (C1-Ph), 149.74 (C6-py*), 149.53 (C4-py), 149.38 (C9), 149.13 (C4-bpy), 149.05 (C6-bpy), 147.90 (C6-py), 143.64 (C2-Ph), 137.19 (C8), 136.89 (C4-py*), 131.84 (C6-Ph), 130.32 (C5-Ph), 129.65 (C10), 124.90 (C12), 124.36 (C3-Ph), 124.26 (C5-py), 122.44 (C5-bpy), 122.17 (C5-py* and C3-bpy), 122.08 (C4-Ph), 120.80 (C3-py*), 120.06 (C3-py), 119.70 (C7), 112.54 (C11), 56.98 (CH2) and 21.29 (CH3). Elemental analysis: C74H62F6N10IrP·0.5CH2Cl2 calc. C, 60.83, H, 4.32, N, 9.52. Found: C, 60.20, H, 4.36, N, 9.18%. HRMS: m/z calc. for C74H62N10Ir: 1283.47883, found: 1283.4793.
References
- (a) I. M. Dixon, J.-P. Collin, J.-P. Sauvage, L. Flamigni, L. S. Encinas and F. Barigelletti, Chem. Soc. Rev., 2000, 29, 385 RSC; (b) M. S. Lowry and S. Bernhard, Chem.–Eur. J., 2006, 12, 7970 CrossRef CAS; (c) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CAS; (d) P.-T. Chou and Y. Chi, Chem.–Eur. J., 2007, 13, 380 CrossRef CAS; (e) J. A. G. Williams, A. J. Wilkinson and V. L Whittle, Dalton Trans., 2008, 2081 RSC.
- M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS; Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Berlin, 2007 Search PubMed.
- (a) Z. Liu, Z. Bian, J. Bian, Z. Li, D. Nie and Huang, Inorg. Chem., 2008, 47, 8025 CrossRef CAS; (b) H. Chen, Q. Zhao, Y. Wu, F. Li, H. Yang, T. Yi and C. Huang, Inorg. Chem., 2007, 46, 11075 CrossRef CAS.
- (a) M. Licini and J. A. G. Williams, Chem. Commun., 1999, 1943 RSC; (b) W. Goodall and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 2000, 2893 RSC; (c) K. Konishi, H. Yamaguchi and A. Harada, Chem. Lett., 2006, 35, 720 CrossRef CAS.
- (a) M.-L. Ho, F.-M. Hwang, P.-N. Chen, Y.-H. Hu, Y.-M. Cheng, K.-S. Chen, G.-H. Lee, Y. Chi and P.-T. Chou, Org. Biomol. Chem., 2006, 4, 98 RSC; (b) Q. Zhao, S. Liu, M. Shi, F. Li, H. Jing, T. Yi and C. Huang, Organometallics, 2007, 26, 5922 CrossRef CAS; (c) Q. Zhao, T. Cao, F. Li, X. Li, H. Jing, T. Yi and C. Huang, Organometallics, 2007, 26, 2077 CrossRef CAS; (d) M. Schmittel and H. Lin, Inorg. Chem., 2007, 46, 9139 CrossRef CAS; (e) K. K.-W. Lo, J. S.-Y. Lau, D. K.-K. Lo and L. T.-L. Lo, Eur. J. Inorg. Chem., 2006, 4054 CrossRef CAS.
- K. K.-W. Lo, D. C.-M. Ng and D. C.-M. Chung, Organometallics, 2001, 20, 4999 CrossRef CAS; K. K.-W. Lo, K. H. K. Tsang, K. S. Sze, C. K. Chung, T. K. M. Lee, K. Y. Zhang, W. K. Hui, C. K. Li, J. S.-Y. Lau, D. C. M. Ng and N. Y. Zhu, Coord. Chem. Rev., 2007, 251, 2292 CrossRef CAS; K. K.-W. Lo, K. Y. Zhang, S.-K. Leung and M.-C. Tang, Angew. Chem., Int. Ed., 2008, 47, 2213 CrossRef CAS.
- See, inter alia: (a) F. O. Garces, K. A. King and R. J. Watts, Inorg. Chem., 1988, 27, 3464 CrossRef CAS; (b) F. Neve, A. Crispini, S. Campagna and S. Serroni, Inorg. Chem., 1999, 38, 2250 CrossRef CAS; (c) E. A. Plummer, J. W. Hofstraat and L. De Cola, Dalton Trans., 2003, 2080 RSC; (d) M. Lepeltier, T. K.-M. Lee, K. K.-W. Lo, L. Toupet, H. Le Bozec and V. Guerchais, Eur. J. Inorg. Chem., 2005, 110 CrossRef CAS; (e) M. Polson, S. Fracasso, V. Bertolasi, M. Ravaglia and F. Scandola, Inorg. Chem., 2004, 43, 1950 CrossRef CAS; (f) F. De Angelis, S. Fantacci, N. Evans, C. Klein, S. M. Zakeeruddin, J.-E. Moser, K. Kalyanasundaram, H. J. Bolink, M. Gratzel and M. K. Nazeeruddin, Inorg. Chem., 2007, 46, 5989 CrossRef.
- Q. Zhao, S. Liu, M. Shi, C. Wang, M. Yu, L. Li, F. Li, T. Yi and C. Huang, Inorg. Chem., 2006, 45, 6152 CrossRef CAS; C. Dragonetti, L. Falciola, P. Mussini, S. Righetto, D. Roberto, R. Ugo, A. Valore, F. De Angelis, S. Fantacci, A. Sgamellotti, M. Ramon and M. Muccini, Inorg. Chem., 2007, 46, 8533 CrossRef CAS.
- (a) M. Lepeltier, H. Le Bozec, V. Guerchais, T. K.-M. Lee and K. K.-W. Lo, Organometallics, 2005, 24, 6069 CrossRef CAS; (b) M. Lepeltier, T. K.-M. Lee, K. K.-W. Lo, L. Toupet, H. Le Bozec and V. Guerchais, Eur. J. Inorg. Chem., 2007, 2734 CrossRef CAS; (c) B. Yin, F. Niemeyer, J. A. G. Williams, J. Jiang, A. Boucekkine, L. Toupet, H. Le Bozec and V. Guerchais, Inorg. Chem., 2006, 45, 8584 CrossRef CAS.
- (a) X. Peng, J. Du, J. Fan, J. Wang, Y. Wu, J. Zhao, S. Sun and T. Xu, J. Am. Chem. Soc., 2007, 129, 1500 CrossRef CAS; (b) L. Zhang, R. J. Clark and L. Zhu, Chem.–Eur. J., 2008, 14, 2894 CrossRef CAS; (c) T. Sakamoto, A. Ojida and I. Hamachi, Chem. Commun., 2009, 141 RSC; (d) A. Takebayashi, S. Shinkai, M. Ikeda and M. Takeuchi, Org. Biomol. Chem., 2008, 6, 493 RSC; (e) X. Huang, Z. Guo, W. Zhu, Y. Xie and H. Tian, Chem. Commun., 2008, 5143 RSC.
- Two examples of metal complexes incorporating the DPA unit have been very recently published: M.-W. Louie, H.-W. Liu, M. H.-C. Lam, T.-C. Lau and K. K.-K. Lo, Organometallics, 2009, 28, 4297 Search PubMed; N. Zhao, Y.-H. Wu, H.-M. Wen, X. Zhang and Z.-N. Chen, Organometallics, 2009, 28, 5603 CrossRef CAS.
- O. Maury, J.-P. Guégan, T. Renouard, A. Hilton, P. Dupeau, N. Sandon, L. Toupet and H. Le Bozec, New J. Chem., 2001, 25, 1553 RSC.
- (a) W. Goodall and J. A. G. Williams, Chem. Commun., 2001, 2514 RSC; (b) W. Leslie, A. S. Batsanov, J. A. K. Howard and J. A. G. Williams, Dalton Trans., 2004, 623 RSC; (c) W. Leslie, R. A. Poole, P. R. Murray, L. J. Yellowlees, A. Beeby and J. A. G. Williams, Polyhedron, 2004, 23, 2769 CrossRef CAS; (d) D. Roberto, F. Tessore, R. Ugo, S. Bruni, A. Manfredi and S. Quici, Chem. Commun., 2002, 846 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental procedures, photophysical data and UV-vis titrations, details of the X-ray structure of 1. CCDC reference number 718415. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/b9nj00515c. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |