Grafting of monoglyceride molecules for the design of hydrophilic and stable porous silicon surfaces†
Stéphanie
Pace
a,
Philippe
Gonzalez
a,
Jean-Marie
Devoisselle
a,
Pierre-Emmanuel
Milhiet
b,
Daniel
Brunel
a and
Frédérique
Cunin
*a
aInstitut Charles Gerhardt Montpellier, UMR 5253 CNRS-ENSCM-UM2-UM1, Ecole Nationale Supérieure de Chimie de Montpellier, 8 rue de l’Ecole Normale, 34296, Montpellier, France. E-mail: frederique.cunin@enscm.fr; Fax: +33 467 163 474; Tel: +33 467 163 444
bCentre de Biochimie Structurale, UMR554 INSERM, UMR5048 CNRS, 29 rue de Navacelles, 34090, Montpellier, France. Fax: +33 467 417 913; Tel: +33 467 417 917
First published on 3rd December 2009
Abstract
Hydrophilic chemically stable porous silicon surfaces are generated by surface functionalisation with polar head terminated lipid biomolecules of the monoglyceride type. Two approaches to anchor the monoglyceride moiety to porous silicon surfaces are presented.
Solid supports are widely used in biology for the immobilisation and study of biological species such as cells, biomolecules or membranes. These include glass, silicon, polystyrene and mica, mainly because they are mechanically and chemically stable. Great attention has been paid to using nanostructured porous substrates that feature an enriched set of physicochemical and physical properties (large surface area, available volume, optical properties), offering increased potential for the development of biomedical devices. Porous silicon, with flexible structural and optical properties, and tunable surface chemistry, has shown high efficiency in chemical and biomolecular sensing,1–8 and is believed to play an important role in application fields that include drug delivery,9–12 cell culture13,14 and tissue engineering.15,16 A major limitation of using porous silicon in biological-like aqueous media is the poor chemical stability of Si–H surface species, which leads, considering the high surface area, to very rapid degradation of the substrate.15 Chemical modification of porous silicon surfaces has proved efficient for protecting them from chemical degradation. Controlled oxidation of the surface, silanisation chemistry and alkylation via Si–C bond chemistry has been developed to passivate and protect the surface of porous silicon.17–20 The most efficient methods for porous silicon surface stabilisation use the attachment of ligands via Si–C bonds, which are more stable towards nucleophilic attack in aqueous media than Si–O bonds.1,20 Chemical functionalisation of porous silicon via surface Si–C bonds leads easily to hydrophobic surfaces due to the presence of hydrocarbon chains. In many cases, including cell adhesion enhancement21 and artificial membrane deposition,22 it is strongly desirable to have highly hydrophilic stable surfaces. In recent work, Sailor and his group reported the important role played by the hydrophilic and hydrophobic properties of porous silicon surfaces on cell adhesion and viability.13 The most hydrophilic porous silicon surfaces were obtained by hydrosilylation reactions using undecanoic acid or oligo(ethylene) glycol ligands. Nevertheless, water contact angle measurements, which were never below 60°, showed the poor relative hydrophilic properties of the surfaces compared to ozone-oxidized porous silicon surfaces (contact angle < 6°). Highly hydrophilic porous silicon surfaces are usually generated by ozone oxidation treatment, but are known not to be stable in aqueous media.1
In this Letter, we report the surface functionalisation of porous silicon with polar head terminated lipid biomolecules in order to generate highly hydrophilic, chemically-stable porous silicon surfaces. Monoglyceride molecules were chosen for this purpose as they contain glycol terminus functions to impart hydrophilic properties and long carbon chains that can play the role of hydrophobic spacers to keep aqueous nucleophiles apart from the surface-reactive silicon hydride species. In the present work, a monoglyceride molecule containing an olefin function, an α-monoglyceryl undecylcarboxylate, is prepared and covalently attached to freshly etched porous silicon surfaces via Si–C bonds (preparation of the α-monoglyceryl undecylcarboxylate is described in the Experimental section). Two approaches to anchor the monoglyceride moiety to porous silicon surfaces are reported.
As a first approach, porous Si was thermally hydrosilylated with α-monoglyceryl undecylcarboxylate. Freshly etched porous silicon was exposed to a few milligrams of the monoglyceride, which was dissolved in anhydrous toluene added to the reaction flask by dynamic distillation under vacuum to avoid the presence of air. The solution was maintained at 120 °C for 17 h under argon for the hydrosilylation reaction according to previously described methods.20 The hydrosilylation reaction between Si–H species and the olefin function of the freshly synthesised monoglyceride is schematically presented in Fig. 1.
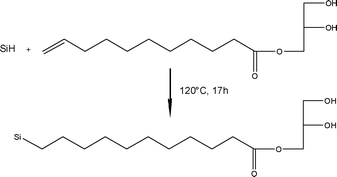 | ||
| Fig. 1 Chemical scheme of α-monoglyceryl undecylcarboxylate grafting onto a porous silicon surface. | ||
After the reaction, the silicon chip was rinsed thoroughly with ethanol to remove excess monoglyceride. Fig. 2 shows FTIR data for (a) a freshly etched porous silicon film and (b) the same porous silicon surface after the reaction with α-monoglyceryl undecylcarboxylate. Spectrum (a) exhibits bands at 912 and 2115 cm−1, assigned to the Si–H2 bending mode and the Si–Hx stretching mode, respectively. After the reaction with α-monoglyceryl undecylcarboxylate, bands are observed in spectrum (b) at 1462, 1743, 2853 and 2930 cm−1 that are due to the δCHtet deformation vibration mode of a CH2, the νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration mode of an ester and the stretching vibration modes of aliphatic C–H bonds, respectively. In addition, the large band centered at 3494 cm−1 is assigned to a νO–H stretching vibration mode of the diol, indicating that the monoglyceride molecule is present at the surface of the porous silicon. Bands are still observed in spectrum (b) at 909 and 2106 cm−1, indicating that silicon hydride sites are still present at the surface of the porous film. Complete reaction is impeded by steric hindrance at the surface, as previously observed in the literature.23 The presence of weak bands at 3076 and at 1637 cm−1 could be assigned, respectively, to the νC–H stretching vibration mode of the arylic C–H bonds and to the νCC stretching vibration mode of the arene CC bonds from toluene that has not been totally desorbed after the reaction. FTIR data of a controlled experiment consisting of porous silicon refluxed in toluene for 17 h without α-monoglyceryl undecylcarboxylate indicates the presence of possible residual toluene within the pores after rinsing and drying of the samples (data not shown). Moreover, the previously mentioned bands at 3076 and 1637 cm−1 could also be assigned to the νC–Htri stretching vibration mode of the vinyl C–H bonds, and the band at 1637 cm−1 assigned to the νCC stretching vibration mode of the CC bonds, indicating that part of the α-monoglyceryl undecylcarboxylate is not covalently grafted and remains physisorbed at the surface of the porous silicon after the rinsing step. A control experiment with lauric monoglyceride (equivalent to α-monoglyceryl undecylcarboxylate but with no CC bond) in the presence of freshly etched porous silicon at 120 °C for 90 min showed that removing the physisorbed lauric monoglyceride by rinsing is difficult. Finally, a competitive reaction with the hydrosilylation is also possible; for example, an etherisation reaction between the diol from the monoglyceride and the silanol groups of the partially oxidized porous silicon surface.
O stretching vibration mode of an ester and the stretching vibration modes of aliphatic C–H bonds, respectively. In addition, the large band centered at 3494 cm−1 is assigned to a νO–H stretching vibration mode of the diol, indicating that the monoglyceride molecule is present at the surface of the porous silicon. Bands are still observed in spectrum (b) at 909 and 2106 cm−1, indicating that silicon hydride sites are still present at the surface of the porous film. Complete reaction is impeded by steric hindrance at the surface, as previously observed in the literature.23 The presence of weak bands at 3076 and at 1637 cm−1 could be assigned, respectively, to the νC–H stretching vibration mode of the arylic C–H bonds and to the νCC stretching vibration mode of the arene CC bonds from toluene that has not been totally desorbed after the reaction. FTIR data of a controlled experiment consisting of porous silicon refluxed in toluene for 17 h without α-monoglyceryl undecylcarboxylate indicates the presence of possible residual toluene within the pores after rinsing and drying of the samples (data not shown). Moreover, the previously mentioned bands at 3076 and 1637 cm−1 could also be assigned to the νC–Htri stretching vibration mode of the vinyl C–H bonds, and the band at 1637 cm−1 assigned to the νCC stretching vibration mode of the CC bonds, indicating that part of the α-monoglyceryl undecylcarboxylate is not covalently grafted and remains physisorbed at the surface of the porous silicon after the rinsing step. A control experiment with lauric monoglyceride (equivalent to α-monoglyceryl undecylcarboxylate but with no CC bond) in the presence of freshly etched porous silicon at 120 °C for 90 min showed that removing the physisorbed lauric monoglyceride by rinsing is difficult. Finally, a competitive reaction with the hydrosilylation is also possible; for example, an etherisation reaction between the diol from the monoglyceride and the silanol groups of the partially oxidized porous silicon surface.
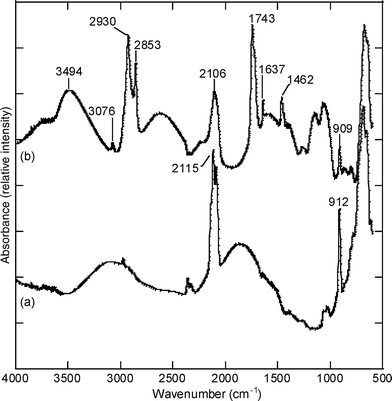 | ||
| Fig. 2 FTIR spectra for (a) a freshly etched porous silicon surface and (b) a porous silicon surface reacted with α-monoglyceryl undecylcarboxylate. | ||
In order to prevent undesirable physisorption and the competitive chemisorption of α-monoglyceryl undecylcarboxylate onto the porous silicon surface, a second approach has been investigated where α-monoglyceryl undecylcarboxylate is grafted onto the porous silicon surface in a two-step reaction. Freshly etched porous silicon was first thermally hydrosilylated with undecenoic acid at 120 °C for 90 min under argon (Fig. 3(a)). Next, the excess of non-grafted undecenoic acid was easily removed by rinsing with dichloromethane. The as-modified porous silicon surface was then exposed to glycidol, which was able to react, in the presence of the triethylamine catalyst, with the carboxylic acid functions of the grafted acid, resulting in the formation of monoglyceride anchored onto the surface of the porous silicon (Fig. 3(b)). After the reaction, the porous silicon chip was rinsed with ethanol to remove the excess glycidol.
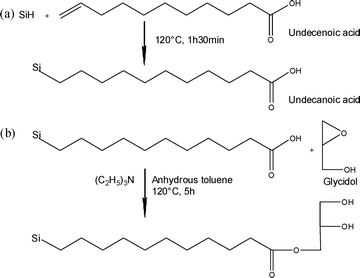 | ||
| Fig. 3 Chemical anchoring of a monoglyceride molecule in a two-step reaction onto a porous silicon surface, including (a) grafting of undecenoic acid onto the porous silicon surface and (b) an addition reaction of glycidol onto the grafted carboxylic acid function. | ||
Fig. 4 shows FTIR data for (a) a freshly etched porous silicon film, (b) the same porous silicon surface grafted with undecanoic acid and (c) the previously undecanoic acid-grafted porous silicon surface after the reaction with glycidol. After modification of the surface with undecenoic acid, spectrum (b) exhibits bands at 1715 cm−1, assigned to the νCO stretching vibration mode of the carboxylic acid, and bands at 1462, 2852 and 2925 cm−1 that are assigned to the deformation and stretching vibration mode of the aliphatic C–H groups. As expected, the absence of characteristic bands for CC bonds (at 1637 and 3076 cm−1) confirms that undecanoic acid is covalently attached to the porous silicon surface and that no undecenoic acid remains physisorbed at the surface after rinsing with dichloromethane. A subsequent reaction between the carboxylic acid and the glycidol leads to a shift in the νCO stretching band to 1740 cm−1, indicating that the carboxylic acid has been converted into an ester group. The presence of a shoulder in the aforementioned band observed at 1710 cm−1 is consistent with unreacted carboxylic acid functions remaining after the reaction with glycidol. Finally, the large band centered at 3398 cm−1 is assigned to the stretching vibration mode of the diol, confirming the presence of the monoglyceride molecule covalently attached to the porous silicon surface. No residual toluene in the pores was observed after rinsing and drying of the samples. This two-step reaction approach allows better control of the grafting of the monoglyceride molecules via Si–C bonds onto the porous silicon.
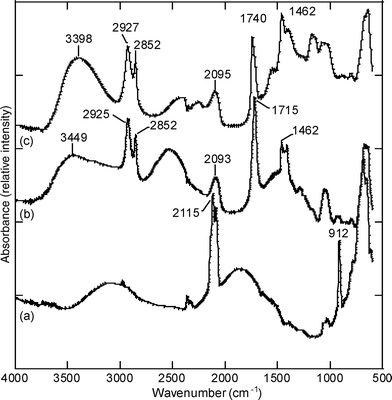 | ||
| Fig. 4 FTIR spectra for (a) the freshly etched porous silicon surface, (b) the undecanoic acid-grafted porous silicon surface and (c) surface (b) reacted with glycidol. | ||
In order to test the hydrophilicity of the monoglyceride modified surface obtained in the two-step reaction process, water contact angle measurements were performed and compared with contact angle values from undecanoic acid-modified porous silicon surfaces. The obtained results are summarised in Table 1. It is important to mention here that contact angle measurements are normally considered for ideal flat surfaces that are traditionally defined as being smooth, rigid, chemically homogeneous and non-reactive.24 In the case of solid surfaces presenting roughness or chemical heterogeneity, the interpretation of contact angle values are more critical and need to be considered individually. Nevertheless, the literature in this field indicates that when the size of the water drop used for the measurement is sufficiently large compared to the scale of roughness or heterogeneity, the effects of surface roughness or heterogeneity are minimized.23 Porous silicon surfaces used in this work exhibit pores of 5 to 8 nm average diameter (obtained by SEM, data not presented). The theoretical diameter of the water drops used in this work were calculated to be 1.56 mm (from the known 2 μL volume of the water drop), which is 2 to 3 million times larger than the pore diameters. Because the two considered surface ligands (undecanoic acid and monoglyceride) are comparable in terms of chain length and conformation, with molecular weights of the same order of magnitude (185 and 254 g mol−1), we consider that their surface rugosities are still comparable after chemical modification, and consequently that the contact angles of the two surfaces are altered in a comparable way.
| Reactant | Θ (standard deviation) (°) | |
|---|---|---|
| Undecylenic acid | 62 (3) |

|
| α-Monoglyceryl undecylcarboxylate | 14 (1) |

|
The contact angle for the modified surface with the monoglyceride (undecanoic acid plus glycidol) was measured as 14°, which is lower than for the modified surface with undecanoic acid only, for which the contact angle was 62°, showing the greater hydrophilicity of the monoglyceride-terminated surface. This is explained by the higher polarity of a diol function compared to that of a carboxylic acid function, as attested by the values of the dipole moments for ethylene glycol (μ = 7.61) compared to acetic acid (μ = 5.6).25 The polarity of solvents is also known to be linked to their dielectric constant, which is 37 and 6.1 for ethylene glycol and acetic acid, respectively.26 Additionally, besides being more polar, the diol function is less pH-dependant than a carboxylic acid function; the pKa value is 4.76 for acetic acid, whereas it is over 14 for ethylene glycol (14.22).25 The diol-modified porous silicon surface presented in this work is expected not to be modified when exposed to varying environmental pH conditions. Such hydrophilic and inert-to-pH-change surfaces can be useful in biological and biosensing applications, where hydrophilic stable surfaces are desirable in various buffered conditions.
The chemical stability to corrosion of the monoglyceride-modified porous silicon surface was tested over time in a phosphate-buffered saline (PBS) solution and compared to a non-modified porous silicon surface and an undecanoic acid-modified porous silicon surface (Fig. 5); it was also compared with the stability of thermally oxidised and ozone-oxidised porous silicon surfaces. Thermal oxidation in air is an efficient method to passivate the surface of porous silicon and produce a stable oxide surface.27–29 Ozone oxidation is known to be a classical and simple way to provide highly hydrophilic surfaces from porous silicon by generating silanol surface groups.1 As expected, the thermal oxidation treatment generates a highly stable surface.27–29 The two Si–C modified surfaces show a slower decrease in their normalised effective optical thickness in time than the non-modified surface and the ozone-oxidised surface, as expected with regards to the kinetic stability properties of the Si–C bond.20
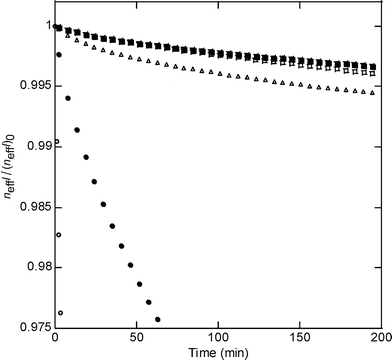 | ||
| Fig. 5 Stability of modified porous silicon surfaces in PBS (pH 7.4) for (○) the silicon hydride terminated surface, (●) the ozone-oxidised surface, (▵) the undecanoic acid-modified surface, (□) the thermally oxidised surface and (▪) the monoglyceride-modified surface, presented as the variation of the normalized effective optical thickness neffl/(neffl)0vs. time (see the Experimental section). | ||
In conclusion, hydrophilic, chemically-stable, porous silicon surfaces have been prepared by the covalent attachment of biocompatible molecules from the monoglyceride family. Two approaches to anchor the monoglyceride to porous silicon surfaces have been developed and compared. In the first approach, the monoglyceride, previously synthesised in the laboratory, was covalently attached to the porous silicon surface using hydrosilylation chemistry. In the second approach, the monoglyceride was covalently attached in a two-step reaction, including the grafting of undecenoic acid onto the porous silicon surface and an addition reaction of glycidol onto the grafted carboxylic acid function. The second approach led to better control of the covalent attachment of the biomolecule. Aqueous intimate wetting without chemical degradation over time was achieved. The obtained surfaces both feature highly hydrophilic properties comparable to silica-type surfaces and have the high chemical stability that is classically observed for more hydrophobic, hydrosilylated porous silicon surfaces. The flexible design of the optical structures of porous silicon is still possible prior to surface modification, providing porous silicon with better potential for biosensing and biological applications, where long term stability in aqueous media is required.
The authors gratefully acknowledge Prof. Michael J. Sailor of the University of California, San Diego, USA, and Profs. Emmanuel Belamie and Michel Granier of the Institut Charles Gerhardt Montpellier, France for helpful discussions. The authors thank the Centre National de la Recherche Scientifique (CNRS) for funding.
Experimental
Synthesis
Porous silicon films were prepared from single-crystal p-type silicon (boron doped, ∼3 Ω cm resistivity, 〈100〉 orientation, from Siltronix, Inc.) in an electrochemical etch at a constant current density of 22.5 mA cm−2 for 5 min in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 48% aqueous hydrofluoric acid–ethanol solution. Prior to etching, the silicon chips were washed successively under sonication with acetone and ethanol for 5 min each. The α-monoglyceryl undecylcarboxylate was prepared according to a previously published procedure in our laboratory.30 The synthesis involved a reaction between undecenoic acid (Aldrich, 98%) and glycidol (Aldrich, 96%) in toluene at 120 °C with a solid catalyst (quaternary ammonium chloride-grafted silica) in anhydrous toluene for 24 h. The quaternary ammonium chloride-grafted silica catalyst preparation was adapted from ref. 26: 0.5 g of (3-(triethoxysilyl)propyl)-trimethylammonium chloride (65 wt% in water, purchased from Aldrich) was added to a suspended silica gel (G5H from Grace Davison with a surface area of 513 m2 g−1) in 30 mL of toluene. The suspension was stirred at RT for 1 h, then heated and stirred at 120 °C for 3 h in a reactor equipped with a Dean–Stark apparatus to remove the ethanol and water from the reaction mixture. The functionalised powder was collected by filtration, and successively rinsed with toluene, ethanol and diethyl ether. It was then extracted with a Soxhlet apparatus by a dichloromethane–diethyl ether mixture (1:1). Prior to the reaction, the catalyst was activated by eliminating water at 150 °C overnight. Following the reaction, the obtained product was filtered and recrystallized from a diethyl ether–hexane solution. The obtained α-monoglyceryl undecylcarboxylate was characterised by mass spectroscopy (FAB: 259 [M + 1]), FTIR spectroscopy (see S1 in the ESI†) and NMR spectroscopy (see S2 in the ESI†). Stability measurements were performed on a highly doped p-type silicon wafer (boron doped, 0.0008–0.0012 mΩ cm resistivity, (100) orientation, from Siltronix, Inc) etched at 22.5 mA cm−2 for 5 min in a 3:1 48% aqueous hydrofluoric acid–ethanol solution. Samples were mounted in a flow cell and exposed to a PBS (pH 7.4) solution at a flow rate of 200 mL min−1. Interference spectra were collected every 30 s for the silicon hydride-terminated, ozone-oxidised, thermally oxidised and undecanoic acid-modified surface samples, and every 1 min for the monoglyceride-modified surface sample. Thermal oxidation was performed in air at 450 °C for 2 h.
:1 48% aqueous hydrofluoric acid–ethanol solution. Prior to etching, the silicon chips were washed successively under sonication with acetone and ethanol for 5 min each. The α-monoglyceryl undecylcarboxylate was prepared according to a previously published procedure in our laboratory.30 The synthesis involved a reaction between undecenoic acid (Aldrich, 98%) and glycidol (Aldrich, 96%) in toluene at 120 °C with a solid catalyst (quaternary ammonium chloride-grafted silica) in anhydrous toluene for 24 h. The quaternary ammonium chloride-grafted silica catalyst preparation was adapted from ref. 26: 0.5 g of (3-(triethoxysilyl)propyl)-trimethylammonium chloride (65 wt% in water, purchased from Aldrich) was added to a suspended silica gel (G5H from Grace Davison with a surface area of 513 m2 g−1) in 30 mL of toluene. The suspension was stirred at RT for 1 h, then heated and stirred at 120 °C for 3 h in a reactor equipped with a Dean–Stark apparatus to remove the ethanol and water from the reaction mixture. The functionalised powder was collected by filtration, and successively rinsed with toluene, ethanol and diethyl ether. It was then extracted with a Soxhlet apparatus by a dichloromethane–diethyl ether mixture (1:1). Prior to the reaction, the catalyst was activated by eliminating water at 150 °C overnight. Following the reaction, the obtained product was filtered and recrystallized from a diethyl ether–hexane solution. The obtained α-monoglyceryl undecylcarboxylate was characterised by mass spectroscopy (FAB: 259 [M + 1]), FTIR spectroscopy (see S1 in the ESI†) and NMR spectroscopy (see S2 in the ESI†). Stability measurements were performed on a highly doped p-type silicon wafer (boron doped, 0.0008–0.0012 mΩ cm resistivity, (100) orientation, from Siltronix, Inc) etched at 22.5 mA cm−2 for 5 min in a 3:1 48% aqueous hydrofluoric acid–ethanol solution. Samples were mounted in a flow cell and exposed to a PBS (pH 7.4) solution at a flow rate of 200 mL min−1. Interference spectra were collected every 30 s for the silicon hydride-terminated, ozone-oxidised, thermally oxidised and undecanoic acid-modified surface samples, and every 1 min for the monoglyceride-modified surface sample. Thermal oxidation was performed in air at 450 °C for 2 h.
Characterisation
FTIR data were collected in diffuse reflectance mode with a Bruker Equinox 55 spectrometer equipped with a DRIFT (diffuse reflectance infrared Fourier transform) Spectratec collector system. Contact angle measurements were performed using Digidrop GBX fast/60 apparatus. Optical reflectivity spectra were obtained using an Ocean Optics USB2000-VIS-NIR miniature fiber optic spectrometer. A tungsten lamp was used for sample illumination. Reflected light from the samples was collected back along an axis coincident with the surface normal. The effective optical thickness, neffl, was obtained by using a fast Fourier transformation of the interference spectra according to a previously published procedure.31References
- A. Janshoff, K. P. S. Dancil, C. Steinem, D. P. Greiner, V. S. Y. Lin, C. Gurtner, K. Moteisharei, M. J. Sailor and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120(46), 12108 CrossRef CAS.
- S. Létant and M. J. Sailor, Adv. Mater., 2000, 12(5), 355 CrossRef CAS.
- S. Chan, P. M. Fauchet, Y. Li, L. J. Rothberg and B. L. Miller, Phys. Status Solidi A, 2000, 182(1), 541 CrossRef CAS.
- S. Chan, S. R. Horner, B. L. Miller and P. M. Fauchet, J. Am. Chem. Soc., 2001, 123(47), 11797 CrossRef CAS.
- M. P. Schwartz, A. M. Derfus, S. D. Alvarez, S. N. Bhatia and M. J. Sailor, Langmuir, 2006, 22(16), 7084 CrossRef CAS.
- S. D. Alvarez, M. P. Schwartz, B. Migliori, C. U. Rang, L. Chao and M. J. Sailor, Phys. Status Solidi A, 2007, 204(5), 1439 CrossRef CAS.
- M. Rocchia, E. Garrone, F. Geobaldo, L. Boarino and M. J. Sailor, Phys. Status Solidi A, 2003, 197(2), 365 CrossRef CAS.
- B. Sciacca, F. Frascella, A. Venturello, P. Rivolo, E. Descrovi, F. Giorgis and F. Geobaldo, Sens. Actuators, B, 2009, 137(2), 467 CrossRef.
- A. B. Foraker, R. J. Walczak, M. H. Cohen, T. A. Boiarski, C. F. Grove and P. W. Swaan, Pharmacol. Res., 2003, 20(1), 110 CrossRef CAS.
- E. J. Anglin, M. P. Schwartz, V. P. Ng, L. A. Perelman and M. J. Sailor, Langmuir, 2004, 20(25), 11264 CrossRef CAS.
- J. Salonen, L. Laitinen, A. M. Kaukonen, J. Tuura, M. Björkqvist, T. Heikkilä, K. Vähä-Heikkilä, J. Hirvonen and V.-P. Lehto, J. Controlled Release, 2005, 108(2–3), 362 CrossRef CAS.
- K. Zhang, S. L. E. Loong, S. Connor, S. W. K. Yu, S.-Y. Tan, R. T. H. Ng, K. M. Lee, L. Canham and P. K. H. Chow, Clin. Cancer Res., 2005, 11(20), 7532 CrossRef CAS.
- S. D. Alvarez, A. M. Derfus, M. P. Schwartz, S. N. Bhatia and M. J. Sailor, Biomaterials, 2009, 30(1), 26 CrossRef CAS.
- V. Chin, B. E. Collins, M. J. Sailor and S. N. Bhatia, Adv. Mater., 2001, 13(24), 1877 CrossRef CAS.
- L. T. Canham, Adv. Mater., 1995, 7(12), 1033 CAS.
- A. H. Mayne, S. C. Bayliss, P. Barr, M. Tobin and L. D. Buckberry, Phys. Status Solidi A, 2000, 182(1), 505 CrossRef CAS.
- J. N. Chazalviel and F. Ozanam, in Properties of Porous Silicon, ed. L. Canham, Short Run Press Ltd., London, 1997, pp. 59 Search PubMed.
- M. J. Sailor and E. Lee, Adv. Mater., 1997, 9, 783 CrossRef CAS.
- R. Boukherroub, S. Maurin, D. D. M. Wayner, F. Bensebaa, G. I. Sproule, J. M. Baribeau and D. J. Lockwood, Chem. Mater., 2001, 13, 2002 CrossRef CAS.
- J. M. Buriak, Chem. Rev., 2002, 102, 1271 CrossRef CAS.
- J. D. Bass, E. Belamie, D. Grosso, C. Boissiere, T. Coradin and C. Sanchez, J. Biomed. Mater. Res., Part A, 2009 DOI:10.1002/jbm.a.32477.
- B. Seantier, M. C. Giocondi, C. Le Grimellec and P. E. Milhiet, Curr. Opin. Colloid Interface Sci., 2008, 13(5), 326 CrossRef CAS.
- R. Boukherroub, J. T. C. Wojtyk, D. D. M. Wayner and D. J. Lockwood, J. Electrochem. Soc., 2002, 149(2), H59 CrossRef CAS.
- A. Marmur, Soft Matter, 2006, 2, 12 RSC.
- M. Chastrette and J. Caretto, Can. J. Chem., 1985, 63, 3492 CrossRef CAS.
- M. Chastrette, M. Rajzmann, M. Chanon and K. F. Purcell, J. Am. Chem. Soc., 1985, 107, 1 CrossRef CAS.
- J. N. Chazalviel and F. Ozanam, MRS Conf. Proc., 1999, 536, 155 Search PubMed.
- R. Boukherroub, D. D. M. Wayner, G. I. Sproule, D. J. Lockwood and L. T. Canham, Nano Lett., 2001, 1(12), 713 CrossRef CAS.
- V. Petrova-Koch, T. Muschik, A. Kux, B. K. Meyer, F. Koch and V. Lehmann, Appl. Phys. Lett., 1992, 61, 943 CrossRef CAS.
- A. Cauvel, G. Renard and D. Brunel, J. Org. Chem., 1997, 62, 749 CrossRef CAS.
- C. Pacholski, M. Sartor, M. J. Sailor, F. Cunin and G. M. Miskelly, J. Am. Chem. Soc., 2005, 127(33), 11636 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: IR and NMR spectra. See DOI: 10.1039/b9nj00469f |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |