DOI:
10.1039/B9NJ00442D
(Paper)
New J. Chem., 2010,
34, 132-136
Multisignaling detection of cyanide anions based on an iridium(III) complex: remarkable enhancement of sensitivity by coordination effect†
Received
(in Montpellier, France)
1st September 2009
, Accepted 16th October 2009
First published on
20th November 2009
Abstract
A new molecule, (Z)-4-(4-(dimethylamino)benzylidene)-3-methyl-1-(pyridin-2-yl)-1H-pyrazol-5(4H)-one (dmpp), and its novel heteroleptic iridium(III) complex, [(ppy)2Ir(dmpp)]PF6, were synthesized and characterized. Both dmpp and [(ppy)2Ir(dmpp)]PF6 could be used as highly selective and sensitive chemodosimeters for the cyanide anion by the naked eye, owing to the addition of the CN− to the vinyl group of dmpp. The addition of CN− to a solution of dmpp induced a change in the solution color from yellow to colorless, and to [(ppy)2Ir(dmpp)]PF6 induced a change in the solution color from pink to colorless. Moreover, [(ppy)2Ir(dmpp)]PF6 could also act as a phosphorescent and electrochemical probe of CN−. The green phosphorescence band at 520 nm and a new irreversible oxidation wave at 0.423 V showed up upon the addition of CN−, indicating that [(ppy)2Ir(dmpp)]PF6 was an excellent multisignaling chemodosimeter of CN−. Importantly, the Ir complex-based cyanide chemodosimeter [(ppy)2Ir(dmpp)]PF6 has much easier and faster detection of CN− than pure organic molecule dmpp.
Introduction
Cyanide is widely used in various industrial processes, such as gold mining, electroplating and organic chemical industries. Due to its high toxicity, there is a growing interest in the development of cyanide chemodosimeters. The methods used for this are mostly colorimetry,1 luminometry,2,3 or luminescence lifetime.4 Different approaches have also been envisaged for the design of chemodosimeters for cyanide detection. Most of them are based on the coupling of dyes to cyanide binding sites1a,2a,3i or the use of specific cyanide reactions1b–e,2e,3f,g which take advantage of the strong nucleophilicity of the cyanide anion. Since the chemodosimeters based on specific cyanide reactions always show better selectivity than other ones, more and more reaction types have been introduced with the aim of developing highly sensitive and selective probes for the cyanide anion. However, there are still several limitations pertinent to these proposed reaction type probes, among which the worst one is that the reactions could take up to tens of minutes at low analytic concentrations.1b–e In some cases, the probe systems even need to be heated3f or added with several hundred-fold excesses of cyanide anions2e in order to speed up the reaction.
Recently, the use of phosphorescent iridium(III) complexes as chemodosimeters has attracted considerable interest5 because of advantageous photophysical properties such as shifts in emission color with changes in the local environment, significant Stokes shifts for easy separation of excitation and emission, and relatively long lifetimes compared to purely organic luminophores. In particular, the long lifetime of phosphorescence can eliminate the interference of the short-lifetime background and scattering light by using a time-resolved luminescence technology. It is well-known that the photophysical properties of iridium(III) complexes are dependent on the chemical structures of ancillary ligands.6 When the ancillary ligand of an iridium(III) complex contains a specific component to interact with the analyte, the presence of this analyte can lead to dramatic changes in the photophysical and electrochemical properties of the iridium(III) complex.
In the present study, on the basis the strong chemical addition of the cyanide anion to an α,β-unsaturated carbonyl compound, a new molecule, dmpp (1), was prepared for CN− detection. Moreover, its novel heteroleptic iridium(III) complex, [(ppy)2Ir(dmpp)]PF6 (2), was also synthesized for multisignaling (UV-vis absorption, phosphorescent emission and electrochemical measurement) cyanide detection. It is worthwhile to note that the electron-withdrawing ability of pyrazol-5(4H)-one moiety of 1, and thus the reactivity toward cyanide anions, could be enhanced by the coordination of the positively-charged Ir(III) center. As a result, the Ir-complex-based cyanide chemodosimeter 2 could have much easier and faster detection for cyanide than 1 and other reported probes based on specific cyanide reactions.1b–e (see Scheme 1)
 |
| | Scheme 1 Structures of probe 1, 2 and the cyanide adduction of 2. | |
Experimental section
Materials
Tetrabutylammonium salts were obtained from Alfa Aesar and Fluka and used without further purification. 4-(Dimethylamino)benzaldehyde, potassium hexafluorophosphate and all the solvents were obtained from Sinopharm Chemical Reagent Beijing Co.
General experiments
The 1H NMR spectra were recorded on an ARX-400 NMR spectrometer. Chemical shift data for each signal are reported in ppm with tetramethylsilane as the internal reference. Elemental analyses (C, H, N) were performed on a VARIO EL instrument. Mass spectra were recorded on a Bruker Apex IV FTMS. UV-vis absorption spectra were measured on a Shimadzu UV-3100 spectrometer. The photoluminescence (PL) spectra were recorded on an Edinburgh Analytical Instruments FLS920 spectrometer.
Electrochemical measurements
Electrochemical measurements were performed with a CHI 600C Chemie’s Autolab. All measurements were carried out in a one-compartment electrolysis cell under Ar gas, equipped with a platinum bottom working electrode, a platinum wire counter electrode, and an Ag/AgCl reference electrode. The supporting electrolyte was tetrabutylammonium hexafluorophosphate (Bu4NPF6) with a concentration of 0.10 mol dm−3. The scan rate was 100 mV s−1. The ferrocene–ferrocenium (Fc+/Fc) potential was measured and used as a reference.
Synthesis of 17
A 100 mL beaker containing a mixture of 3-methyl-1-(pyridin-2-yl)-1H-pyrazol-5(4H)-one (3 mmol) and 4-(dimethylamino)benzaldehyde (3.4 mmol) was irradiated in a microwave oven for 6 min at 400 W, and then the dark red oil was dissolved in CHCl3 with a little CH3OH. After a few days for volatilization, the red solid was separate out and chromatographed by using ethyl acetate to afford a red solid in 82% yield. Melting point: 174–176 °C. 1H NMR (400 MHz, CDCl3 + CD3CN): δ 8.59 (d, 2H), 8.51 (d, 2H), 8.20 (d, 1H), 7.84 (t, 1H), 7.42 (s, 1H), 7.16 (dd, 1H), 6.79 (d, 2H), 3.16 (s, 6H), 2.37 (s, 3H). Anal. calcd for C18H18N4O: C, 70.57; H, 5.92; N, 18.29. Found: C, 70.31; H, 5.95; N, 18.17. MS (ESI+): m/z = 306.2. The NOESY spectrum shows the molecular 1 is (Z)-4-(4-(dimethylamino)benzylidene)-3-methyl-1-(pyridin-2-yl)-1H-pyrazol-5(4H)-one.
Synthesis of 2
A mixture of CH2Cl2 and ethanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was added to a flask containing [(ppy)2IrCl]2 (1 mmol) and compound 1 (1 mmol). The mixture was refluxed for 6 h. The dark red solution was cooled to room temperature and then a 10-fold excess of potassium hexafluorophosphate was added. The suspension was stirred overnight and then filtered to remove insoluble inorganic salts. The solution was evaporated to dryness under reduced pressure. It was chromatographed by CH2Cl2 to afford a brown solid in 70% yield. 1H NMR (400 MHz, CDCl3 + CD3CN): δ 8.91(d, 1H), 8.26 (s, 1H), 7.89–8.02 (m, 6H), 7.72 (d, 2H), 7.65 (s, 1H), 7.58 (d, 1H), 7.49 (s, 1H), 7.46 (m, 1H), 7.18 (t, 1H), 6.97–7.11 (m, 4H), 6.82–6.88 (m, 4H), 6.25 (d, 1H), 6.14 (d, 1H), 3.25 (s, 6H), 1.81 (s, 3H). Anal. calcd for IrC40H34F6N6OP: C, 50.47; H, 3.60; N, 8.83. Found: C, 50.23; H, 3.66; N, 8.79. MS (ESI+): m/z = 807.3 (M − PF6).
:1, v/v) was added to a flask containing [(ppy)2IrCl]2 (1 mmol) and compound 1 (1 mmol). The mixture was refluxed for 6 h. The dark red solution was cooled to room temperature and then a 10-fold excess of potassium hexafluorophosphate was added. The suspension was stirred overnight and then filtered to remove insoluble inorganic salts. The solution was evaporated to dryness under reduced pressure. It was chromatographed by CH2Cl2 to afford a brown solid in 70% yield. 1H NMR (400 MHz, CDCl3 + CD3CN): δ 8.91(d, 1H), 8.26 (s, 1H), 7.89–8.02 (m, 6H), 7.72 (d, 2H), 7.65 (s, 1H), 7.58 (d, 1H), 7.49 (s, 1H), 7.46 (m, 1H), 7.18 (t, 1H), 6.97–7.11 (m, 4H), 6.82–6.88 (m, 4H), 6.25 (d, 1H), 6.14 (d, 1H), 3.25 (s, 6H), 1.81 (s, 3H). Anal. calcd for IrC40H34F6N6OP: C, 50.47; H, 3.60; N, 8.83. Found: C, 50.23; H, 3.66; N, 8.79. MS (ESI+): m/z = 807.3 (M − PF6).
Results and discussion
Synthesis
The preparation of 1 is simple. The precursor, 3-methyl-1-(pyridin-2-yl)-1H-pyrazol-5(4H)-one,8 was easily obtained in good yield according to literature procedures by a reaction of 2-hydrazinylpyridine with ethyl acetoacetate. Ligand 1 was synthesized in a yield of 82% by the microwave-assisted Knoevenagel condensation.7 The dinuclear cyclometalated iridium(III) chloro-bridged precursor [ppy2IrCl]2 was synthesized using the same methods as those reported by Nonoyama.9 By bridge splitting reactions of [ppy2IrCl]2, and subsequent complexation with 1, complex 2 was synthesized as a brown solid in a yield of 70% and characterized by elemental analysis, 1H NMR spectroscopy, and ESI-MS spectrometry.
The nature of the reaction between 1 (2) and CN−
The probes 1 and 2 are yellow (λmax = 461 nm, log ε = 4.72) and pink (λmax = 526 nm, log ε = 4.92) dyes, respectively, while both of their cyanide adducts are transparent in dilute solution. It is possible to use probe 2 in ppm concentrations for naked eye detection of cyanide anions due to its extremely large ε.1
The nature of the reaction of cyanide anions with 1 and 2 is putative,10 and we monitored the 1H NMR spectral and ESI-MS changes produced via the addition of ∼2 equiv. cyanide anions (as tetrabutylammonium salts) at room temperature. As shown in Fig. 1, upon the addition of cyanide anions, the vinylic protons of 1 (Ha) at 7.42 ppm completely disappear, while a new signal grows in at 5.12 ppm and the molecular weight of 1 increases by 25 (CN−-1) (see ESI†). Similar changes were found for probe 2 (see both Fig. 1 and ESI†). These observations clearly indicate that the cyanide anions are added to the vinyl groups, breaking the conjugation of the probes.
 |
| | Fig. 1
1H NMR spectral changes of 1 and 2 produced via the addition of cyanide anions in a mixture of CDCl3 and CD3CN. | |
The reaction rate (detection sensitivity)
In order to confirm the high sensitivity to CN− of probe 2, time-dependent absorption spectral changes of 1 and 2 (20 μM) in the presence of excess cyanide anions (3.0 equiv. to 1 and 1.6 equiv. to 2) are monitored. As shown in Fig. 2, the addition of cyanide anions to 1 was rather slow. It took almost 80 min for the complete bleaching of 1 at room temperature, and the calculated pseudo first order rate constant for cyanide adduction is 1.32 × 10−3 s−1 (24 °C, inset b in Fig. 2). However, the addition of cyanide anion to 2 was too quick to detect, and the pink color faded out immediately when they were mixed together. (inset a in Fig. 2). As is expected, the sensitivity to cyanide anion of probe 2 is improved so significantly due to the coordination of Ir(III) center, that it meets the on-line cyanide anion detection.
 |
| | Fig. 2 Time-dependent UV-vis spectral changes of probes 1 and 2 (inset a) at 20 μM upon the addition of 3 equiv. cyanide for 1 and 1.6 equiv. for 2 in CH3CN. The inset b shows the calculation of the rate constant for cyanide addition to probe 1. | |
Photophysical titration tests
A detailed analysis of the absorption changes of 1 and 2 produced upon the addition of CN− is shown in Fig. 3. The probes 1 and 2 are in 20 and 2.0 μM concentrations, respectively, with 2.0 μM being among the lowest analytic concentrations for colorimetric cyanide detection.1 Upon the addition of 2.0 equiv. of cyanide anion to 1 and 1.6 equiv. of cyanide anion to 2, the absorbance of the solution bleached completely and the solutions became colorless. The bleaching of 2 was completed immediately after the addition of cyanide anion, but the bleaching of 1 was completed only after several hours. The presence of clear isosbestic points (λ = 353 nm for 1 and λ = 396 nm for 2) further confirms the simple reaction process. Hence, both 1 and 2 can act as excellent colorimetric probes for CN−.
Fig. 4 illustrates the emission spectral changes of 2 upon the addition of cyanide anions. The rising broad emissive band around 518 nm (19300 cm−1) is characteristic of emission from the (ppy)2Ir moiety at room temperature.11 The inset there shows a schematic representation of the mechanism for the luminescence intensity changes in the system of probe 2 upon the addition of cyanide anions. The cyanide adduction breaks the conjugation and elevates the triplet state energy levels (T1) of the dmpp moiety from below 19200 to 23400 cm−1 (Fig. 5), which is obviously higher than the excited state of the (ppy)2Ir moiety (19300cm−1) and, thus, the luminescence of this moiety is no longer quenched. Hence, complex 2 can act as an excellent phosphorescent probe for CN−.
 |
| | Fig. 4 Changes observed in luminescence spectra of 2 (2.0 μM) upon addition of cyanide at room temperature in acetonitrile. Excitation is at 396 nm. Inset: schematic representation of the cause of the rising luminescence. | |
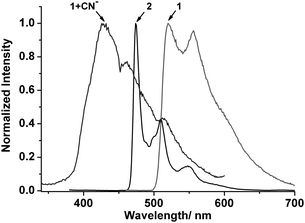 |
| | Fig. 5 The emission spectra of 1, 2, and 1 + CN− (∼4 equiv.) at 77 K in a EtOH glass in 20 μM concentrations. | |
Electrochemical titration tests
The electrochemical response of 2 to CN− was studied by cyclic voltammetry. In the absence of CN−, complex 2 shows one reversible oxidation wave at 0.72 V, which can be assigned to the metal-centered IrIII/IrIV and cyclometalated ligand oxidation processes,11 one quasi-reversible oxidation wave at 1.05 V, which can be assigned to the dimethylamino group and one irreversible reduction wave at −1.31 V , which can be assigned to the ancillary ligand. A significant modification in the cyclic voltammetry was observed upon addition of increasing amounts of CN− to an acetonitrile solution of 2 (Fig. 6). The oxidation waves at 1.05 and 0.72 V were shifted to 1.17 and 0.86 V, respectively. The reduction wave at −1.31 decreased gradually. Importantly, a new irreversible oxidation wave at 0.423 V increased linearly (insert in Fig. 6), which, obviously, can be assigned to the newborn molecule 2 + CN−. Hence, complex 2 can act as an excellent electrochemical probe for CN−.
 |
| | Fig. 6 Changes in the cyclic voltammograms of 2 (2 mM) in CH3CN solution with various amounts of CN− (0.14–2.4 mM). Inset: electrochemical titration profile of 2 at 0.423 V with CN−. | |
Selectivity tests of 1 and 2 for various anions.
Selectivity is also of paramount importance for probes. To test the selectivity of our probes, mixtures of 1 (20 μM) or 2 (2.0 μM) with various anions were monitored using absorption and naked-eye measurements (see ESI†). Among the 10 anions tested in CH3CN, namely, CN−, F−, Cl−, Br−, I−, NO3−, AcO−, H2PO4−, HSO4− and ClO4−, both 1 and 2 responded to CN−, resulting in color changes from yellow and pink, respectively, to colorless (Fig. 7). Even 12 equiv. of F−, AcO− and H2PO4− can only slightly bleach the probe 1.
 |
| | Fig. 7 Color changes of 1 (top, 20 μM) and 2 (bottom, 2.0 μM) upon adding 12 equiv. and 1.6 equiv., respectively, of various anions in CH3CN. | |
Aqueous cyanide detection
In order to examine the potential use of the excellent probe 2 for the detection of cyanide in aqueous solutions, further experiments were carried out in the water–acetonitrile 1:1 v/v solution (concentration of 2 in the mixture was 10 μM, pH ∼ 6.0). As in acetonitrile solutions, it was observed that the variation in the absorbance and emission intensity were related to the cyanide concentration (Fig. 8 and Fig. 9). However, the bleaching for 2 is much slower in the aqueous solution (heated at 50 °C for 55 min before tests) than that in acetonitrile, since water can weaken the nucleophilicity of CN−via hydration.1b To circumvent the problem brought on by water, we tried the chloroform solution of 2 to extract CN− from water. Though it was reported that a biphasic fluoride (TBAF) capture is very difficult,12 it should be easier for the large cyanide ions based on Hofmeister effects.13 For example, shaking a biphasic mixture containing 4.0 μM probe 2 in 5.0 mL CHCl3 and 10 equiv. of TBACN, 0.118 mM MgSO4, 0.689 mM NaCl and 0.182 mM KNO3 in 5.0 mL deionized water for 1 min results in distinct bleaching of probe 2 (Fig. 10), which makes it possible to easily and quickly detect levels as low as 40 μM cyanide in pure water. We feel that this protocol could be usefully applied to all other reaction-type cyanide probes which exhibit high sensitivity for CN− in organic solutions.
 |
| | Fig. 9 Emission spectral changes of the probe 2 (10 μM) seen upon the addition of cyanide anions in water–acetonitrile 1:1 v/v solution after heating at 50 °C for 55 min. Inset: emission titration profile at 486 nm versus equivalents of cyanide anions; λexi = 270 nm. | |
 |
| | Fig. 10 Color changes of 2 (40 μM) in 5 mL CHCl3 (bottom phase) after shaking for 1 min. Top phase (5 mL deionized water containing 0.118 mM MgSO4, 0.689 mM NaCl and 0.182 mM KNO3): (a) no CN−, (b) 8.0 μM CN−, (c) 16 μM CN−, (d) 24 μM CN−, (e) 32 μM CN−, (f) 40 μM CN−. | |
Conclusion
In summary, probe 1 is a specific chromo chemodosimeter and probe 2 is a specific lumino-chromo-electro chemodosimeter for cyanide anions. Probe 2 shows much larger ε at λmax and much faster detection of cyanide anions due to the strong electron-withdrawing ability of the iridium(III) center than probe 1 and most previously reported probes based on specific cyanide reactions.1b–e,2e,3f We also achieved detection of CN− in pure water based on extraction. We also believe this work will provide a practical solution to the problem of relatively slow cyanid detection in specific reaction-based approaches.
Acknowledgements
We thank the National Basic Research Program (2006CB601103) and the NNSFC (20821091, 20671006, 50772003, 90922004 and 20971006) for financial support, Hong-Wei Li for help in NOESY text and Dr Kun Hou for language editing.
Notes and references
-
(a) Y.-H. Kim and J.-I. Hong, Chem. Commun., 2002, 512 RSC;
(b) J. V. Ros-Lis, R. Martínez-Máñez and J. Soto, Chem. Commun., 2002, 2248 RSC;
(c) F. García, J. M. García, B. García-Acosta, R. Martínez-Máñez, F. Sancenón and A. Soto, Chem. Commun., 2005, 2790 RSC;
(d) D.-G. Cho, J. H. Kim and J. L. Sessler, J. Am. Chem. Soc., 2008, 130, 12163 CrossRef CAS;
(e) S.-J. Hong, J. Yoo, S.-H. Kim, J. S. Kim, J. Yoon and C.-H. Lee, Chem. Commun., 2009, 189 RSC;
(f) K. Poland, E. Topoglidis, J. R. Durrant and E. Palomares, Inorg. Chem. Commun., 2006, 9, 1239 CrossRef CAS;
(g) J. V. Ros-Lis, R. Martínez-Máñez and J. Soto, Chem. Commun., 2005, 5260 RSC;
(h) M. Tomasulo, S. Sortino, A. J. P. White and F. M. Raymo, J. Org. Chem., 2006, 71, 744 CrossRef CAS;
(i) Y. Chung and K. H. Ahn, J. Org. Chem., 2006, 71, 9470 CrossRef CAS;
(j) M. Tomasulo and F. M. Raymo, Org. Lett., 2005, 7, 4633 CrossRef CAS;
(k) J. Ren, W. Zhu and H. Tian, Talanta, 2008, 75, 760 CrossRef CAS;
(l) H. Liu, X.-B. Shao, M.-X. Jia, X.-K. Jiang, Z.-T. Lia and G.-J. Chenb, Tetrahedron, 2005, 61, 8095 CrossRef CAS;
(m) F. H. Zelder, Inorg. Chem., 2008, 47, 1264 CrossRef CAS;
(n) C. Männel-Croisé and F. Zelder, Inorg. Chem., 2009, 48, 1272 CrossRef CAS.
-
(a) S.-S. Sun and A. J. Lees, Chem. Commun., 2000, 1687 RSC;
(b) W. J. Jin, M. T. Fernández-Argüelles, J. M. Costa-Fernández, R. Pereiro and A. Sanz-Medel, Chem. Commun., 2005, 883 RSC;
(c) Y. M. Chung, B. Raman, D.-S. Kim and K. H. Ahn, Chem. Commun., 2006, 186 RSC;
(d) R. Badugu, J. R. Lakowicz and C. D. Geddes, J. Am. Chem. Soc., 2005, 127, 3635 CrossRef CAS;
(e) K.-S. Lee, H.-J. Kim, G.-H. Kim, I. Shin and J.-I. Hong, Org. Lett., 2008, 10, 49 CrossRef CAS;
(f) Z. Li, X. Lou, H. Yu, Z. Li and J. Qin, Macromolecules, 2008, 41, 7433 CrossRef CAS.
-
(a) E. Palomares, M. V. Martínez-Díaz, T. Torres and E. Coronado, Adv. Funct. Mater., 2006, 16, 1166 CrossRef CAS;
(b) R. Badugu, J. R. Lakowicz and C. D. Geddes, Dyes Pigm., 2005, 64, 49 CrossRef CAS;
(c) C.-F. Chow, M. H. W. Lam and W.-Y. Wong, Inorg. Chem., 2004, 43, 8387 CrossRef CAS;
(d) J. L. Sessler and D.-G. Cho, Org. Lett., 2008, 10, 73 CrossRef CAS;
(e) C.-L. Chen, Y.-H. Chen, C.-Y. Chen and S.-S. Sun, Org. Lett., 2006, 8, 5053 CrossRef CAS;
(f) Y.-K. Yang and J. Tae, Org. Lett., 2006, 8, 5721 CrossRef CAS;
(g) Z. Ekmekci, M. D. Yilmaz and E. U. Akkaya, Org. Lett., 2008, 10, 461 CrossRef CAS;
(h) T. W. Hudnall and F. P. Gabba, J. Am. Chem. Soc., 2007, 129, 11978 CrossRef CAS;
(i) S. K. Kwon, S. Kou, H. N. Kim, X. Chen, H. Hwang, S.-W. Nam, S. H. Kim, K. M. K. Swamy, S. Park and J. Yoon, Tetrahedron Lett., 2008, 49, 4102 CrossRef CAS;
(j) R. Badugu, J. R. Lakowicz and C. D. Geddes, Anal. Biochem., 2004, 327, 82 CrossRef CAS.
- P. Anzenbacher, D. S. Tyson, K. Jurskov and F. N. Castellano, J. Am. Chem. Soc., 2002, 124, 6232 CrossRef.
-
(a) Q. Zhao, F. Li, S. Liu, M. Yu, Z. Liu, T. Yi and C. Huang, Inorg. Chem., 2008, 47, 9256 CrossRef CAS;
(b) Q. Zhao, S. Liu, F. Li, T. Yi and C. Huang, Dalton Trans., 2008, 3836 RSC;
(c) M.-L. Ho, Y.-M. Cheng, L.-C. Wu, P.-T. Chou, G.-H. Lee, F.-C. Hsu and Y. Chi, Polyhedron, 2007, 26, 4886 CrossRef CAS.
- Y. You and S. Y. Park, J. Am. Chem. Soc., 2005, 127, 12438 CrossRef CAS.
- Y.-J. Bai, M. Li, J. Lu, Z.-J. Wang and Z. Shi, Chinese J. Org. Chem., 2004, 24, 616 Search PubMed.
- A. Cingolani, Effendy, F. Marchetti, C. Pettinari, R. Pettinari, B. W. Skelton and A. H. White, Inorg. Chem., 2004, 43, 4387 CrossRef CAS.
- K. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767 CAS.
- E. D. Bergmann, D. Ginsburg and R. Pappo, Org. React., 1959, 10, 179 CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H. E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304 CrossRef CAS.
- C.-W. Chiu and F. P. Gabbaï, J. Am. Chem. Soc., 2006, 128, 14248 CrossRef CAS.
- R. Custelcean and B. A. Moyer, Eur. J. Inorg. Chem., 2007, 1321 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |
Click here to see how this site uses Cookies. View our privacy policy here. 
