DOI:
10.1039/B9NJ00400A
(Paper)
New J. Chem., 2010,
34, 123-131
Received
(in Gainesville, FL, USA)
12th August 2009
, Accepted 29th September 2009
First published on
5th November 2009
Abstract
A series of N-glycosylamine based organogelators were synthesised from three different 4,6-O-protected saccharides. All these compounds were characterized using different spectral techniques. The effect of substituents present in the N-glycosylamines on gelation was studied from NMR and computation. Among the eighteen different gelators studied, 3b, bearing fluorine as a substituent, was observed to gelate at very low concentrations (CGC: 1%). Further, it is revealed that dipole–dipole interactions play an important role in the case of N-glycosylamine-based gelators. The presence of π–π stacking and H-bonding, as inferred from the reported X-ray diffraction data, are responsible for the gelation. Various possible modes of interaction are identified from computational studies. The gelation properties of these compounds were studied with regard to their molecular structure by scanning electron microscopy, differential scanning calorimetry, FT-IR and NMR studies.
Introduction
In recent years, low molecular weight organogelators (LMOGs) have played an important role in the field of materials.1 Some of the sugar-based derivatives possessing strong H-bond-forming segments are prone to form gels even at low concentration.2 Molecular self assembly is becoming a popular tool to construct different types of micro- and nanostructured materials.3 LMOGs are known as distinct soft materials and can self-assemble into various types of fibrils, strands, and tapes in organic solvents via weak intermolecular interactions.4 In an organogel medium, one dimensional (1D) supramoleculer fibers are bundled up together and entangled at nodes, so-called “junction points”, to form three-dimensional (3D) network structures, which entrap the solvent molecules. The nanostructured functional molecular assembly thus created is a promising candidate for organic devices with intriguing photo- and electrochemical functions.5 In the present case, H-bonding, CH–π, π–π stacking and dipole–dipole interactions seem to play an important role in self assembly. In order to obtain further insight into the structure–function relationship of sugar-based organogelators, we have generated a library of N-glycosylamines.
Results and discussion
Synthesis and characterisation of N-glycosylamines
4,6-O-Butylidene-D-glucopyranose (BGP) 2a, 4,6-O-ethylidene-D-glucopyranose (EGP) 2b and 4,6-O-benzylidene-D-glucopyranose (BzGP) 2c were synthesised from D-glucose (1)6–9 and characterised by adopting literature procedures. N-Glycosylamines were synthesised by reacting the primary amine in the aromatic moiety and the active hydroxyl group of the 4,6-O-protected D-glucose.10–12N-Glycosylamine compounds (3–5) were identified through spectral techniques. During the synthesis of different N-glycosylamines using partially-protected saccharides, gel formation was observed and this observation prompted us to study the gelation property. 1H NMR spectra of N-glycosylated products 3–5 showed glycosidic NH at around 5.0–6.0 ppm, the anomeric proton at around 4.5 ppm and the acetal proton changing with respect to the protecting group, whereas in the case of 3f, 4f and 5f, the glycosidic NH appeared below 5.0 ppm. The existence of the β-anomeric form was identified from coupling constant values given in Table 1.
Table 1 Synthesis of N-glycosylamines
| R |
Compound |
Substituents |
CGC %/g mL−1 |
δ (ano-H), 3JH1,H2/Hz |
δ (gly-NH), 3JH1,H2/Hz |
Yield (%) |
| X |
Y |
|
Denotes compound does not form gel. For all gelators, except gelators 4b, 4c and 4e, 1,2-dichlorobenzene was used to measure the CGC. For 4b and 4c, o-xylene was used and for 4e benzene was used.
|
| C3H7 |
a |
H |
Cl |
1.5 |
4.51, 8.4 |
5.71, 6.9 |
89 |
|
3
|
b |
H |
F |
1.0 |
4.47–4.59, — |
5.33, 6.3 |
81 |
| |
ca |
F |
H |
— |
4.52–4.56, — |
5.63, 5.1 |
78 |
| |
da |
Cl |
H |
— |
4.60, 8.4 |
5.59, — |
92 |
| |
e |
H |
Br |
1.5 |
4.50, 7.5 |
5.84, 6.9 |
85 |
| |
fa |
CH3 |
H |
— |
4.56–4.63 |
4.56–4.63 |
91 |
| CH3 |
a |
H |
Cl |
1.3 |
4.51, 8.4 |
5.60, 6.6 |
85 |
|
4
|
b |
H |
F |
1.5 |
4.47, 7.2 |
5.22, 6.6 |
86 |
| |
c |
F |
H |
1.1 |
4.59, 5.1 |
5.93–5.95 |
81 |
| |
da |
Cl |
H |
— |
4.60–4.74, — |
5.60, 7.2 |
87 |
| |
e |
H |
Br |
1.5 |
4.77, 5.1 |
5.68, 6.6 |
84 |
| |
fa |
CH3 |
H |
— |
4.60, 7.4 |
4.68, 6.0 |
87 |
| C6H5 |
aa |
H |
Cl |
— |
5.36, 6.3 |
5.54, — |
81 |
|
5
|
b |
H |
F |
1.5 |
5.54, 6.6 |
5.51, — |
83 |
| |
c |
F |
H |
1.5 |
4.66, 8.8 |
5.99, 6.0 |
79 |
| |
d |
Cl |
H |
1.5 |
4.69, 8.5 |
5.63, 7.3 |
91 |
| |
e |
H |
Br |
2.0 |
4.56, 7.5 |
6.10, 6.6 |
89 |
| |
fa |
CH3 |
H |
— |
5.54, 8.7 |
4.63–4.75 |
92 |
All gel samples were prepared by dissolving the gelator in a solvent in such a way that it forms a homogenous solution. The solution was allowed to cool down to room temperature, whereby the gel is formed. The gelation ability of N-glycosylamines [3(a–f)–5(a–f)] has been assessed by using “stable to inversion of the container” method. The study includes eighteen substituted aromatic-based saccharide derivatives [3(a–f)–5(a–f)] in fourteen different organic solvents and the results of gelation are summarised in Table 2. Recently, we have reported4 the gelation of a novel class of 4,6-O-protected-β-C-glycosides and sugar-pyridyl derivatives and the present studies show gel formation of N-glycosylamines. In general, protecting groups such as ethylidene, butylidene and benzylidene present on the D-glucose moiety and the substituents present on the aromatic ring showed remarkable changes to the gelation process. Among the three different 4,6-O-protected N-glycosylamines studied, BGP and EGP derivatives possessing a halogen at the para position form gels in both polar and non-polar solvents. Whereas in case of BzGP, both ortho- and para-halogen substituted derivatives form gels. The difference in gelation ability is mainly due to the effect of the protecting group and it is as follows: 4c > 5c > 3a, 3b, 3e, 4a, 4b, 4e > 5b, 5d, 5e. The critical gelation concentrations (CGC) of these compounds are given in Table 1. Among the various polar and non-polar solvents used for gelation, aromatic solvents viz., 1,2-dichlorobenzene (1,2-DCB), o-xylene, m-xylene, benzene and toluene were found to be the best solvents for the gelation process, which may be attributed to a strong solute–solvent interaction. Of all the gelators, N-glycosylamine 3b acts as an efficient organogelator and gelates at lower concentrations (CGC: 1%). The gelation ability of the organogelator has a significant dependence on the protecting group and the substitutents present on the aromatic ring. The greater gelation ability of BGP and EGP is due to higher London dispersion forces13 which exist between the alkyl chain groups. However, in BzGP, π–π interactions seem to be largely responsible for the gelation property. These results support the fact that partially protected N-glycosylamines with halogen substituted at the para position of the aromatic ring have a greater ability to undergo gelation compared to the substituents present at the ortho position. Moreover, anisotropic interactions through H-bonding, CH–π, π–π stacking and dipole–dipole interactions were also responsible for the linear aggregation of LMOGs, allowing gel formation. A pictorial representation of the groups in different N-glycosylamines, which are responsible for gel formation is shown in Fig. 1.
 |
| | Fig. 1 Rationalization of different groups responsible for gelation in different N-glycosylamines. | |
Table 2 Gelation studies of N-glycosylamines
| Solvent |
3 (R = C3H7) |
4 (R = CH3) |
5 (R = C6H5) |
| a |
b |
e |
a |
b |
c |
e |
b |
c |
d |
e |
| Note: G—gelator, PG—partial gelator, S—solution, P—precipitation, I—insoluble. |
| C6H6 |
G |
I |
G |
G |
G |
PG |
G |
G |
PG |
G |
G |
| CH3C6H5 |
G |
P |
G |
G |
G |
G |
G |
G |
G |
G |
S |
| NO2C6H5 |
G |
G |
S |
S |
S |
G |
G |
S |
PG |
S |
S |
|
p-Xylene |
G |
G |
G |
PG |
S |
G |
PG |
G |
G |
G |
G |
|
o-Xylene |
G |
G |
G |
PG |
G |
G |
PG |
PG |
G |
G |
G |
|
m-Xylene |
G |
G |
G |
G |
G |
G |
PG |
PG |
G |
G |
G |
| 1,2-DCB |
G |
G |
G |
G |
PG |
G |
G |
G |
G |
G |
G |
| CHCl3 |
P |
P |
S |
S |
G |
S |
S |
S |
S |
S |
P |
| CCl4 |
P |
P |
G |
PG |
PG |
PG |
S |
S |
PG |
G |
G |
|
i-PrOH |
G |
G |
PG |
S |
S |
S |
S |
S |
S |
S |
S |
| 1,2-DCE |
P |
G |
P |
G |
G |
PG |
PG |
PG |
S |
S |
P |
| CH3CN |
S |
G |
S |
S |
S |
S |
S |
S |
P |
P |
S |
| MeOH |
S |
P |
S |
S |
S |
P |
P |
P |
S |
S |
PG |
| EtOH |
S |
P |
S |
S |
S |
PG |
P |
P |
PG |
S |
S |
Single crystal X-ray diffraction (XRD) data of compounds 3d11 and 4d12 helps us to understand the π–π stacking and H-bonding interactions that exists in different N-glycosylamines. Free hydroxyl groups present at the C-2 and C-3 positions of the glucopyranose ring are involved in the strong hydrogen bonding, thereby forming a chain of molecules (Fig. 2 and Fig. 3). Although protecting groups are different in 3d and 4d, the hydrogen bonding interactions are the same and one may presume that just changing the protecting group and substituents may not affect the hydrogen bonding. However, H-bonding of the core sugar moiety remains unaltered in all the N-glycosylamines. Thus, self-assembly of a gelator molecule depends upon the nature of the protecting group and also the substituents present in the aromatic ring.
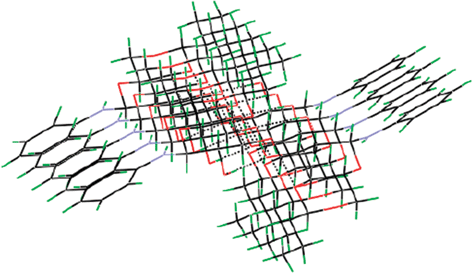 |
| | Fig. 2 π–π Stacking and H-bonding interactions of compound 3d.11 | |
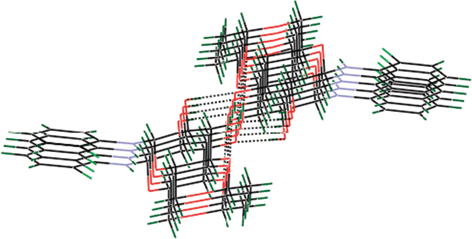 |
| | Fig. 3 π–π Stacking and H-bonding interactions of compound 4d. 12 | |
 |
| | Fig. 4 SEM images of organogel formed from (a), 5d in 1,2-DCB; (b), 3b in 1,2-DCE; (c), 5c in 1,2-DCB; (d), 4e in benzene; (e), 4e in benzene (magnified) and (f), 4e in benzene (fiber and bore are labeled). | |
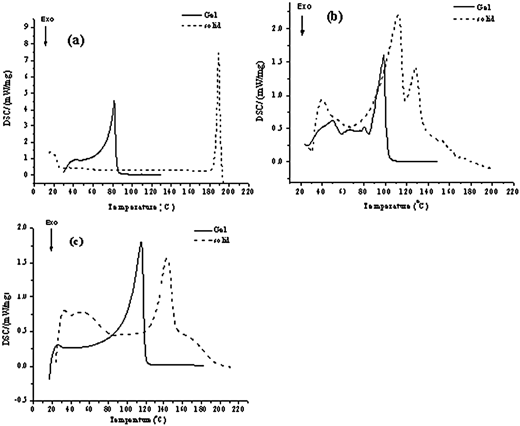
Thermoanalysis
To study the thermal properties of the organogelators and gels, differential scanning calorimetry (DSC) experiments for three different N-glycosylamines of 4,6-O-protected derivatives (3a, 4b and 5c) were performed. In the case of 3a, the enthalpy values and the melting point peak values are found to be 191 J g−1 and 189 °C, and 248 J g−1 and 82 °C in the solid and gel phases, respectively (Fig. 5). The enthalpy values and the melting point peaks for compound 4b are 35 J g−1 and 128 °C in the solid phase, and 74 J g−1 and 99 °C in the gel phase. The peak appearing at around 112 °C (ΔH = 138 J g−1) is due to the liberation of moisture. The enthalpy values and melting point peak for compound 5c and its gel were found to be 113.4 J g−1 and 143 °C, and 186 J g−1 and 115 °C, respectively. These results indicate the thermal stabilities of both gel and organogelator. DSC studies further reveal that in the gel phase, the molecules were loosely packed and the system moves from the most ordered state to a less ordered state. The phase transition temperatures of compounds, 3a, 4b and 5c and their corresponding gels are given in Table 3. The melting temperatures of gelator and gel are obtained from DSC experiments.
Table 3 Melting temperatures of 3a, 4b and 5c in the solid and gel phases
| Compound |
Phase transition temperature of gelator/°C |
Phase transition temperature of gel/°C |
|
3a
|
189 |
82 |
|
4b
|
128 |
99 |
|
5c
|
143 |
115 |
NMR studies
1H NMR studies were carried out for solution of 1.0–1.5% wt of compounds 4a and 4f. In the case of compound 4a (Fig. 6) the formation of the gel at 27 °C was accompanied by a broadening and shifting of both aromatic and core sugar proton signals. The 1H NMR spectrum of non-gelator 4f shows slight broadening due to D2O exchange and not due to gelation (Fig. 7); gelator molecules are assembled in the gel network by means of hydrogen bonding, π–π stacking and dipole–dipole interactions, which results in the broadening of signals and due to long correlation times. The entire broadening and shifting of signals in the organogelator 4a shows the involvement of the aromatic ring, the sugar part and also the protecting group, whereas, in the case of non-gelator molecule 4f, these effects were not observed. Moreover, the involvement of H-bonding in gelation can also be explained by FT-IR and single crystal XRD studies.11,12
 |
| | Fig. 6
1H NMR of compound, 4a recorded at 27 °C in (a) CDCl3 + 2 drops of DMSO-d6 and (b) CDCl3 + 2 drops of DMSO-d6 + 4 drops of D2O. | |
 |
| | Fig. 7
1H NMR of compound, 4f recorded at 27 °C in (a) CDCl3 + 2 drops of DMSO-d6 and (b) CDCl3 + 2 drops of DMSO-d6 + 4 drops of D2O. | |
Computational studies
In the present study, gelators 3b, 4b and 5b and non-gelators 3f, 4f, and 5f have been taken into account. All the systems were optimized (without any geometrical constraint) by using the M05-2X hybrid density functional method.15 The optimized geometries were characterized as the minima on the potential energy surface by frequency analysis. The stabilization energies (SEs) are obtained using the equation,
| SE = −[(Ecomplex − (Emonomer1 + Emonomer2)] |
where Ecomplex, Emonomer1, and Emonomer2 are the total energies of the complex and the monomers. The SE are corrected for basis set superposition error (BSSE) using the method adopted by Boys and Bernardi.16 All the calculations were performed using the GAUSSIAN 03 (revision E.01) suite of programs.17 Due to the large system size and limited computational facility, two approaches were used for calculation here: (i) the complete system with the 6-31G* basis set; (ii) only the active part of the system with the 6-311++G* basis set.
All the optimized dimeric complexes are shown in Fig. 9 and their corresponding BSSE-corrected SE are depicted in Table 4. Since the binding energies for gelator complexes are comparatively larger than those of non-gelators, one can predict that the dipole–dipole interactions (Fig. 9) play a vital role in the gel formation. Fluorine substitution plays a significant role in the high SE values because it interacts with the glycosidic NH group of N-glycosylamines. In the case of non-gelators, very weak CH–π interactions are observed, causing the lower SE.
 |
| | Fig. 9 Optimized geometries of the complete dimer: grey = C, blue = N, red = O, white = H, yellow = F | |
Table 4 Stabilization energy due to dipole interactions obtained at the M05-2X level of calculation using 6-31G* basis set
| S. No. |
Compound |
BSSE-corrected SE/kcal mol−1 |
| 1 |
3b
|
9.66948 |
| 2 |
4b
|
6.98644 |
| 3 |
5b
|
8.39269 |
| 4 |
3f
|
2.54499 |
| 5 |
4f
|
1.52937 |
| 6 |
5f
|
3.25684 |
Other interactions are also important in gel formation, therefore analysis of just the active part of the system has been considered. Since this system size is small, a larger basis set has been employed for the calculations. All the SE and optimized geometries are shown in Table 5 and Fig. 10. In the case of the benzene dimer (Bz–Bz), only the stable parallel-displaced conformation is taken into account, the other conformation, parallel, is less stable.4b The calculation suggests that, apart from the dipole–dipole interacting sites, various other possible interactions play a significant role in the gel formation (Fig. 10). The Bz–Bz stacking energy is 1.67 kcal mol−1 which reflects in the weak tail–tail interactions in 5b, whereas in the case of 3b and 4b, head–head and head–tail interactions were observed. Two different basis sets were used here, but in the first case (i.e. complete system consideration) results show good agreement with the experimental results. Moreover, cis-Me–C6H4–NH2 and cis-F–C6H4–NH2 have larger SE values than their corresponding trans arrangements, as presented in Table 5, but practically (i.e. considering the complete molecule) this arrangement is highly unstable due to the repulsion among the various groups.
 |
| | Fig. 10 Optimized geometries of the various interacting active parts of the gelators and non-gelators: grey = C, blue = N, red = O, white = H, yellow = F. | |
Table 5 Stabilization energies due to all possible interactions obtained from M05-2X level of calculation using the 6-311++G* basis set
| Entry |
Interacting species |
Nature of interaction |
BSSE-corrected SE/kcal mol−1 |
| 1 |
Bz (parallel displaced) |
Dimer |
1.6739 |
| 2 |
trans-Me–C6H4–NH2 |
Dimer |
3.3971 |
| 3 |
cis-Me–C6H4–NH2 |
Dimer |
5.8217 |
| 4 |
trans-F–C6H4–NH2 |
Dimer |
5.1062 |
| 5 |
cis-F–C6H4–NH2 |
Dimer |
6.8986 |
| 6 |
Bz–F–C6H4–NH2 |
— |
2.8867 |
| 7 |
Bz–Sug |
— |
4.7878 |
| 8 |
F–C6H4–NH2–Sug |
— |
5.9550 |
| 9 |
Me–C6H4–NH2–Sug |
— |
6.8595 |
Conclusion
In summary, we have reported the synthesis and gelation properties of various sugar-based N-glycosylamines derived from aromatic amines. The presence of π–π stacking and H-bonding in N-glycosylamines was identified from single crystal XRD studies.11,12 The gelation nature of these sugar N-glycosylamine derivatives depends on the protecting groups such as 4,6-O-butylidene, 4,6-O-ethylidene, 4,6-O-benzylidene and halogen substituents. Variation in the protecting groups and the position of the halogen atom alters the morphology and the gelation abilities to a considerable extent as observed from SEM analysis. Thermoanalysis of these gelators reveals that molecules move from the most ordered state to a less ordered state by forming a gel, whereby the melting temperature becomes lower than the corresponding gelator. Moreover, the presence of H-bonding was established from FT-IR and of molecular aggregation by NMR studies. Computational studies show the existance of dipole–dipole interactions, CH–π and π–π stacking which play a vital role in gel formation. Experimental results were reinforced with theoretical prediction. From these results, even a small tuning of molecules can drastically alter their gelation properties. Based on the present studies, one could conclude that the presence of aliphatic or aromatic rings present at the 4,6-O-positions of the D-glucose unit of N-glycosylamines result in gelation. The presence of halogen atoms is needed for dipole–dipole interaction, whereas the methyl-substituted do not form gels.
Experimental
General details
All reagents were purchased from Sigma Aldrich (benzaldehyde dimethyl acetal), SRL (D-glucose) and Loba chemie (butyraldehyde, paraldehyde, aromatic amines). Solvents used for gelation studies were analytical grade and were purchased from Qualigens, Sd-fine chemicals and SRL, India. NMR spectra were recorded with a Bruker Avance 300 (300 MHz) using TMS as internal standard. FT-IR studies were carried out using a Perkin Elmer PE257 IR spectrometer. Thermal transitions for gelators and gels were determined on a NETZSCH DSC 204 instrument. The measurments were carried out under nitrogen atmosphere using 50 μL sealed aluminium sample pans. The temperature calibration for the DSC was done using two standard materials (n-decane, indium) and energy calibration by an indium standard (28.45 J g−1). The gels were imaged with a HITACHI-S-3400W scanning electron microscope. The samples were gold-coated in such a way that the gold coating was less than 1 nm thick on average. Elemental analysis was performed by using a Perkin-Elmer 2400 series CHNS/O analyser.
BGP, EGP and BzGP were synthesised by adopting the literature procedures.6–9N-Glycosylamines 3c, 3d, 4c, 4d, 5c and 5d were synthesised and their purity confirmed using different spectral techniques.10–12 Synthetic procedures and spectral data for all other compounds are provided below.
Synthesis
General synthetic procedure for the synthesis of compounds 3–5 is as follows:
To a solution of 1 mol of the 4,6-O-protected-β-D-glucopyranose, (2a–c) in 10 ml of ethanol, 1.2 mol of the substituted aniline was added. The reaction mixture was then stirred at 50 °C for 10 min and at room temperature for 24 h. The reaction was thereby followed through TLC. The solid, which separates, was filtered off, washed with ethanol and dried using diethyl ether.
Synthesis of gels
The gelation ability of N-glycosylamines was determined by the “stable to inversion of the container” method.13 The gelation properties have been tested with fourteen different solvents as follows: gelator (1.5 mg) was mixed in a close capped test tube with 1 ml of solvent to result in a concentration of 1.5% (gm L−1) and the mixture was heated until the solid was dissolved. By this procedure, the solvent boiling point becomes higher than that under standard atmospheric pressure. The sample vial was cooled in air to 25 °C, left for 12 h at this temperature and then turned upside down. When the gelator formed a clear or slightly opaque gel by immobilizing the solvent at this stage, it was denoted by “G”. Some of them form a partial gel, denoted PG. Some of them are insoluble and some get precipitated, denoted by I and P, respectively.
Spectral characterization of gelators
Abbreviations, such as Sac and Ar, correspond to the saccharide and aromatic groups, respectively.
4,6-O-Butylidene-N-(p-chlorophenyl)-β-D-glucopyranosylamine (3a).
Mp 186–188 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.09 (d, J = 8.4 Hz, 2H, Ar–H), 6.68 (d, J = 8.4 Hz, 2H, Ar–H), 5.71 (d, J = 6.9 Hz, 1H, –NH), 5.10 (s, 1H, Sac-OH), 4.80 (s, 1H, Sac-OH), 4.57 (d, J = 4.8 Hz, 1H, Sac-H), 4.51 (t, J = 8.4 Hz, 1H, Sac-H), 4.12–4.16 (dd, J = 3.9 Hz, J = 9.6 Hz, 1H, Sac-H), 3.69 (t, J = 8.7 Hz, 1H, Sac-H), 3.26–3.51 (m, 4H, Sac-H), 1.60–1.67 (m, 2H, –CH2), 1.37–1.50 (m, 2H, –CH2), 0.92 (t, J = 7.2 Hz, 3H, –CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 144.7, 128.3, 122.4, 114.6, 101.7, 86.2, 80.0, 73.6, 73.4, 67.9, 66.7, 35.8, 16.9, 13.5 ppm. Anal. calcd for C16H22NO5Cl: C 55.90, H 6.45, N 4.07. Found C 56.18, H 6.62, N 4.19.
4,6-O-Butylidene-N-(p-fluorophenyl)-β-D-glucopyranosylamine (3b).
Mp 147–149 °C; 1H NMR (CDCl3 + DMSO-d6): δ 6.84–6.90 (m, 2H, Ar–H), 6.67–6.71 (m, 2H, Ar–H), 5.33 (d, J = 6.3 Hz, 1H, –NH), 4.99 (d, J = 4.2 Hz, 1H, Sac-H), 4.47–4.59 (m, 3H, Sac-H), 4.14–4.19 (dd, J = 4.2 Hz, J = 9.6 Hz, 1H, Sac-H), 3.69–3.73 (m, 1H, Sac-H), 3.28–3.54 (m, 4H, Sac-H), 1.61–1.67 (m, 2H, –CH2), 1.41–1.48 (m, 2H, –CH2), 0.92 (t, J = 6.9 Hz, 3H, –CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 142.5, 115.4, 115.1, 114.8, 114.7, 102.1, 87.3, 80.3, 74.1, 73.9, 68.3, 67.1, 36.1, 17.2, 13.8 ppm. Anal. calcd for C16H22NO5F: C 58.71, H 6.77, N 4.28. Found C 58.47, H 7.06, N 4.51.
4,6-O-Butylidene-N-(p-bromophenyl)-β-D-glucopyranosylamine (3e).
Mp 118–120 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.21 (d, J = 8.7 Hz, 2H, Ar–H), 6.64 (d, J = 8.7 Hz, 2H, Ar–H), 5.84 (d, J = 6.9 Hz, 1H, –NH), 5.22 (d, J = 4.2 Hz, 1H, Sac-H), 4.95 (d, J = 3.3 Hz, 1H, Sac-H), 4.56 (t, J = 5.1 Hz, 1H, Sac-H), 4.50 (d, J = 7.5 Hz, 1H, Sac-H), 4.11–4.15 (dd, J = 4.2 Hz, J = 9.4 Hz, 1H, Sac-H), 3.65–3.72 (m, 1H, Sac-H), 3.29–3.50 (m, 4H, Sac-H), 1.59–1.67 (m, 2H, –CH2), 1.37–1.49 (m, 2H, –CH2), 0.92 (t, J = 7.3 Hz, 3H, –CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 145.1, 131.1, 115.1, 109.5, 101.7, 90.0, 80.04, 73.6, 73.4, 67.9, 66.7, 35.8, 16.9, 13.5 ppm. Anal. calcd for C16H22NO5Br: C 49.50, H 5.71, N 3.61. Found C 49.27, H 5.92, N 3.45.
4,6-O-Butylidene-N-(o-methylphenyl)-β-d-glucopyranosylamine (3f).
Mp 101–103 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.01–7.11 (m, 2H, Ar–H), 6.69–6.78 (m, 2H, Ar–H), 4.56–4.63 (m, 3H, Sac-H), 4.15–4.19 (dd, J = 4.2 Hz, J = 9.2 Hz, 1H, Sac-H), 3.73–3.79 (m, 1H, Sac-H), 3.41–3.55 (m, 3H, Sac-H), 3.34 (t, J = 8.9 Hz, 1H, Sac-H), 3.03 (s, 2H, Sac-H), 2.19 (s, 3H, Ar–CH3),1.62–1.68 (m, 2H, –CH2), 1.41–1.48 (m, 2H, –CH2), 0.92 (t, J = 7.4 Hz, 3H, –CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 143.5, 129.7, 126.5, 122.4, 118.4, 111.4, 101.8, 86.1, 79.9, 73.8, 73.6, 68.0, 66.8, 35.8, 17.0, 16.87, 13.4 ppm. Anal. calcd for C17H25NO5: C 63.14, H 7.79, N 4.33. Found C 62.89, H 7.87, N 4.50.
4,6-O-Ethylidene-N-(p-chlorophenyl)-β-D-glucopyranosylamine (4a).
Mp 123–125 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.09 (d, J = 8.7 Hz, 2H, Ar–H), 6.71 (d, J = 8.7 Hz, 2H, Ar–H), 5.60 (d, J = 6.6 Hz, 1H, –NH), 5.06 (d, J = 3.9 Hz, 1H, Sac-H), 4.74 (t, J = 4.0 Hz, 2H, Sac-H), 4.48–4.53 (dd, J = 6.9 Hz, J = 8.4 Hz, 1H, Sac-H), 4.11–4.16 (dd, J = 4.2 Hz, J = 9.9 Hz, 1H, Sac-H), 3.67–3.74 (m, 1H, Sac-H), 3.40–3.53 (m, 4H, Sac-H), 1.36 (d, J = 5.1 Hz, 3H, –CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 149.8, 133.6, 127.8, 119.8, 104.2, 91.5, 85.2, 78.9, 78.7, 73.0, 71.8, 25.2 ppm. Anal. calcd for C14H18NO5Cl: C 53.25, H 5.75, N 4.44. Found C 53.46, H 5.63, N 4.61.
4,6-O-Ethylidene-N-(p-fluorophenyl)-β-D-glucopyranosylamine (4b).
Mp 124–126 °C; 1H NMR (CDCl3 + DMSO-d6): δ 6.85–6.90 (d, J = 8.6 Hz, 2H, Ar–H), 6.67–6.72 (d, J = 8.5 Hz, 2H, Ar–H), 5.22 (d, J = 6.6 Hz, 1H, –NH), 5.01 (d, J = 3.9 Hz, 1H, Sac-H), 4.73–4.76 (m, 2H, Sac-H), 4.49–4.45 (t, J = 7.2 Hz, 1H, Sac-H), 4.14–4.17 (m, 1H, Sac-H), 3.73–3.75 (m, 1H, Sac-H), 3.35–3.53 (m, 4H, Sac-H), 1.38 (d, J = 5.1 Hz, 3H, –CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 147.4, 120.3, 120.0, 119.6, 119.5, 104.1, 92.6, 85.2, 78.99, 78.7, 73.1, 71.8, 25.2 ppm. Anal. calcd for C14H18NO5F: C 56.18, H 6.06, N 4.68. Found C 55.87, H 6.24, N 4.75.
4,6-O-Ethylidene-N-(p-bromophenyl)-β-D-glucopyranosylamine (4e).
Mp 127–129 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.26 (d, J = 8.7 Hz, 2H, Ar–H), 6.62 (d, J = 8.7 Hz, 2H, Ar–H), 5.68 (d, J = 6.6 Hz, 1H, –NH), 5.20 (d, J = 3.9 Hz, 1H, Sac-H), 4.84 (d, J = 3.4 Hz, 1H, Sac-H), 4.77 (d, J = 5.1 Hz, 1H, Sac-H), 4.52–4.58 (t, J = 7.8 Hz, 1H, Sac-H), 4.15–4.19 (dd, J = 4.2 Hz, J = 10.2 Hz, 1H, Sac-H), 3.32–4.19 (m, 5H, Sac-H), 1.40 (d, J = 4.8 Hz, 3H, –CH3); 13C NMR (CDCl3 + DMSO-d6): δ 145.3, 131.6, 115.4, 110.1, 99.3, 80.2, 73.9, 73.7, 68.2, 66.9, 20.3. Anal. calcd for C14H18NO5Br: C 46.68, H 5.04, N 3.89. Found C 46.45, H 5.26, N 4.03.
4,6-O-Ethylidene-N-(o-methylphenyl)-β-D-glucopyranosylamine (4f).
Mp 129–131 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.02–7.11(m, 2H, Ar–H), 6.69–6.79 (m, 2H, Ar–H), 4.98 (d, J = 4.2 Hz, 1H, Sac-H), 4.73–4.78 (dd, J = 4.8 Hz, J = 9.9 Hz, 1H, Sac-H), 4.68 (d, J = 6.0 Hz, 1H, Sac-H), 4.60 (t, J = 7.4 Hz, 1H, Sac-H), 4.50 (d, J = 2.7 Hz, 1H, Sac-H), 4.15–4.19 (m, 1H, Sac-H), 3.73–3.79 (m, 1H, sac-H), 3.50–3.57 (m, 4H, Sac-H), 2.19 (s, 3H, Ar–CH3), 1.39 (d, J = 4.8 Hz, 3H, –CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 143.9, 130.1, 126.9, 122.8, 118.8, 111.8, 99.4, 86.3, 80.3, 74.3, 74.1, 68.3, 67.0, 20.3, 17.4 ppm. Anal. calcd for C15H21NO5: C 61.00, H 7.17, N 4.74. Found C 61.13, H 7.36, N 4.60.
4,6-O-Benzylidene-N-(p-chlorophenyl)-β-D-glucopyranosylamine (5a).
Mp 140–143 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.51 (d, J = 3.3 Hz, 2H, Ar–H), 7.26 (d, J = 3.3 Hz, 3H, Ar–H), 7.12 (d, J = 8.4 Hz, 2H, Ar–H), 6.70 (d, J = 8.7 Hz, 2H, Ar–H), 5.54 (s, 1H, –NH), 5.36 (d, J = 6.3 Hz, 1H, Sac-H), 4.91 (s, 1H, Sac-OH), 4.56–4.61 (m, 2H, Sac-H), 4.28–4.33 (m, 1H, Sac-H), 3.59–3.84 (m, 5H, Sac-H) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 114.7, 137.3, 129.1, 128.9, 128.1, 126.4, 123.5, 115.2, 101.8, 86.8, 80.9, 74.1, 73.9, 68.8, 67.1 ppm. Anal. Cald for C19H20NO5Cl: C 60.40, H 5.34, N 3.71. Found C 60.65, H 5.19, N 3.94.
4,6-O-Benzylidene-N-(p-fluorophenyl)-β-D-glucopyranosylamine (5b).
Mp 117–119 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.46–7.51 (m, 2H, Ar–H), 7.34–7.36 (m, 3H, Ar–H), 6.82–6.90 (m, 2H, Ar–H), 6.69–6.73 (dd, J = 4.5 Hz, J = 11.4 Hz, 2H, Ar–H), 5.53 (m, 1H, –NH), 5.18 (s, 1H, Sac-OH), 4.48–5.04 (m, 2H, Sac-H), 5.54 (d, J = 6.6 Hz, 1H, Sac-H), 4.30–4.34 (m, 1H, Sac-H), 3.45–3.95 (m, 5H, Sac-H) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 142.4, 133.8, 132.9, 131.3, 120.3, 120.0, 119.6, 119.5, 106.4, 92.2, 86.0, 78.9, 78.6, 73.6, 71.8 ppm. Anal. calcd for C19H20NO5F: C 63.15, H 5.58, N 3.88. Found C 62.80, H 5.77, N 3.71.
4,6-O-Benzylidene-N-(p-bromophenyl)-β-D-glucopyranosylamine (5e).
Mp 135–137 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.50 (d, J = 2.4 Hz, 2H, Ar–H), 7.35 (d, J = 2.9 Hz, 2H, Ar–H), 6.88 (d, J = 8.7 Hz, 2H, Ar–H), 7.22 (d, J = 8.7 Hz, 1H, Ar–H), 6.67 (d, J = 8.7 Hz, 1H, Ar–H), 6.10 (d, J = 6.6 Hz, 1H, –NH), 5.53 (d, J = 5.7 Hz, 1H, Sac-H), 4.56 (t, J = 7.5 Hz, 1H, Sac-H), 4.21–4.30 (m, 2H, Sac-H), 4.10 (d, J = 2.4 Hz, 1H, Sac-H), 3.12–3.72 (m, 5H, Sac-H) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 150.7, 142.6, 136.4, 133.8, 132.9, 131.4, 120.3, 114.3, 106.3, 91.3, 86.1, 83.4, 78.6, 73.6, 71.8 ppm. Anal. calcd for C19H20NO5Br: C 54.04, H 4.77, N 3.32. Found C 54.36, H 4.71, N 3.42.
4,6-O-Benzylidene-N-(o-methylphenyl)-β-D-glucopyranosylamine (5f).
Mp 99–101 °C; 1H NMR (CDCl3 + DMSO-d6): δ 7.51 (d, J = 4.5 Hz, 2H, Ar–H), 7.36 (d, J = 3.9 Hz, 3H, Ar–H), 7.02–7.12 (m, 2H, Ar–H), 6.70–6.81 (m, 2H, Ar–H), 5.54 (d, J = 8.7 Hz, 1H, –NH), 5.02 (s, 1H, Sac-OH), 4.63–4.75 (m, 3H, Sac-H), 4.32–4.37 (m, 1H, Sac-H), 3.48–3.85 (m, 4H, Sac-H), 2.87 (m, 1H, Sac-H), 2.20 (s, 3H, Ar-CH3) ppm; 13C NMR (CDCl3 + DMSO-d6): δ 148.2, 141.6, 134.2, 133.1, 132.2, 131.0, 130.6, 126.9, 122.9, 116.0, 105.8, 91.0, 85.3, 78.3, 78.1, 73.0, 71.2, 21.6 ppm. Anal. calcd for C20H23NO5: C 67.21, H 6.49, N 3.92. Found C 67.55, H 6.23, N 3.78.
Acknowledgements
T.M.D. acknowledges financial support from the Department of Science and Technology, New Delhi, India. We thank the Department of Science and Technology, New Delhi for the 300 MHz NMR facility under FIST scheme to the Department of Organic Chemisty, University of Madras, Chennai, India. S.N. thank UGC, New Delhi for research fellowship.
References
-
(a) J. Peng, K. Liu, Q. Zhang, X. Feng and Y. Fang, Langmuir, 2008, 24, 2992 CrossRef CAS;
(b) M. George, S. L. Snyder, P. Terech, C. J. Glinka and R. G. Weiss, J. Am. Chem. Soc., 2003, 125, 10275 CrossRef CAS;
(c) M. George and R. G. Weiss, Chem. Mater., 2003, 15, 2879 CrossRef CAS;
(d) X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am. Chem. Soc., 2005, 127, 4336 CrossRef CAS;
(e) X. Huang, S. R. Raghavan, P. Terech and R. G. Weiss, J. Am. Chem. Soc., 2006, 128, 15341 CrossRef CAS;
(f) M. George, G. P. Funkhouser and R. G. Weiss, Langmuir, 2008, 24, 3537 CrossRef CAS;
(g) T. Klawonn, A. Gansauer, I. Winkler, T. Lauterbach, D. Franke, R. J. M. Nolte, M. C. Feiters, H. Borner, J. Hentschel and K. H. Dotz, Chem. Commun., 2007, 1894 RSC;
(h) L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS;
(i) S. Bhattacharya and S. N. G. Acharya, Chem. Mater., 1999, 11, 3504 CrossRef CAS;
(j) M. de Loos, J. H. van Esch, R. M. Kellogg and B. L. Feringa, Tetrahedron, 2007, 63, 7285 CrossRef CAS;
(k) M. George, G. Tan, V. T. Jhon and R. G. Weiss, Chem.–Eur. J., 2005, 11, 3243 CrossRef CAS;
(l) M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489 CrossRef CAS;
(m) C. Baddeley, Z. Yan, G. King, P. M. Woodward and J. D. Badjic, J. Org. Chem., 2007, 72, 7270 CrossRef CAS;
(n) A. Pal, Y. K. Ghosh and S. Bhattacharya, Tetrahedron, 2007, 63, 7334 CrossRef CAS;
(o) K. Tanaka, S. Hayashi and M. R. Caira, Org. Lett., 2008, 10, 2119 CrossRef CAS.
-
(a) O. Gronwald, K. Sakurai, R. Luboradzki, T. Kimura and S. Shinkai, Carbohydr. Res., 2001, 331, 307 CrossRef CAS;
(b) O. Gronwald and S. Shinkai, J. Chem. Soc., Perkin Trans. 2, 2001, 2, 1933 Search PubMed;
(c) R. J. H. Hafkamp, B. P. A. Kokke, I. M. Danke, H. P. M. Geurts, A. E. Rowan, M. C. Feiters and J. M. Notle, Chem. Commun., 1997, 545 RSC;
(d) R. Ghosh, A. Chakrabarty, D. K. Maiti and V. G. Puranik, Org. Lett., 2006, 8, 1061 CrossRef CAS;
(e) B. Xing, C.-W. Yu, K.-H. Chow, P.-L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846 CrossRef CAS;
(f) S. Kiyonaka, K. Sugiyasu, S. Shinkai and I. Hamachi, J. Am. Chem. Soc., 2002, 124, 10954 CrossRef CAS;
(g) J. H. Jung, J. A. Rim, E. J. Cho, S. J. Lee, I. Y. Jeong, N. Kameda, M. Masuda and T. Shimizu, Tetrahedron, 2007, 63, 7449 CrossRef CAS;
(h) S. Cheuk, E. D. Stevens and G. Wang, Carbohydr. Res., 2009, 344, 417 CrossRef CAS and references cited therein.
-
(a) S. Zhang, Nat. Biotechnol., 2003, 21, 1171 CrossRef CAS;
(b) X. Y. Gao and H. Matsui, Adv. Mater., 2005, 17, 2037 CrossRef CAS;
(c) D. T. Bong, T. D. Clark, J. R. Granja and M. R. Ghadiri, Angew. Chem., Int. Ed., 2001, 40, 988 CrossRef CAS;
(d) V. Percec, A. E. Dulcey, V. S. K. Balagurusamy, Y. Miura, J. Smidrkal, M. Peterca, S. Nummelin, U. Edlund, S. D. Hudson, P. A. Heiney, H. Duan, S. N. Maganov and S. A. Vinogradov, Nature, 2004, 430, 764 CrossRef CAS.
-
(a) S. Nagarajan, T. Mohan Das, P. Arjun and N. Raaman, J. Mater. Chem., 2009, 19, 4587 RSC;
(b) K. Karthik Kumar, M. Elango, V. Subramanian and T. Mohan Das, New J. Chem., 2009, 33, 1570 RSC;
(c) S. Nagarajan and T. Mohan Das, Carbohydr. Res., 2009, 334, 1028 CrossRef.
- K. Sugiyasu, S.-I. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863 CrossRef CAS.
- P. L. Barili, G. C. Berti, G. Catelani, C. Cini, F. D. Andran and F. Mastrorilli, Carbohydr. Res., 1995, 278, 43 CrossRef CAS.
- P. L. Mellies, C. L. Mehltretter and E. C. Rist, J. Am. Chem. Soc., 1951, 73, 294 CrossRef.
- G. T. Bonner, J. E. Bourne and D. Lewis, J. Chem. Soc., 1965, 7453 RSC.
- R. Barker and D. L. Mac Donald, J. Am. Chem. Soc., 1960, 82, 2301 CrossRef CAS.
- T. Mohan Das, C. P. Rao and E. Kolehmainen, Carbohydr. Res., 2001, 334, 261 CrossRef CAS.
- G. Rajsekhar, C. P. Rao, P. K. Saarenketo, E. Kolehmainen and K. Rissanen, Carbohydr. Res., 2002, 337, 187 CrossRef CAS.
- A. K. Sah, C. P. Rao, P. K. Saarenketo, E. K. Wegelius, K. Rissanen and E. Kolehmainen, J. Chem. Soc., Dalton Trans., 2000, 3681 RSC.
- H. Yu, H. Kawanishi and H. Koshima, J. Photochem. Photobiol., A, 2006, 178, 62 CrossRef CAS.
- A. V. Stuart and G. B. B. M. Sutherland, J. Chem. Phys., 1956, 24, 559 CAS.
- Y. Zhao, N. E. Schultz and D. G. Truhlar, J. Chem. Theory Comput., 2006, 2, 364 CrossRef.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al–Laham, C. Y. Peng, A. Nanayakkara, M. hallacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03, (Revision E.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
|
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.