Metal ions binding to NAD-glycohydrolase from the venom of Agkistrodon acutus: Regulation of multicatalytic activity
Xiaolong
Xu
*a,
Liyun
Zhang
b,
Zhaofeng
Luo
c,
Dengke
Shen
a,
Hao
Wu
a,
Lili
Peng
a,
Jiajia
Song
a and
Yan
Zhang
a
aDepartment of Chemistry, University of Science and Technology of China, Hefei, 230026, P. R. China. E-mail: xuxl@ustc.edu.cn; Fax: 86-551-3603388; Tel: 86-551-3603214
bNational Synchrotron Radiation Laboratory, University of Science and Technology of China, Hefei, 230029, P. R. China
cSchool of lifesciences, University of Science and Technology of China, Hefei, 230026, P. R. China
First published on 3rd June 2010
Abstract
AA-NADase from Agkistrodon acutus venom is a unique multicatalytic enzyme with both NADase and AT(D)Pase activities. Among all identified NADases, only AA-NADase contains Cu2+ ions that are essential for its multicatalytic activity. In this study, the interactions between divalent metal ions and AA-NADase and the effects of metal ions on its structure and activity have been investigated by equilibrium dialysis, isothermal titration calorimetry, fluorescence, circular dichroism, dynamic light scattering and HPLC. The results show that AA-NADase has two classes of Cu2+ binding sites, one activator site with high affinity and approximately six inhibitor sites with low affinity. Cu2+ ions function as a switch for its NADase activity. In addition, AA-NADase has one Mn2+ binding site, one Zn2+ binding site, one strong and two weak Co2+ binding sites, and two strong and six weak Ni2+ binding sites. Metal ion binding affinities follow the trend Cu2+ > Ni2+ > Mn2+ > Co2+ > Zn2+, which accounts for the existence of one Cu2+ in the purified AA-NADase. Both NADase and ADPase activities of AA-NADase do not have an absolute requirement for Cu2+, and all tested metal ions activate its NADase and ADPase activities and the activation capacity follows the trend Zn2+ > Mn2+ > Cu2+ ∼ Co2+ > Ni2+. Metal ions serve as regulators for its multicatalytic activity. Although all tested metal ions have no obvious effects on the global structure of AA-NADase, Cu2+- and Zn2+-induced conformational changes around some Trp residues have been observed. Interestingly, each tested metal ion has a very similar activation of both NADase and ADPase activities, suggesting that the two different activities probably occur at the same site.
Introduction
NAD-glycohydrolase (NADase; EC 3.2.2.5), the enzyme that catalyzes the hydrolysis of NAD to ADP-ribose and nicotinamide, is ubiquitously present from microorganisms to mammals.1–3 Most eukaryotic NADases are bifunctional enzymes with both ADP ribosyl cyclase (ADPR cyclase) and cyclic ADP-ribose hydrolase (cADPR hydrolase) activities.4,5 These NADases that have an ADP-ribosyl cyclase activity belong to the family of ADP-ribosyl cyclases (ADPRCs). NADases/ADPRCs play a critical role in regulation of intracellular Ca2+ concentration via cADPR, a second messenger that can mobilize Ca2+ from intracellular stores and regulate Ca2+ signaling.6–9 The classical NADases, such as A. californica cyclase from Aplysia californica, CD38 and CD157 from mammals, share high homologous sequences with very similar structures and they are all composed of two identical chains without disulfide linkage between two chains,10–14 and none of them contain any metal ion.Although NADases/ADPRCs are non-metalloproteins, some metal ions have been reported to serves as a regulator of the enzyme activities. Zn2+, Cd2+ and Cu2+ stimulate the ADPR cyclase activity of CD38, but inhibit its NADase activity.15,16 Zn2+ and Mn2+ increase the activities of both ADPR cyclase and cADPR hydrolase of BST-1 ADPR-cyclase, whereas Cu2+ inhibit both cyclase and hydrolase activities of the enzyme.17 Zn2+ and Cu2+ inhibit the ADPR cyclase activities of both plasma membrane ADPR-cyclase and VSMC ADPR-cyclase.18,19 Zn2+ also inhibit the ADPR cyclase activity of cardiac ADPR-cyclase.20
NADases have been found in some snake venoms.21–23Agkistrodon acutus is a monotypic Viperiae inhabiting South China and Taiwan.24 It is one of the most dangerous snakes in China due to its hemorrhagic venom. AA-NADase was first purified from the venom of Agkistrodon acutus by Huang et al.22 Previously, we have showed that AA-NADase differs from other known NADases/ADPRCs in that it consists of two chains linked with disulfide-bond(s) and contains Cu2+ ions.25,26 Cu2+ ions are essential for catalyzing the hydrolysis of NAD and nicotinamide guanine dinucleotide (NGD), but the inhibition of AA-NADase by EDTA is irreversible.22 In contrast to mammalian NADases, AA-NADase is very stable. Reactions catalyzed by AA-NADase, therefore, represent an ideal experimental system for mechanistic studies of ADPR transfer reactions. Our recent studies indicated that AA-NADase is a unique multicatalytic enzyme with both NADase and ATPase/ADPase-like activities,27 because it is not only able to cleave the C–N glycosyl bond of NAD to produce ADPR and nicotinamide, but also able to cleave the P–O–P bond of ATP, ADP and adenosine 5′-(β, γ-imido) triphosphate (AMP-PNP) to produce AMP. Cu2+ ions are also required for the ATPase/ADPase-like activities of the enzyme. It remains unclear whether the activity of AA-NADase is absolute requirement for Cu2+ ions and why the inhibition of AA-NADase by ethylene-diaminetetraacetic acid (EDTA) is irreversible. Isothermal titration calorimetry (ITC) and equilibrium dialysis have been used to quantify the stoichiometry, affinity, and thermodynamics of metal ions binding to AA-NADase. The effects of metal ions on the activity and structure of the enzyme have been also investigated in this paper.
In the present study, two classes of binding sites have been identified for Cu2+ ions in the enzyme, which correspond to the activator and the inhibitor sites, respectively. Mn2+, Co2+, Ni2+ and Zn2+ ions all bind to the enzyme with different affinities. The NADase and ADPase activities of AA-NADase do not have an absolute requirement for Cu2+. Mn2+, Co2+, Zn2+ and Ni2+ ions are all activators for its NADase and ADPase activities.
Experimental
Chemicals
Lyophilized venom powder was provided by the TUN-XI Snakebite Institute (AnHui, P. R. China). EDTA and β-NAD were purchased from Roche. All metals were the highest purity chloride salts available and produced by Sigma Chemical Company. Chelex-100 was purchased from Bio-Rad Laboratories (Richmond, CA, USA). The products for high performance liquid chromatography were obtained from Shimadzu. All other reagents were of analytical reagent grade. Milli-Q purified water was used throughout.Solutions
The solutions of metal ions were prepared from metal chloride salts in Milli-Q water, respectively, and standardized by ITC titration with standard EDTA solution. Tris buffer and water was free from any possible contamination of multivalent cations by passage through a column (25 × 3 cm) of Chelex-100.Purification of AA-NADase
The purification of AA-NADase was performed as previously described.25 Protein purity was confirmed using SDS–PAGE analysis and the concentration of AA-NADase was calculated from the absorption coefficient (A1%1cm = 0.66) at 280 nm and the relative molecular weight (Mr = 100 kDa). Apo-AA-NADase was prepared by dialysis of the purified AA-NADase extensively against a suspension of Chelex-100 in 20 mM Tris-HCl (pH 7.4).Equilibrium dialysis
10 μM purified AA-NADase solution was dialyzed against 0.02 M Tris-HCl buffer (pH 7.4) containing 10 μM CuCl2, and then the concentrations of Cu2+ ion in the sample and the solution outside of dialysis bag were measured respectively with inductively coupled plasma-mass spectrometry (ICP-MS), PlasmaQuad 3 (VG Elemental, USA).Assays for NADase and ADPase activities
The NADase and ADPase activities were assayed by HPLC.27 Reaction mixtures were maintained at 37 °C and contained 20 mM Tris-HCl buffer, pH 7.4, 1.0 μg AA-NADase (apo-type or holo-type), 0.5 mM substrate (NAD or ADP) and 1.0 mM Cu2+, Co2+, Mn2+, Ni2+, or Zn2+ in a final volume of 2.0 ml. At timed intervals, 10 μL aliquots were immediately injected into the HPLC using a 10-μL sample syringe. High performance liquid chromatography of products was performed on a LC-20AD (Shimadzu, Japan) HPLC equipped with a SPD-M20A spectrophotometric detector. Separations were made on a Shim-pack VP-ODS (250 × 4.6 mm) column. The isocratic mobile phase contained 10 mM ammonium phosphate, pH 5.5, acetonitrile (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v). Product concentrations were calculated by a LC-20AD integrator previously calibrated with known concentrations of the products to be analyzed.
:2 v/v). Product concentrations were calculated by a LC-20AD integrator previously calibrated with known concentrations of the products to be analyzed.
Isothermal titration calorimetry (ITC) measurements
ITC experiments were carried out at 25 °C on a MicroCal VP-ITC microcalorimeter (MicroCal, LLC), as described previously.31 All buffers and solutions were filtered through a 0.22 μm sterile filter membrane (Millex GP, Millipore) and were degassed immediately before loading into the ITC cell and syringe. Reference titrations (injecting metal to buffer) was carried out first to establish good thermal history and baselines. The solution in the cell was stirred by the syringe at 307 rpm, which ensured rapid mixing but did not cause foaming of the protein solution. A typical injection schedule included the addition of 5 μL aliquots of 2.5 mM metal ion (Cu2+, Co2+, Mn2+, Ni2+, or Zn2+) to 1 mg ml−1 apo-AA-NADase solution in 10 mM Tris-HCl, pH 7.4, with 50 injections spaced between 2-min intervals. The heat of dilution was subtracted from experimental titrations by subtracting a background titration, consisting of the identical titrant solution but only the buffer solution in the sample cell. ITC results are presented by showing the baseline adjusted experimental titration data (heat flow versus time) on the top and the peak-integrated concentration-normalized molar heat flow per aliquot versus the titrant-to-sample molar ratio on the bottom.Thermodynamic parameters N (stoichiometry), KA (association constant), and ΔH (enthalpy change) were obtained by nonlinear least-squares fitting of the experimental data by the Origin software package (version 7.0) provided with the instrument. The free energy of binding (ΔG) and entropy change (ΔS) were obtained using the following equations.
| ΔG = −RT lnKA | (1) |
| ΔG = ΔH − TΔS | (2) |
Circular dichroism (CD) measurements
CD measurements were carried out with a Jasco J-810 spectropolarimeter. The instrument was calibrated with d-10-camphorsulfonic acid. All the CD measurements were made at 25 °C with a thermostatically controlled cell holder. Far-UV CD spectra were collected between 200 nm and 250 nm with a scan speed of 20 nm min−1 and a response time of 1 s, at a protein concentration of 0.4 mg mL−1, in quartz cells of 1 mm path length. The obtained values were normalized by subtracting the baseline recorded for the buffer having same concentration of salts under similar conditions. The data were expressed in mean residue ellipticity [θ] in deg cm2 dmol−1, which is defined as [θ] = 100θobs(lc)−1, where θobs is the observed ellipticity in degrees, c is the concentration in residue moles per litre, and l is the length of the light path in centimetre.Dynamic light scattering (DLS)
The hydrodynamic radii (Rh) of apo-AA-NADase (0.5 mg ml−1) in 20 mM Tris-HCl buffer with 0.2 M NaCl at pH 7.4 in the absence and presence of 1.0 mM Cu2+, Co2+, Mn2+, Ni2+, or Zn2+ were determined at 25.0 °C by a DynaPro MSTC800 DLS instrument (Wyatt Technology Corporation, America) fitted with a 624.4 nm, 50 mW laser by measuring scattering at 90° angle. Protein samples were filtered through a 100 nm membrane before the experiment. The data were analyzed using Dynamics V6.2 software.Steady-state fluorescence measurements
All fluorescence measurements were performed on a Shimadzu RF-5000 spectrofluorometer using a 10 mm quartz cuvette. The sample temperature was kept at 25.0 °C with a circulating water bath. In all experiments, the samples were excited at 295 nm, and the bandwidths for excitation and emission were both set to 2 nm. Each spectrum is the average of three consecutively acquired spectra. All spectra were corrected by subtracting the spectrum of the blank, lacking the protein but otherwise identical to the sample.UV absorption measurements
The UV spectra were taken on a UV-3700 spectrophotometer (Shimadzu, Japan), using an optical path of 1 cm at 25.0 °C in duplicate.Results
Cu2+ binding to AA-NADase
AA-NADase has been purified from the crude venom powder by a three-step chromatography procedure25 and is shown to be homogeneous as judged by SDS-polyacrylamide gel electrophoresis. The content of Cu2+ ions in the purified AA-NADase has been determined by inductively coupled plasma-mass spectrometry (ICP-MS) and the result shows that the moles of bound Cu2+ ions/mol of AA-NADase is 0.95 ± 0.02 (mean ± SE, n = 3), indicating that each purified AA-NADase molecule contains one Cu2+ ion. This observation is in agreement with the result of Huang et al.22 The purified AA-NADase molecule still binds with one Cu2+ ion after the purification process, suggesting that AA-NADase possesses one specific Cu2+-binding site with high affinity. Equilibrium dialysis has been performed to analyze the number of Cu2+-binding sites in AA-NADase. The moles of bound Cu2+ ions/mol of AA-NADase has been determined to be 6.43 ± 0.07 (mean ± SE, n = 3) at pH 7.4 in the presence of 10 μM Cu2+, suggesting that there are at least five weak Cu2+-binding sites in AA-NADase besides the high affinity Cu2+-binding site.ITC was used to study Cu2+ binding to apo-AA-NADase. Fig. 1 displays a representative thermogram of Cu2+ titrated into apo-AA-NADase at pH 7.4 in 10 mM Tris buffer. The titration of Cu2+ into apo-AA-NADase exhibits a complex behavior involving both exo- and endothermic reactions. The endothermic reaction indicates the presence of additional weak Cu2+ binding to the protein. An excellent fit to the data can be achieved using a model of two classes of binding sites (n1 and n2) in the Origin 7.0 software provided with the MicroCal titration calorimeter, and the average best-fit apparent parameters (n, K, ΔH) are summarized in Table 1. Apo-AA-NADase displays an exothermic reaction for the strong Cu2+ binding with a binding stoichiometry of n1 = 0.76 ± 0.01 and an endothermic reaction for the weak Cu2+ binding with a binding stoichiometry of n2 = 6.1 ± 0.6 with a binding affinity of approximately 50 times weaker than that of the high-affinity binding site (Table 1). The binding stoichiometry of 0.76 ± 0.01 and 6.1 ± 0.6 for the first and second classes of sites further indicates that AA-NADase possesses one specific Cu2+-binding site with high affinity and about six weak Cu2+-binding sites with low affinity, which is essentially in agreement with the above result obtained by equilibrium dialysis. The negative ΔH and ΔS values for the strong Cu2+-binding indicate that this specific binding is mainly enthalpy-driven and the entropy is unfavorable for it. In contrast, the positive ΔH and ΔS values for the weak Cu2+-binding indicate that the weak Cu2+-bindings are mainly entropy-driven and the enthalpy is unfavorable for them.
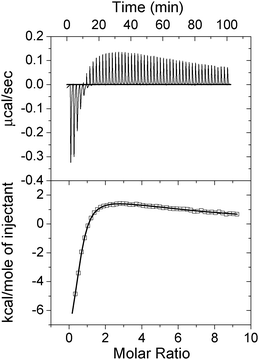 | ||
| Fig. 1 Isothermal titration calorimetry (ITC) measurement of Cu2+ binding to apo-AA-NADase. (Upper panel) Raw ITC data for injecting 1 mM Cu2+ in 20 mM Tris-HCl (pH 7.4) into 25 μM apo-AA-NADase in the same buffer at 25 °C. (Lower panel) Normalized ITC data for titrations plotted vs. molar ratio of Cu2+/apo-AA-NADase. Data analysis using Origin 7.5 software indicates that the binding data fit well to a model of two classes of binding sites. | ||
| Ligant | Site | n | K A | ΔG | ΔH | ΔS | TΔS |
|---|---|---|---|---|---|---|---|
| (M−1) | (kcal mol−1) | (kcal mol−1) | cal/(mol K) | (kcal mol−1) | |||
| 1 All values represent the average of triplicate determinations. | |||||||
| Cu2+ | I | 0.76 ± 0.01 | (1.19 ± 0.23)×106 | −8.28 ± 0.11 | −9.90 ± 0.43 | −5.43 ± 0.65 | −1.68 ± 0.19 |
| II | 6.38 ± 0.55 | (1.47 ± 0.46)×104 | −5.68 ± 0.18 | 4.06 ± 0.55 | 32.7 ± 2.2 | 9.74 ± 0.66 | |
| Mn2+ | 1.12 ± 0.07 | (2.53 ± 0.24)×105 | −7.37 ± 0.15 | −2.44 ± 0.18 | 16.5 ± 1.4 | 4.92 ± 0.42 | |
| Zn2+ | 1.23 ± 0.17 | (7.33 ± 0.43)×104 | −6.63 ± 0.05 | −7.85 ± 0.11 | 19.6 ± 1.5 | 5.84 ± 0.45 | |
| Ni2+ | I | 1.71 ± 0.09 | (5.66 ± 0.03) ×105 | −7.84 ± 0.01 | −2.23 ± 0.02 | 18.8 ± 0.4 | 5.60 ± 0.12 |
| II | 6.43 ± 0.15 | (1.57 ± 0.02) ×103 | −4.36 ± 0.02 | −3.40 ± 0.10 | 3.22 ± 0.8 | 0.96 ± 0.24 | |
| Co2+ | I | 1.02 ± 0.03 | (1.83 ± 0.07) ×105 | −7.18 ± 0.03 | 1.84 ± 0.06 | 30.2 ± 0.8 | 9.00 ± 0.24 |
| II | 2.12 ± 0.11 | (3.05 ± 0.13) ×104 | −6.11 ± 0.04 | 2.76 ± 0.22 | 29.8 ± 1.1 | 8.88 ± 0.33 |
Effects of metal ions on the NADase and ADPase activities of AA-NADase
Huang et al. reported that apo-AA-NADase can not recover its NADase activity after dialysis of apo-AA-NADase against 100 mM CuCl2 for 48 h.22 However, our recent result indicated that apo-AA-NADase can recover its NADase activity after addition of one equivalent of Cu2+. This inconsistency may be caused by that the Cu2+ ions in low-affinity sites may affect the NADase activity of AA-NADase. Fig. 2A shows the effects of the concentration of Cu2+ on the NADase activity of the purified AA-NADase. Although Cu2+ ions are essential for catalyzing the hydrolysis of NAD, they are also able to inhibit its NADase activity in a concentration-dependent manner. These data suggest that Cu2+ ions in low-affinity binding sites inhibit its NADase activity.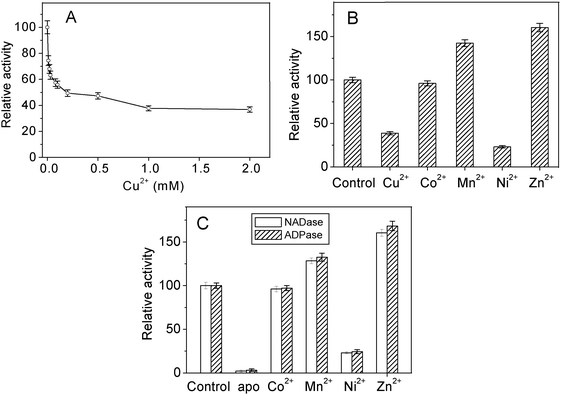 | ||
| Fig. 2 Effects of metal ions on the NADase and ADPase activities of AA-NADase. (A) Inhibition of the NADase activity of the purified AA-NADase by Cu2+. NAD (0.5 mM) was incubated with 0.5 mg AA-NADase in 20 mM Tris-HCl (pH 7.4) in the increasing concentrations of Cu2+ at 37 °C in a final volume of 2.0 ml. (B) Effects of metal ions on the NADase activity of the purified AA-NADase. NAD (0.5 mM) was incubated with 0.5 mg AA-NADase in 20 mM Tris-HCl (pH 7.4) in the presence of 1 mM Cu2+, Co2+, Mn2+, Ni2+ and Zn2+, respectively, at 37 °C in a final volume of 2.0 ml. (C) Activation of NADase and ADPase activities of apo-AA-NADase by metal ions. NAD (0.5 mM) or ADP (0.5 mM) was incubated with 0.5 mg apo-AA-NADase in 20 mM Tris-HCl (pH 7.4) in the presence of 1 mM Co2+, Mn2+, Ni2+ and Zn2+, respectively, at 37 °C in a final volume of 2.0 ml. All products were analyzed by HPLC as described under Experimental. The NADase or ADPase activity of the purified AA-NADase was taken as 100%. Each value represents the mean of three independent determinations. | ||
The effects of Co2+, Mn2+, Ni2+, and Zn2+ ions on the NADase activity of the purified AA-NADase have been determined and the results are shown in Fig. 2B. Among the metal ions tested, Mn2+ and Zn2+ obviously exert a stimulatory effect on its NADase activity, while Ni2+ inhibits its NADase activity, and Co2+ has no obvious effect on its NADase activity. Fig. 2C shows the activation of apo-AA-NADase by Co2+, Mn2+, Ni2+ and Zn2+. Apo-AA-NADase can acquire 96%, 128%, 23% and 160% of the NADase activity in the presence of 1.0 mM Co2+, Mn2+, Ni2+ and Zn2+, respectively. Similarly, Apo-AA-NADase can acquire 97%, 132%, 24% and 168% of the ADPase activity in the presence of 1.0 mM Co2+, Mn2+, Ni2+ and Zn2+, respectively. This result indicates that the multicatalytic activity of AA-NADase does not have an absolute requirement for Cu2+. Mn2+, Co2+, Zn2+ and Ni2+ ions are all effective for its NADase and ADPase activities, among which Zn2+ is the most effective for its NADase and ADPase activities.
Metal ions binding to apo-AA-NADase
ITC was used to quantify metal ions binding to apo-AA-NADase. Fig. 3 shows the representative thermograms of metal ions titrated into apo-AA-NADase in 10 mM Tris-HCl, pH 7.4. The exothermic evolution of heat upon Mn2+ injection shown in the upper panel in Fig. 3A illustrates a specific binding of Mn2+ to apo-AA-NADase. A good fit to the data for calorimetric titration can be achieved using a single-site binding model, and the average best-fit values of apparent thermodynamic parameters (KA, ΔG, ΔH and ΔS) are also summarized in Table 1. The ITC data fitting indicates that the binding ratio between Mn2+ and apo-AA-NADase is 1.12 ± 0.07, which suggests that apo-AA-NADase has one specific binding site for Mn2+. The negative ΔH and positive ΔS values of the Mn2+ binding indicate that the binding is both enthalpy- and entropy-driven. Similarly, the ITC data for Zn2+ binding to apo-AA-NADase can be best fitted with a single-site binding model (Fig. 3B). The binding stoichiometry of n = 1.23 ± 0.17 indicates one Zn2+ ion binding to the enzyme (Table 1). Zn2+ binding is also both enthalpy- and entropy-driven. The binding affinity for Zn2+ [KA = (7.33 ± 0.43) × 104) M−1] is less than that for Mn2+. The ITC profiles of Co2+ and Ni2+ binding to apo-AA-NADase are different from that of Mn2+ or Zn2+ binding. The Co2+ binding event exhibits endothermic heats of reaction (Fig. 3C). A good fit of the ITC data can be obtained with a model of two classes of binding sites. As shown in Table 1, AA-NADase has one high affinity Co2+-binding site (n1 = 1.02 ± 0.03, KA1 = (1.83 ± 0.07) ×105 M−1) and two low affinity Co2+-binding sites (n2 = 2.12 ± 0.11, KA2 = (3.05 ± 0.13) ×104 M−1). The positive ΔS values of the two classic Co2+ bindings indicate that all Co2+ bindings are entropy-driven. As shown in Fig. 3D, the raw ITC data and the integrated heats from Ni2+ binding to apo-AA-NADase also suggest the presence of two classes of binding sites and were therefore fitted using a model of two classes of binding sites. Apo-AA-NADase displays the exothermic reactions for both strong Ni2+ binding (n1 = 1.71 ± 0.09) and weak Ni2+ binding (n2 = 6.43 ± 0.15). The negative ΔH and positive ΔS values for both classic bindings indicate that all Ni2+ bindings are both enthalpy- and entropy-driven. Interestingly, the derived binding stoichiometry of n2 for the weak Ni2+-binding sites is similar to that for the weak Cu2+-binding sites, indicating that AA-NADase has same number of weak binding-sites for either Cu2+ or Ni2+.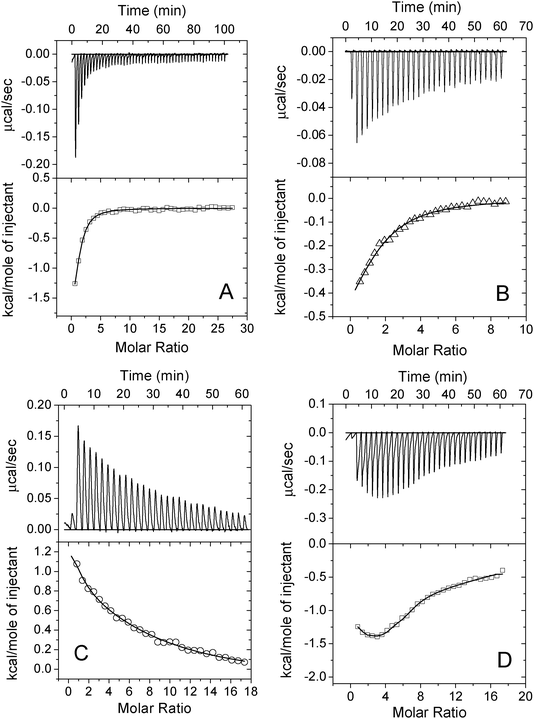 | ||
| Fig. 3 Isothermal titration calorimetry (ITC) measurements of Mn2+ (A), Zn2+ (B), Co2+(C) and Ni2+ (D) binding to apo-AA-NADase. The upper panels show raw ITC data for injecting 1 mM each metal ion in 20 mM Tris-HCl (pH 7.4) into 25 μM apo-AA-NADase in the same buffer at 25 °C. The lower panels show normalized ITC data for titrations plotted vs. molar ratio of metal ion/apo-AA-NADase. The solid line through each data set represents the best fit of the data to a single-site binding model for Mn2+ or Zn2+ and a model of two classes of binding sites for Co2+ or Ni2+. | ||
Effects of metal ions binding on the structure of AA-NADase
Circular dichroism (CD) measurements were carried out to study the effects of Cu2+, Mn2+, Zn2+, Ni2+ and Co2+ on the secondary structure of AA-NADase. As shown in Fig. 4A, apo-AA-NADase and the purified AA-NADase show a very similar CD spectrum. No obvious changes have been observed for the CD spectrum of apo-AA-NADase after addition of 1.0 mM Cu2+, Mn2+, Zn2+, Ni2+ or Co2+, suggesting that these metal ions have no effect on the secondary structure of AA-NADase. Dynamic light scattering (DLS) studies show that the hydrodynamic radius of apo-AA-NADase is 4.2 ± 0.2 nm corresponding to the molecular weight of 96 kD (Fig. 4B). The hydrodynamic radius of apo-AA-NADase does not change in the presence of 1 mM Co2+, and slightly increases from 4.2 ± 0.2 nm to 4.4 ± 0.3 nm, 4.3 ± 0.2 nm, 4.6 ± 0.2 nm and 4.5 ± 0.3 nm in the presence of 1.0 mM Cu2+, Mn2+, Ni2+ and Zn2+ respectively. This result indicates that AA-NADase does not oligomerize in the presence of 1.0 mM each tested metal ion.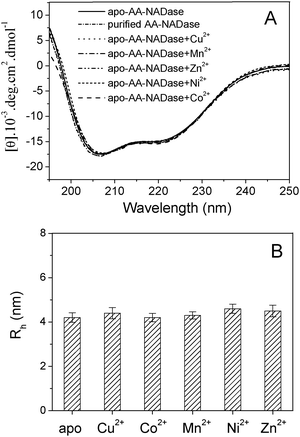 | ||
| Fig. 4 Effects of metal ions binding on the structure of AA-NADase. (A) Far-UV CD spectra of 0.2 mg ml−1 purified AA-NADase in 20 mM Tris-HCl (pH 7.4) in the absence of metal ion and 0.2 mg ml−1 apo-AA-NADase in 20 mM Tris-HCl (pH 7.4) in the absence and presence of 1 mM Cu2+, Mn2+, Co2+, Ni2+ or Zn2+, respectively. (B) The mean hydrodynamic radii (Rh) of 0.5 mg ml−1 apo-AA-NADase in 20 mM Tris-HCl (pH 7.4) in the absence and in the presence of 1 mM Cu2+, Mn2+, Co2+, Ni2+ or Zn2+ determined by DLS experiments. Error bars for three experimental data are shown. | ||
Effects of metal ions binding on the fluorescence of AA-NADase
To examine the interaction of AA-NADase with metal ions, the fluorescence measurements of the purified AA-NADase and apo-AA-NADase in the presence of 1.0 mM Zn2+, Mn2+, Co2+, Ni2+ or Cu2+ were performed at an excitation wavelength of 295 nm at 25 °C. Upon excitation at 295 nm only the Trp residue emission is observed. As shown in Fig. 5A, the maximum emission of the purified AA-NADase is at 340 nm by excitation at 295 nm. Removal of Cu2+ from the purified AA-NADase induces an obvious quenching (13.5%) of the Trp fluorescence of AA-NADase with no obvious shift in the emission maximum, indicating that the binding of Cu2+ to the high affinity site increases the Trp fluorescence in AA-NADase. In contrast, addition of 1.0 mM Cu2+ ions to apo-AA-NADase induces a significant quenching (94.2%) of the Trp fluorescence of AA-NADase with a blue-shift in the emission maximum from 340 nm to 325 nm. A linear Stern–Volmer plot (F0/F versus [Cu2+]) of the fluorescence quenching of apo-AA-NADase by Cu2+ is observed (Fig. 5C), yielding a Stern–Volmer quenching constant (KS) value of (1.02 ± 0.30)×104 M−1 (mean ± SE, n = 3). The linear Stern–Volmer plot indicates involvement of one type quenching process: either static or dynamic quenching. Apo-AA-NADase fluorescence slightly decreases with no obvious shift in the emission maximum in the presence of 1 mM Mn2+, Co2+ and Ni2+, and obviously increases with a blue-shift in the emission maximum from 340 nm to 337 nm in the presence of 1 mM Zn2+ (Fig. 5A).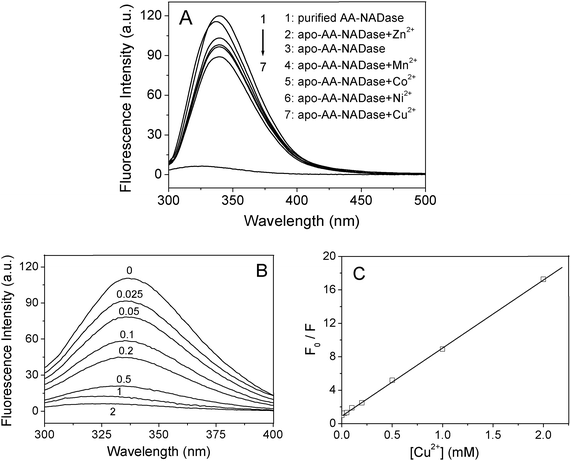 | ||
| Fig. 5 Effects of metal ions on the fluorescence of AA-NADase. (A) The fluorescence spectra of the purified AA-NADase (5 μM) in 20 mM Tris-HCl (pH 7.4) in the absence of metal ion and apo-AA-NADase (5 μM) in 20 mM Tris-HCl (pH 7.4) in the absence and presence of 1 mM Cu2+, Mn2+, Co2+, Ni2+ or Zn2+ were recorded by exciting at 295 nm. (B) The fluorescence spectra of apo-AA-NADase (5 μM) in 20 mM Tris-HCl (pH 7.4) at the indicated concentration (in micromolar) of Cu2+ were recorded by exciting at 295 nm. (C) Stern–Volmer plot of the fluorescence quenching of apo-AA-NADase by Cu2+. F0 and F are the fluorescence intensities of apo-AA-NADase in the absence and presence of Cu2+, respectively. | ||
Effects of Cu2+ on the UV absorption spectrum of AA-NADase
Collisional quenching only affects the excited states of the fluorophores, and does not affect the absorption spectra. In contrast, ground state complex formation will frequently result in perturbation of the absorption spectrum of the fluorophore.28 To distinguish static and dynamic quenching of AA-NADase by Cu2+, the UV absorption spectrum of the purified AA-NADase has been measured in the absence and presence of Cu2+, respectively. As shown in Fig. 6, addition of Cu2+ to the purified AA-NADase results in a strong absorbance in the UV region of the electronic spectrum, indicating that the purified AA-NADase has formed a complex with Cu2+ ions. The difference spectrum shows increases in the absorption around 248 nm and 299 nm upon binding with Cu2+ ions. In a control experiment, addition of Cu2+ to Tris buffer alone does not results in any change in the UV absorption spectrum. The absorptions of proteins at 248 nm and 299 nm are due to Phe and Trp residues, respectively.28 This result indicates that the binding of Cu2+ to the purified AA-NADase enhances the UV absorption spectrum of Phe and Trp residues.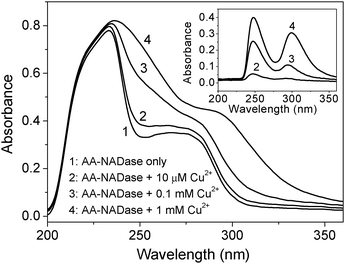 | ||
| Fig. 6 UV absorption spectra of the purified AA-NADase in the absence and presence of Cu2+, respectively for 0.5 mg ml−1 enzyme in 20 mM Tris-HCl, pH 7.4, and an optical path of 1 cm. The difference absorption spectra of the purified AA-NADase in the absence and presence of Cu2+ are shown in the inset. | ||
Discussion
AA-NADase is a unique NADase with multicatalytic activity, because among all identified NADases, only AA-NADase contains Cu(II) ions which are essential for its multicatalytic activity. The goal of the present study was to characterize the thermodynamics of metal ions binding to AA-NADase and the effects of metal ions on its structure and activity. The results indicate that AA-NADase has two classes of Cu2+ binding sites, one activator site with high binding affinity and approximate six inhibitor sites with low binding affinity. The multicatalytic activity of AA-NADase does not have an absolute requirement for Cu2+. Mn2+, Co2+, Zn2+ and Ni2+ ions are all effective for its multicatalytic activity.The purified AA-NADase contains one Cu2+ as determined by ICP-MS. Equilibrium dialysis and ITC measurements demonstrate that AA-NADase has two classes of Cu2+ binding sites: one specific Cu2+-binding site with high affinity and approximate six weak Cu2+-binding sites with low affinity. The sum of n1 (0.76 ± 0.01) and n2 (6.38 ± 0.55) results in 7.14 total Cu2+ ions that bind to AA-NADase, which is close to the value determined by equilibrium dialysis. The high-affinity binding is mainly enthalpy-driven, while the low-affinity bindings are mainly entropy-driven. The Cu2+ in the high affinity site is essential for its multicatalytic activity.22 In contrast, the Cu2+ ions in low-affinity binding sites inhibit its NADase activity (Fig. 2A). Therefore Cu2+ ions function as a switch for its NADase activity. As mentioned above, Cu2+ was reported to stimulate the ADPR cyclase activity of CD38, but inhibit its NADase activity.15,16 Cu2+ also inhibits the ADPR cyclase activity of both plasma membrane ADPR-cyclase and VSMC ADPR-cyclase.18,19 Although Cu2+ is not essential for the activity of CD38, plasma membrane ADPR-cyclase and VSMC ADPR-cyclase, it can regulate their activity.
Currently, the three dimensional structure of AA-NADase is not yet available. Electron paramagnetic resonance spectroscopy revealed that the Cu2+ ion in the high-affinity site in AA-NADase may be coordinated with three N atoms and one water molecule.26 Cu2+ ions in both high and low affinity sites have no effects on the second structure of AA-NADase (Fig. 4A). Cu2+ ions also do not induce AA-NADase to oligomerize (Fig. 4B). Removal of Cu2+ from the high-affinity site in AA-NADase decreases the Trp fluorescence in AA-NADase by 13.5%, suggesting that the binding of Cu2+ in the high-affinity site induces a conformational change around some Trp residues and increases their fluorescence. In contrast, the bindings of Cu2+ in the low-affinity sites markedly decrease the fluorescence of AA-NADase and induce a blue-shift of the maximum emission from 340 nm to 325 nm. Cu2+ ions-induced changes in the UV absorption spectrum of the purified AA-NADase demonstrate that the fluorescence quenching by Cu2+ ions is a static quenching. The observed quenching of Trp fluorescence of AA-NADase is specific–due to the binding of Cu2+ in the vicinity of some Trp residues of AA-NADase. For static quenching, Stern–Volmer quenching constant represents the association constant between the fluorophore and quencher. Stern–Volmer quenching constant (KS) value of (1.02 ± 0.30)×104 M−1 is close to the association constant of (1.47 ± 0.46)×104 M−1 for the weak Cu2+ binding, indicating that the fluorescence quenching appears to represent the binding of Cu2+ ions to the low-affinity sites. The binding constants for the weak Cu2+ binding obtained by the fluorescence quenching and the ITC methods are consistent. Although the binding of Cu2+ in the high affinity site slightly increases the fluorescence of AA-NADase, a marked decrease in the fluorescence of AA-NADase was observed upon occupation of Cu2+ ions in both high and low affinity sites. These results suggest that Cu2+ ions in low affinity sites also quench the fluorescence of some Trp residues that can be enhanced by the Cu2+ in high affinity site.
Three amino acid residues (Tyr, Trp, and Phe) contribute to protein UV absorption spectra from 240 to 310 nm. The binding of Cu2+ to the purified AA-NADase enhances the UV absorption spectrum of Phe residues at 248 nm and Trp residues at 299 nm, suggesting that some Phe and Trp residues may be close to the weak Cu2+-binding sites. The fluorescence quenching by Cu2+ ions in the low-affinity sites further suggests that some Trp residues may be close to the weak Cu2+-binding sites.
The Cu2+ as an activator may bind in catalytic site and play an important role in catalysis, while the Cu2+ ions as inhibitors may bind in the vicinity of catalytic site. Cu2+ ions-induced significant fluorescence quenching with an obvious blue-shift of the maximum emission and UV absorption enhancement of AA-NADase suggest that (1) a dramatically conformational change of AA-NADase is induced upon Cu2+ binding to the low-affinity sites; (2) this conformational change may affect on the microenvironment of catalytic site or mask the catalytic site which results in the inhibition of the enzyme. Although we can not infer the detailed picture of the pathway of the activation and inhibition by Cu2+ ions from the present data, it is certain that Cu2+ ions function as a switch for the NADase activity of AA-NADase.
Apo-AA-NADase has one specific Mn2+-binding site and one specific Zn2+-binding site (Fig. 3A and B). Both Mn2+ and Zn2+ bindings are enthalpy- and entropy-driven. It is interesting to note that compared with the purified AA-NADase, Mn2+-loaded AA-NADase can acquire 128% of the NADase activity and 132% of the ADPase activity, while Zn2+-loaded AA-NADase can acquire 160% of the NADase activity and 168% of the ADPase activity. Therefore, both Mn2+ and Zn2+ ions are more effective to activate the multicatalytic activity of AA-NADase than Cu2+. Although Zn2+ has the lowest affinity for AA-NADase, Zn2+-loaded AA-NADase has the highest multicatalytic activity. As mentioned above, Mn2+ was reported to increase the activities of both ADPR cyclase and cADPR hydrolase of BST-1 ADPR-cyclase.17 Zn2+ inhibited the ADPR cyclase activity of plasma membrane ADPR-cyclase, VSMC ADPR-cyclase and cardiac ADPR-cyclase.18–20 These results together indicate that Mn2+ activates or stimulates the activity of NADases/ADPRCs, while Zn2+ activates or inhibits the activity of NADases/ADPRCs.
Like Cu2+, both Mn2+ and Zn2+ neither affect on the second structure of AA-NADase (Fig. 4A) nor induce an oligomerization of AA-NADase (Fig. 4B). The binding of Mn2+ to AA-NADase slight decreases the fluorescence, indicating that Mn2+ binding has a little effect on the environments of Trp residues. The binding of Zn2+ to AA-NADase not only obviously increases the fluorescence of Trp residues but also induces a blue-shift in the emission maximum from 340 nm to 337 nm, suggesting that Zn2+ binding has a large effect on the environments of Trp residues, which results in decrease in the polarity of the environments of some Trp residues. Zn2+-induced large conformational change of the enzyme may explain why Zn2+ is most effective to activate the multicatalytic activity.
AA-NADase has one strong and two weak Co2+-binding sites and Co2+-loaded AA-NADase has a similar multicatalytic activity to that of the purified AA-NADase. AA-NADase has two strong and six weak Ni2+-binding sites and Ni2+-loaded AA-NADase exhibits much lower multicatalytic activity than that of the purified AA-NADase.
Apo-AA-NADase does not show any NADase and ADPase activities, indicating that the multicatalytic activity of AA-NADase relies on metal ions. Cu2+, Mn2+, Zn2+ and Co2+ are all the activators of AA-NADase, while Ni2+ has a weak activation of the multicatalytic activity. These five divalent metal ions are all trace elements that are required for biology and distributed in different organs. These metal ions serve as regulators for the multicatalytic activity of AA-NADase. Comparison of association constants (KA) for high affinity binding of metal ions to apo-AA-NADase obtained from ITC measurements shows that the Cu2+ has the highest binding affinity for AA-NADase among the tested metal ions, which accounts for the presence of one Cu2+ in the purified AA-NADase.
It has been reported that the active-site structural requirements for the glycohydrolase reactions are different from that for the ADPase reactions.12,29 Our recent study showed that the hydrolysis reactions of NAD and ADP catalyzed by AA-NADase are mutually competitive27 and the inhibition curves of both ADPase and NADase activities by each reductant [dithiothreitol, glutathione, tris(2-carboxyethyl)phosphine or L-ascorbate] share a similar trend to the inhibition,30 which suggests that both ADPase and NADase activities of AA-NADase probably occur at the same site. Interestingly, as shown in Fig. 2C, each tested metal ion has a very similar activation of both NADase and ADPase activities. For example, Mn2+ activates both NADase and ADPase activities to a similar extent, and Zn2+ also activates both NADase and ADPase activities to a similar extent, while Ni2+ has a similar weak activation of both NADase and ADPase activities. This result further supports that both ADPase and NADase activities occur at the same site. Without the structure of AA-NADase, it is difficult to address how AA-NADase cleave the C–N glycosyl bond of NAD and the P–O–P bond of ADP in the same active site and how metal ions activate the multicatalytic activity. Further investigation is necessary to clarify these issues.
Conclusions
The present study reveals that AA-NADase has two classes of Cu2+ binding sites, one activator site with high affinity and approximately six inhibitor sites with low affinity. Cu2+ ions function as a switch for its NADase activity. AA-NADase has one Mn2+ binding site, one Zn2+ binding site, one strong and two weak Co2+ binding sites, and two strong and six weak Ni2+ binding sites. Metal ions binding affinities follow the trend Cu2+ > Ni2+ > Mn2+ > Co2+ > Zn2+, which accounts for the existence of one Cu2+ in the purified AA-NADase. Both NADase and ADPase activities of AA-NADase do not have an absolute requirement for Cu2+. Mn2+, Co2+, Zn2+ and Ni2+ ions all activate its NADase and ADPase activities and the activation capacity follows the trend Zn2+ > Mn2+ > Cu2+ ∼ Co2+ > Ni2+. Metal ions serve as regulators for its multicatalytic activity. Although all tested metal ions have no obvious effects on the global structure of AA-NADase, Cu2+- and Zn2+-induced conformational changes around some Trp residues have been observed. Interestingly, each tested metal ion has a very similar activation of both NADase and ADPase activities, suggesting that the two different activities probably occur at the same site.Acknowledgements
We thank PhD Shouye Wang for helpful suggestions. This work was supported by grants from the National Natural Science Foundation of China (Grant No. 20871111, 20571069).References
- I. Tatsuno, J. Sawai, A. O. Oto, M. Matsumoto, M. Minami, M. Isaka, M. Ohta and T. Hasegawa, Microbiology, 2007, 153, 4253–4260 CrossRef CAS.
- F. Preugschat, G. H. Tomberlin and D. J. T. Porter, Arch. Biochem. Biophys., 2008, 479, 114–120 CrossRef CAS.
- V. Polzonetti, S. Pucciarelli, A. Vita, S. Vincenzetti and P. Natalini, J. Membr. Biol., 2009, 227, 105–110 CrossRef CAS.
- R. Graeff, Q. Liu, I. A. Kriksunov, M. Kotaka, N. Oppenheimer, Q. Hao and H. C. Lee, J. Biol. Chem., 2009, 284, 27629–27636 CrossRef CAS.
- R. Gul, J. H. Park, S. Y. Kim, K. Y. Jang, J. K. Chae, J. K. Ko and U. H. Kim, Cardiovasc. Res., 2008, 81, 582–591 CrossRef.
- G. Orsomando, V. Polzonetti and P. Natalini, Comp. Biochem. Physiol., 2000, 126, 89–98 Search PubMed.
- M. Ziegler, D. Jorcke and M. Schweiger, Biochem. J., 1997, 326, 401–405 CAS.
- S. Bruzzone, I. Moreschi, L. Guida, C. Usai, E. Zocchi and A. De Flora, Biochem. J., 2006, 393, 697–704 CrossRef CAS.
- J. B. Yue, W. J. Wei, C. M. C. Lam, Y. J. Zhao, M. Dong, L. R. Zhang, L. H. Zhang and H. C. Lee, J. Biol. Chem., 2009, 284, 29335–29342 CrossRef CAS.
- G. S. Prasad, D. E. McRee, E. A. Stura, D. G. Levitt, H. C. Lee and C. D. Stout, Nat. Struct. Biol., 1996, 3, 957–964 CrossRef CAS.
- Q. Liu, I. A. Kriksunov, R. Graeff, C. Munshi, H. C. Lee and Q. Hao, Structure, 2005, 13, 1331–1339 CrossRef CAS.
- S. Yamamoto-Katayama, M. Ariyoshi, K. Ishihara, T. Hirano, H. Jingami and K. Morikawa, J. Mol. Biol., 2002, 316, 711–723 CrossRef CAS.
- W. Kou, S. Banerjee, J. Eudy, L. M. Smith, R. Persidsky, K. Borgmann, L. Wu, N. Sakhuja, M. S. Deshpande, T. F. Walseth and A. Ghorpade, J. Neurosci. Res., 2009, 87, 2326–2339 CrossRef CAS.
- Q. Liu, R. Graeff, I. A. Kriksunov, H. Jiang, B. Zhang, N. Oppenheimer, H. N. Lin, B. V. L. Potter, H. C. Lee and Q. Hao, J. Biol. Chem., 2009, 284, 27637–27645 CrossRef CAS.
- I. Kukimoto, S. Hoshino, K. Kontani, K. Inageda, H. Nishina, K. Takahashi and T. Katada, Eur. J. Biochem., 1996, 239, 177–182 CrossRef CAS.
- W. Zielinska, H. Barata and E. N. Chini, Life Sci., 2004, 74, 1781–1790 CrossRef CAS.
- Y. Hirata, N. Kimura, K. Sato, Y. Ohsugi, S. Takasawa, H. Okamoto, J. Ishikawa, T. Kaisho, K. Ishihara and T. Hirano, FEBS Lett., 1994, 356, 244–248 CrossRef CAS.
- F. G. S. de Toledo, J. F. Cheng, M. Y. Liang, E. N. Chini and T. P. Dousa, Circ. Res., 2000, 86, 1153–1159 CAS.
- M. Liang, E. N. Chini, J. Cheng and T. P. Dousa, Arch. Biochem. Biophys., 1999, 371, 317–325 CrossRef CAS.
- G. H. Xie, S. Y. Rah, S. J. Kim, T. S. Nam, K. C. Ha, S. W. Chae, M. J. Im and U. H. Kim, Biochem. Biophys. Res. Commun., 2005, 330, 1290–1298 CrossRef CAS.
- D. A. Yost and B. M. Anderson, J. Biol. Chem., 1981, 256, 3647–3653 CAS.
- W.-Z. Huang, C. Wang, L.-Q. Luo and Z.-X. Lu, Toxicon, 1988, 26, 535–542 CrossRef CAS.
- T. Tatsuki, S. Iwanaga, G. Oshima and T. Suzuki, Toxicon, 1975, 13, 211–220 CrossRef CAS.
- Y. M. Wang, S. R. Wang and I. H. Tsai, Biochem. J., 2001, 354, 161–168 CrossRef CAS.
- S. D. Wu, Y. L. Liu, X. L. Xu and Z. G. Zhu, Protein Expression Purif., 2002, 25, 319–322 CrossRef CAS.
- S. D. Wu, X. L. Xu, Y. S. Xie, L. X. Chen, Z. L. Chen and Q. L. Liu, Chem. Lett., 2002, 354–355 CrossRef CAS.
- L. Zhang, X. Xu, Z. Luo, D. Shen and H. Wu, Biochimie, 2009, 91, 240–251 CrossRef CAS.
- Principles of fluorescence spectroscopy, ed. J. R. Lakowicz, Plenum Press, New York, 1983 Search PubMed.
- S. Y. Rah, K. H. Park, T. S. Nam, S. J. Kim, H. Kim, M. J. Im and U. H. Kim, J. Biol. Chem., 2006, 282, 5653–5660 CrossRef.
- L. Y. Zhang, X. L. Xu, Z. F. Luo, H. Wu, D. K. Shen, L. L. Peng and Y. Z. Liu, Biopolymers, 2010, 93, 141–149 CrossRef CAS.
- X. L. Xu, L. Y. Zhang, D. K. Shen, H. Wu, L. L. Peng and J. H. Li, J. Biol. Inorg. Chem., 2009, 14, 559–571 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |