Metallomics approach for the identification of the iron transport protein transferrin in the blood of harbour seals (Phoca vitulina)†
Mechthild
Grebe
*ab,
Daniel
Pröfrock
a,
Antje
Kakuschke
a,
Jose A.C.
Broekaert
b and
Andreas
Prange
a
aGKSS Forschungszentrum Geesthacht GmbH, Institute for Coastal Research, Marine Bioanalytical Chemistry, Max-Planck Str. 1, 21502 Geesthacht, Germany. E-mail: Mechthild.Grebe@gkss.de
bUniversity of Hamburg, Department of Chemistry, Institute for Inorganic and Applied Chemistry, Martin-Luther King Platz 6, 20146 Hamburg, Germany
First published on 17th August 2010
Abstract
The health status of marine mammals such as harbour seals (Phoca vitulina) represents an indirect but powerful way for the assessment of environmental changes. The present work illustrates the first investigation and characterisation of Tf isolated from blood samples of North Sea harbour seals with a view to using changes in Tf isoform patterns as an additional parameter in extended studies of their health status. Therefore, an HPLC-ICP-MS approach has been developed which allows the highly resolved separation and fractionation of up to eight different Tf isoforms, as well as their sensitive and specific detection on the basis of their characteristic iron content. Molecule-specific detection techniques such as nanoLC-ESI-QTRAP-MS or MALDI-TOF-MS were used as complementary techniques to unambiguously identify the isolated proteins as Tf via cross species protein identification and to further characterise the molecular weight as well as the sialic acid content, which is responsible for the elution behaviour of the different isoforms during their ion exchange separation. A molecular mass above 80 kDa has been measured for the different seal Tf isoforms, which is in good agreement with the known molecular mass in other mammalian species, while the estimated pI of the different isoforms indicates some differences in comparison to other species. A number of homologies to known Tf sequences have been observed, which finally allows the cross species protein identification. The combined metallomics orientated analytical approach, which includes the complementary application of element and molecule-specific detection techniques, opens up interesting possibilities for the fast and targeted isolation and identification of a diagnostically relevant metal containing protein from an un-sequenced mammalian species prior to its utilisation in extended studies.
Introduction
The ongoing, increasing commercial activity, in terms of shipping, fishery or more recently the operation of offshore wind parks, as well as the sustained input of various kinds of pollutants still strongly influences the North Sea ecosystems. Due to their role as top predators within the marine food web, marine mammals such as harbour seals (Phoca vitulina) can be used as indicators for the medium and long term changes in the ecosystem. The Trilateral Monitoring and Assessment Program (TMAP) between Germany, the Netherlands and Denmark specifies seals as a biological parameter for changes in the ecosystem North Sea. Behind investigations to population trends and seal migration, seal population parameters as survival, age structure, sex ratio, and in particular the health status are sensitive parameters for the assessment of the ecosystem. Such studies involve several factors, e.g. haematological and immunological investigations, biochemical analysis of enzymes and metabolites, bacteriological screenings, as well as the analysis of inorganic trace elements and organic pollutants.Most projects aiming at ecosystem or environmental assessment are still based on the quantification of pollution levels in different environmental compartments or measurements of body burdens in selected organisms such as harbour seals.1–5 Bioaccumulation of environmental contaminants in the tissues of marine mammals is suspected to cause suppression of the immune system, which increases the vulnerability of these organisms to pathogens or infectious diseases.6–8 However, the diagnostic relevance of such pure concentration measurements is often not sufficient to describe a complex and “vague” parameter such as the health status. On the other hand, it is proven that an individual biomarker can only provide limited information and do not give complete diagnostic information. Therefore, it is recommended to use a set of biomarkers in conjunction with other chemical and biological measurements.9
Several studies concerning the health/immune status10–12 and/or contaminant body burdens13–16 of harbour seals were conducted in the Wadden Sea. The list of investigated parameters includes trace elements14 and selected element species,16 organic contaminants,17 lymphocyte proliferation11,18 and several acute phase proteins (APP),17,19,20 as well as standard clinical chemistry parameters.17
Our current work is focused on the extension of the parameter list, regarding the analysis of selected proteins and their isoforms directly at the protein level. The APP were synthesised in the liver during an acute phase reaction, which represents a central physiological, fast acting process during e.g. inflammation, trauma or infections. This process is characterised by a significant concentration change in the APP involved in the blood plasma.21 In veterinary medicine, APP represent an important diagnostic tool to monitor the progress of various diseases and stress at the cellular level as well as acute pathogenic effects.22–24
Within this context, transferrin (Tf) represents a further important diagnostic APP beside the ones mentioned, which is in addition the most important iron-transporting glycoprotein in the blood of mammalian species. In clinical diagnosis, altered distributions of Tf Isoforms are used as biomarkers e.g. for the impairment of the liver as well as various liver diseases,25–27 which represents one of the most frequently observed pathological indications in harbour seals of the North Sea.12 Therefore, veterinary medicine utilises Tf as a helpful diagnostic parameter in combination with other APP for assessing the progression of disease states or the health status of mainly domestic but also wildlife animals.28
Unfortunately, marine mammal species such as Phoca vitulina are poorly sequenced animal species with only a few protein entries in the databases commonly used for protein identification such as SWISSProt or NCBI. This hampers the fast identification and further analysis of this protein in a complex matrix such as blood.
In this study, high performance liquid chromatography (HPLC) coupled to inductively-coupled plasma mass spectrometry (ICP-MS) has been used for the iron-specific detection of the different Tf isoforms. Protein identification has been performed using two-dimensional sodium dodecyl sulfate polyacrylamide gel-electrophoresis (2D-SDS-PAGE) and either matrix-assisted laser desorption/ionisation time-of-flight mass spectrometry (MALDI-TOF) or nano-HPLC coupled to electro-spray ionisation linear ion trap mass spectrometry (ESI-QTRAP-MS).
Therefore, as a first step a combined analytical approach has been developed, which utilises the characteristic iron content of transferrin for its targeted isolation from either seal serum or plasma samples before its identification via cross species protein identification. The unambiguous identification of Tf as well as its additional characterisation are a prerequisite for its future absolute quantification and further application in extended studies with respect to the analysis of the health status of harbour seals.
Experimental
Chemicals and materials used
Ultra pure water (18 MΩ cm) was prepared using a Millipore Elix 3/Milli-Q element water purification system (Millipore, Milford, MA, USA). Single element standards of Ge and Y, as well as Na2CO3, FeCl3, acetonitrile, methanol, acetic acid, ammonia solution and glycerol were obtained from Merck at the highest available quality (Merck, Darmstadt, Germany). MgCl2, Dextransulfate, Bis-Tris buffer and urea were purchased from Fluka (Fluka, Buchs, Switzerland). The reagent for the Bradford protein assay and the enzymes Trypsin, PNGaseF and Neuraminidase, as well as Iodoacetamide (IAA), Dithiotreitol (DTT) and Tris-base were obtained from Sigma (Sigma, Deisenhofen, Germany). All components used for the two-dimensional gel-electrophoresis, immobilised pH gradient (IPG) strips, 10% Criterion Tris-HCL gels, agarose, paper wicks, mineral oil, bromphenol blue, sodium dodecyl sulfate (SDS), glycine and Roti-Blue solution were purchased from Bio Rad (Bio Rad Laboratories GmbH, Munich, Germany). CHAPS was supplied by Roth (Carl Roth GmbH + Co. KG, Karlsruhe, Germany), while phosphoric acid was purchased from Riedel de Haen (Riedel de Haen GmbH, Seelze, Germany). Α-cyano-4-hydroxycinnamic acid (HCCA) and sinapinic acid (SA), which were used as MALDI matrix as well as the peptide and the protein calibration standard for the near neighbour calibration, were purchased from Bruker (Bruker Daltonics, Bremen, Germany). Ultra-filtration units (Vivaspin 6, MWCO 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were obtained from Sartorius (Sartorius, Göttingen, Germany). Argon 5.0 (99.999% purity) was used as plasma gas. Hydrogen 5.0 (99.999% purity) was used as cell gas inside the octopole reaction system during all experiments. Both gases were obtained from Air Liquide (Air Liquide, Lübeck, Germany). Solutions, standards and samples were prepared under clean room conditions (class 1000) on a clean bench (class 100) to avoid contamination. To reduce possible sample degradation, all the handling and preparation steps of the plasma and serum samples were performed under cooled conditions if not otherwise stated (4 °C).
000) were obtained from Sartorius (Sartorius, Göttingen, Germany). Argon 5.0 (99.999% purity) was used as plasma gas. Hydrogen 5.0 (99.999% purity) was used as cell gas inside the octopole reaction system during all experiments. Both gases were obtained from Air Liquide (Air Liquide, Lübeck, Germany). Solutions, standards and samples were prepared under clean room conditions (class 1000) on a clean bench (class 100) to avoid contamination. To reduce possible sample degradation, all the handling and preparation steps of the plasma and serum samples were performed under cooled conditions if not otherwise stated (4 °C).
Instrumentation
All connections were made of 1/16′′ o.d., 125 μm i.d. PEEK tubings and zero dead volume fittings (Upchurch Scientific/GAT Analysen Technik, Bremen, Germany). All LC modules were arranged in order to achieve the lowest possible dead volume between the LC setup and the ICP-MS. An adjustable flow splitter was used to allow parallel UV detection (at 280 and 460 nm, respectively) as well as on-line fraction collection of the separated Tf species. Detailed chromatographic conditions as well as further instrumental parameters are summarised in Table 1.
| HPLC Agilent 1100 | |
| SAX Column | PorosHQ, 2.1 mm × 100 mm, 10 μm particles |
| Mobile Phase | A: 20 mM Bis-Tris, pH 6.5, 10 μg L−1 Ge |
| B: 20 mM Bis-Tris, 500 mM Ammonium acetate, pH 6.5, 10 μg L−1 Ge, Y | |
| Injection volume | 50 μL |
| Flow rate | 0.75 mL min−1 |
| Gradient | 0–2 min 0% B |
| 2–26 min 0% B to 12% B | |
| 26–28 min 12% B to 100% B | |
| 28–33 min 100% B | |
| 33–34 min 100% B to 0% B | |
| 34–40 min 0% B | |
| DAD detection | 280 nm, 460 nm |
| ICP-MS Agilent 7500cs | |
| RF power | 1600 W |
| Carrier gas | 0.85 L min−1 |
| Makeup gas | 0.31 L min−1 |
| Extraction lens 1 | 6 V |
| Extraction lens 2 | −180 V |
| Octopole bias | −18 V |
| Quadrupole bias | −16 V |
| Cell gas | 5 mL min−1 H2 |
| Spray chamber Temperature | 4 °C |
| Measured isotopes | 54Fe, 56Fe, 57Fe, 72Ge, 89Y |
| Dwell time | 0.1 s |
To reduce the background, especially for 56Fe, which is mainly interfered by polyatomic ions such as 40Ar16O+, H2 with an optimum flow-rate of 5 mL min−1 was used in all experiments. In addition, kinetic energy discrimination obtained by the settings of the octopole and quadrupole bias were applied to further reduce the background on the main isotope of iron. A description of the principles of kinetic energy discrimination can be found elsewhere.29–31 A PFA 100 micro concentric nebuliser (Elemental Scientific, Omaha, Nebraska, USA) combined with the standard quartz double pass Scott spray chamber, as supplied with the ICP-MS instrumentation, was used for the hyphenation of the HPLC system to the ICP-MS. Details of the ICP-MS settings are also given in Table 1.
Α-cyano-4-hydroxycinnamic acid (HCCA) as well a sinapinic acid (SA) was used as MALDI matrix during all experiments. A near neighbour mass calibration was performed using a mixture of peptides containing Angiotensin II, Angiotensin I, Substance P, Bombesin, ACTH clip 1–17, ACTH clip 18-39 and Somatostatin 28. For mass calibration during protein analysis a mixture of Insulin, Ubiquitin I, Cytochrom C and Myoglobin or Trypsinogen, Protein A and Bovine Serum Albumin were used.
ESI analysis was performed using an Applied Biosystems 4000 QTRAP linear ion trap system. Intact protein analysis was carried out using static nano spray, while the tryptic digests were analysed using a nano-LC based hyphenated setup. For static nano-spray the desalt protein fraction was diluted 10-fold with a 10% acetonitrile in 0.1% formic acid solution. These samples were applied to the ESI system in a constant nano flow using single-use metal coated borosilicate spray capillaries (Proxeon Biosystems, Odense, Denmark). For the nano-LC separation of the tryptic peptides a Dionex PepMap C18 column with 75 μm i.d. and l = 150 mm (Dionex GmbH, Idstein, Deutschland) was used combined with a gradient of 5–50% acetonitrile in 0.1% formic acid over 40 min.
Sample preparation
Blood samples of two male and juvenile (about 7 month old) harbour seals of the North Sea were used in this study. The animals were euthanized because of serious illness by authorised persons following the established protocol. Immediately after this, blood samples were taken in lithium-heparin monovettes specified for trace element analysis (Sarstedt AG & Co, Nümbrecht, Germany) and in Serum Gel S monovettes (Sarstedt AG & Co). After centrifugation, aliquots of plasma and serum were separated and frozen until further use.Since the element guided fractionation requires a uniform iron stoichiometry in the different Tf isoforms, an iron saturation procedure was developed based on a method described by del Castillo Busto et al.33,34 Briefly, 100 μL plasma or serum were mixed with 5 μL of a 500 mM Na2CO3 solution as well as 5 μL of a 10 mM FeCl3 solution followed by an incubation time of 30 min at room temperature. To improve the long term stability of the chromatographic column as well as to remove additional interfering matrix components from the plasma or serum samples, the original method was further optimised by the implementation of a lipoprotein precipitation step similar to that described by Jeppsson et al.35 Therefore, 5 μL of the precipitation reagent (2 g MgCl2 and 1 g Dextransulfate in 10 mL H2O) were added to the iron saturated sample. After a further incubation time of additional 30 min at 4 °C, the sample was centrifuged for 10 min with 18000 g at 4 °C. 100 μL of the supernatant were diluted with 400 μL of a 20 mM, pH 6.5 Bis-Tris buffer followed by an additional centrifugation step (conditions as mentioned before) for 5 min. The resulting supernatant was stored under cooled conditions for the short-term and used in subsequent further investigations.
Sample desalting and preconcentration after fraction collection
To allow their further analysis with MALDI-TOF and ESI-MS respectively, the samples obtained from the ICP-MS guided, iron specific fractionation were desalted in order to remove the high salt load caused by the elution buffer B and pre-concentrated using Vivaspin 6 ultra filtration units with a molecular weight cut off of 10 kDa (6000 g at 4 °C).Gel electrophoresis (GE)
For the determination of the protein content of the sample prior to loading the IPG stripes, a Bradford assay was performed. The Bradford test was conducted as described in the product information provided with at least 3 replicates. The 96 well plate assay protocol was carried out with detection of the protein dye complex at 595 nm.IPG strips with a length of 11 cm and a linear pH gradient of 5–8 were used. For this strip length, 100–200 μg protein load in 200 μl is recommended. The samples were diluted with rehydration buffer (8 M urea, 2% CHAPS (w/v), 0.2% Bio-Lyte® 3/10 (w/v), 0.001% bromphenol blue (v/v), 70 mM DTT) and the final 200 μl were pipetted into the channel of the isoelectric focusing (IEF) tray, before peeling the cover sheet from the IPG strip and placing the strip with its gel side down into the channel. The strips were covered with mineral oil followed by an active rehydration step at constant 50 V for 16 h. Before starting the IEF the mineral oil was removed, so as to allow the positioning of a water soaked paper wick under each strip end on top of the electrodes. Afterwards, the strips were again overlaid with fresh mineral oil. The focusing occurs during a one step ramping in the rapid mode up to 8000 V with 30000 V*h. After focusing, the stripes were incubated for 15 min in 2 ml equilibration buffer I (0.375 M Tris, 6 M urea, 2% SDS (w/v), 30% glycerol (v/v), 130 mM DTT), 2 ml equilibration buffer II (0.375 M Tris, 6 M urea, 2% SDS (w/v), 30% glycerol (v/v), 130 mM IAA) followed by an equilibration in running buffer (25 mM Tris-base, 192 mM glycine, 0.1% SDS (w/v)). Afterwards, every strip was fixed with fused agarose in the slot of a suitable 10% pre-cast Criterion Tris-HCl gel. Additionally, 5 μl of a protein molecular weight marker (PageRuler™, Fermentas GmbH, St. Leon-Rot, Germany) was added to the gel. The separation in the second dimension occurs at a constant 200 V generated by a PowerPac 300 (Bio Rad laboratories GmbH) with maximum settings of 400 mA and 250 W for about 55 min.
Gel staining was performed after fixation of the gels (1 h incubation in a solution containing 20% methanol, 1% phosphoric acid), by using a coomassie solution (20% methanol, 20% Roti-Blue solution, overnight) followed by the final washing of the gel using a 25% methanol solution for at least 15 min.
Enzymatic digestion of the cut out protein spots
The protein in the gel spot was denatured and the cysteins were carbamidomethylated. The digestion was performed as described in the product information supplied with the GlycoProfil™I kit from Sigma. First the spots were destained, with a 200 mM ammonium bicarbonate buffer in 40% acetonitrile at 37 °C for 30 min. This procedure was repeated twice with fresh destaining solution until the disappearance of the blue dye. Afterwards, the clear gel spot was dried in a speed vac (Thermo Electron Corporation, Karlsruhe, Germany) followed by the addition of 5 units of PNGaseF and overnight incubation at 37 °C. The supernatant, which contains the carbohydrates in solution was removed and stored at −20 °C for further glyco analysis, while the protein is restrained in the gel spot. After an additional washing and drying step, trypsin solubilised in a 1 mM HCl solution, was added to the spot together with a 40 mM ammonium bicarbonate buffer containing 9% acetonitrile to allow the overnight digestion at 37 °C. Afterwards, the peptide containing supernatant was removed and the peptides left in the spot were extracted with a 0.1% trifluoroacetic acid in 50% acetonitrile solution, which is then combined with the removed supernatant. The volume of the pooled peptide extract was reduced to a final volume of about 10 μl using a speed vac.For digestion with α(2 → 3,6,8,9)Neuraminidase, 5 units of the reconstituted enzyme was directly added to a 100 μl aliquot of a seal serum sample. The digestion was carried out for different time periods at 37 °C. The reaction was stopped by adding 5 μl of the 500 mM Na2CO3 solution to the sample, which directly allows the necessary additional sample preparation steps for HPLC-ICP-MS analysis, such as iron saturation and lipoprotein precipitation as described above.
Database searches
Peptide mass fingerprinting (PMF) for searching in sequence databases with search algorithms such as Mascot, enables the identification of proteins or their homologous counterparts derived from other animal species, if they are already sequenced. The MALDI-MS data of the tryptic peptides from seal Transferrin were used for such a Mascot search with the following search parameter setting: Taxonomy—other mammalian; database—SwissProt; global modification—Carbamidomethyl (C); variable modification—Oxidation (M); missed Cleavages—1; mass tolerance—50 ppm; mass values—MH+, monoisotopic.ESI-MS-MS data were analysed using the ProteinPilot Software supplied with the QTRAP system.
Results and discussion
The following results outline the fast and targeted isolation and identification of a diagnostically relevant protein from an un-sequenced mammalian species prior to its utilisation in extended studies, using a combined analytical, metallomics36–38 orientated approach, which includes the complementary application of ICP-MS and molecule-specific detection techniques such as ESI or MALDI-MS.Investigation of the sample preparation procedure
All methodological developments to optimising the initial isolation and fractionation procedures were performed with plasma or serum samples derived from two North Sea seals (male, age ca. 7 months), which had to be euthanized (here named seal 1 and 2 respectively).To further characterise the sample preparation procedure, the remaining protein concentration after each step was analysed using the Bradford protein assay.39 In Fig. 1 the average protein concentration of three replicate measurements of (a) the pure plasma as well as a serum sample of seal 1, (b) after the iron saturation/lipoprotein precipitation as well as (c) after the final dilution step are shown. The lipoprotein precipitation results in protein depletion of approximately 20% for plasma compared to a protein depletion of only 3% when analysing serum. Since the sample indicates an alkaline pH of 11 after the lipoprotein precipitation due to the application of Na2CO3 for the iron saturation, the final dilution step adjusted the pH back to 7–7.5, which causes an additional protein precipitation resulting in an overall protein depletion of approximately 25 to 30%. To visualise the effect of the different procedures, the samples were additionally separated by two-dimensional gel-electrophoresis using IPG strips with a pH range from 5–8 followed by the separation on 10% pre-cast Tris-HCL gels. As indicated in Fig. 2a–c, different spots disappeared from the gel and also the intensity of the highly abundant albumin spot, which often masks less abundant proteins during the analysis of plasma or serum sample was reduced. When comparing the different steps of the plasma sample preparation procedure, the spot intensity within the 80 kDa, pH 6.5–8 region, where seal Tf is anticipated, remains nearly unchanged. Also, no Tf was detected when reanalysing the precipitate. To conclude, it can be stated that this result demonstrates the effective removal of the lipoproteins from the sample, which often influence the separation efficiency and long term stability of the separation column used. However, it has to be mentioned, that the quality of the gel-electrophoretic separation is affected by the still high abundance of albumin in the plasma and serum samples used. In principle both sample types are useful and delivers reliable results. The preference for serum is given, because of the comparability to other studies, because mostly serum samples are used.
![Mean protein concentrations determined using the Bradford assay after different stages of the sample preparation procedure for samples of seal 1, [plasma—grey pillar; serum—white pillar]: (a) pure seal plasma or serum, (b) after iron saturation and lipoprotein precipitation, (c) after the final dilution and centrifugation step.](/image/article/2010/MT/c0mt00009d/c0mt00009d-f1.gif) | ||
| Fig. 1 Mean protein concentrations determined using the Bradford assay after different stages of the sample preparation procedure for samples of seal 1, [plasma—grey pillar; serum—white pillar]: (a) pure seal plasma or serum, (b) after iron saturation and lipoprotein precipitation, (c) after the final dilution and centrifugation step. | ||
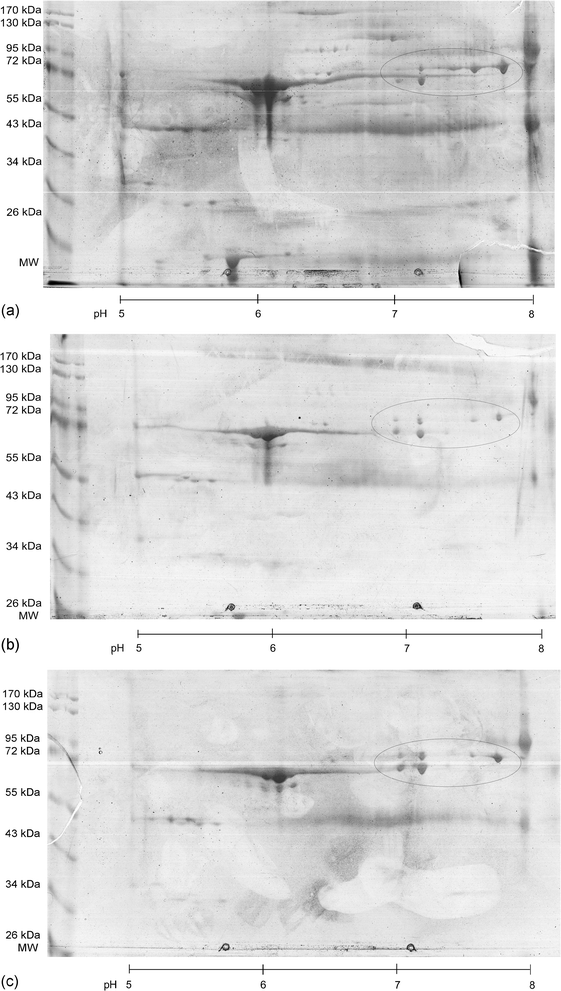 | ||
| Fig. 2 Visualisation of the effects of the sample preparation procedure using a 2D gel electrophoretic separation of a plasma sample at different stages of the sample preparation procedure for a plasma sample of seal 1: (2a) separation of an unprocessed plasma sample (75 μg protein), (2b) separation after iron saturation and lipoprotein precipitation (51 μg protein), (2c) separation after the final dilution and centrifugation step (92 μg protein). The anticipated Tf region on the gel is marked with a circle. | ||
HPLC-ICP-MS assisted fractionation of Tf isoforms in plasma samples
Since the depleted plasma samples still contain a number of proteins as shown in Fig. 2, HPLC-ICP-MS was used for the further analysis of the depleted plasma or serum samples in order to separate and fractionate the Tf isoforms utilising their characteristic iron content for highly specific detection.33,34 In addition, on-line UV detection at 280 and 460 nm, respectively, was performed (data not shown) to monitor the proteins eluting from the column as well as the Tf iron complex, which shows a characteristic absorption at 460 nm.35 Different anion exchange columns as well as chromatographic conditions were evaluated so as to obtaining a highly resolved separation of the possible Tf isoforms. Best results were obtained using a PorosHQ column, while applying an ammonium acetate gradient (500 mM, pH 6.5) from 0 to 12% over a period of 26 min followed by a washing and equilibration step at 100% and 0% solvent B, respectively. Further chromatographic parameters are described in the experimental section. Under these conditions it was possible to separate up to eight different isoforms depending on the sample used in less than 30 min. Reproducible separations with retention times and peak area RSDs below 1.4% and 10%, respectively, were obtained, which is a prerequisite for the further accurate iron guided fractionation of the different Tf isoforms. Detailed results are summarised in Table S-1 of the supplementary material. In Fig. 3 a typical chromatogram of a serum sample from seal 2 is shown, which indicates the presence of a number of iron containing species besides the dominant species. To further characterise the purity of the iron containing isoforms, a fraction containing all separated compounds were collected and reanalysed using 2D gel-electrophoresis (24 cm IPG strips with a linear pH gradient from 3–10, 12% acrylamid/bis-acrylamid separation gel). In Fig. S-1 of the supplementary material a typical gel-electrophoretic separation of the combined collected iron proteins is shown. As illustrated by the gel picture, the initial HPLC step effectively separates the iron containing isoforms from the remaining protein matrix, as indicated by the nearly total absence of further protein spots. This is underlined by the purity of the gel compared to those obtained during the direct analysis of the depleted plasma samples (see Fig. 2c). The gels contained only protein spots in the anticipated pI and molecular weight region of seal Tf, as shown in the small insert of Fig. S-1. The pI of the different seal Tf isoforms ranges from about 6.9 to 7.9 (Fig. 2). For further mass spectrometric analysis after PNGaseF and tryptic digestion, respectively, the separated spots were cut out from the gel matrix.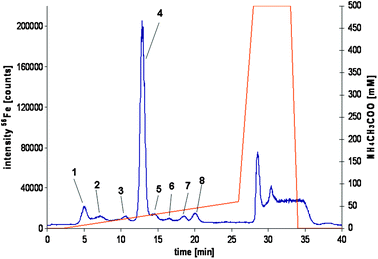 | ||
| Fig. 3 A typical chromatogram of a depleted seal plasma sample using anion exchange HPLC-ICP-MS for the separation and for element specific detection (56Fe isotope). | ||
Characterisation of the intact protein using MALDI-TOF and ESI-MS
As listed in Table 2, the Tf of different mammalian species show quite similar molecular masses of around 80 kDa, taking into account an average glycan content of about 6%.40 To characterise the fractionated iron containing protein species with respect to the presence of transferrin and in order to get first molecular mass information on seal Tf, MALDI-TOF-MS was used. The fractions of the different Tf isoforms were desalted and pre-concentrated using ultra filtration units with a molecular weight cut of 10 kDa, as pointed out in the experimental section. As one example, in Fig. 4 the MALDI-TOF spectra of the main fraction (see Fig. 3, peak 4) is shown. The fractions have been obtained during the iron specific fractionation of a seal plasma sample using HPLC-ICP-MS. Corresponding figures for some of the remaining peaks have been included into the supplementary material section (Fig. S-2). For the early eluting peaks insufficient spectra have been obtained mainly as a result of their lower abundance. Due to the limited sample availability, further repetitive fractionation and pooling with regard to providing a more concentrated sample for analysis was not possible. During all experiments, molecular masses of about 80.7 kDa were detected for the fractionated protein isoforms, which is in good agreement with the known mass of Tf from other species (see Table 2), similar results have been found for human Tf.33 However, the observed mass accuracy was highly compromised by the heterogeneity of the protein, leading to a relatively large peak width. To obtain additional mass information on the intact protein, static nano-spray ESI-QTRAP-MS was used as a complementary technique for the analysis of the main isoform of seal Tf. In Fig. 5a the spectra obtained during the static nano-spray analysis of the main fraction (see Fig. 3, peak 4) is shown. Data deconvolution (Fig. 5b) results in a molecular mass of the main Tf isoform of about 81.138 kDa for seal 2. | ||
| Fig. 4 Annotated MALDI-TOF spectra of the main iron containing protein species (peak 4) obtained by element guided fractionation using anion exchange HPLC-ICP-MS. | ||
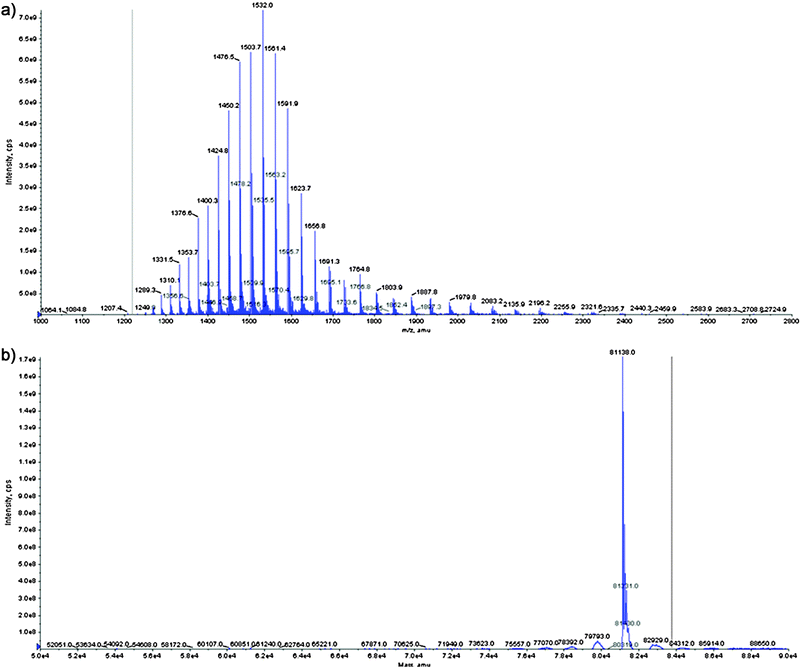 | ||
| Fig. 5 (a) Static nano ESI-QTRAP spectra of the main iron containing protein species obtained during the element guided fractionation using anion exchange HPLC-ICP-MS, (b) Data deconvolution revealed a molecular mass for the main isoform of about 81 kDa. | ||
| Species | SwissProt entry | Number of amino acids | Mass (Da) | Theoretical pI |
|---|---|---|---|---|
| a Sequenz entry in NCBI, mass and theoretical pI calculated with a program available on http://www.encorbio.com/protocols/Prot-MW.htm. | ||||
| Rabbit (Oryctolagus cuniculus) | P19134 | 676 | 74886.1 | 6.35 |
| Mouse (Mus musculus) | Q921I1 | 678 | 74881.4 | 6.81 |
| Human (Homo sapiens) | P02787 | 679 | 75181.4 | 6.70 |
| Chimpanzee (Pan troglodytes) | A5A6I6 | 679 | 75195.4 | 7.02 |
| Dog (Canis lupus familiaris) | XP_864568.1 (NCBI)a | 684 | 75797.2 | 8.5 |
| Bovine (Bos taurus) | Q29443 | 685 | 75829.6 | 6.50 |
| Horse (Equus caballus) | P27425 | 687 | 76141.0 | 6.61 |
| Pig (Sus scrofa) | P09571 | 696 | 76967.9 | 6.93 |
Enzymatic digestion of the Glycoprotein with Neuraminidase
To further characterise the separated iron containing proteins with respect to their glycostructure a serum sample of seal 2 was digested with α(2 → 3,6,8,9)Neuraminidase, prior to the sample preparation procedure and chromatographic separation of the Tf glycoforms. The mixture of different neuraminidase enzymes assured, that every possible connection of the sialic acid residues to the other sugars of the glycosilation structure are cleaved during the incubation. This reaction continues until the appearance of asialo Tf (S0). The cleavage reaction was carried out for different time periods at 37 °C. In Fig. 6a the separation of the Tf glycoforms after a 3 h incubation time without the enzyme neuraminidase is shown. This chromatography allows the separation of up to eight different isoforms under optimised chromatographic conditions. The elution order is particularly defined by the number of terminal sialic acids, which influences the charge state of the molecule. In comparison to Fig. 6a, Fig. 6b and c show the results for an incubation time of the sample with neuraminidase of 3 h (b) and 24 h (c), respectively. As indicated in the chromatograms (6b and c), the late eluting isoforms 5, 6, 7 and 8 disappeared after an incubation time of 3 or 24 h, respectively, while the abundance of the less sialinated isoforms increases due to the ongoing loss of terminal sialic acids, which finally affects their chromatographic retention. The enzymatic digestion continues until the appearance of an additional isoform without any sialic acid residue, the asialo Tf (S0) in Fig. 6c. The results prove that the separated isoforms differ in the degree of their sialic acid content. As a consequence, the original pattern of the isoforms, which indicates one dominant glycoform, changes to a different pattern, which shows the less sialinated glycoforms with lower chromatographic retention times at higher abundance depending on the duration of the incubation. Similar results were obtained by del Castillo Busto et al. for human Tf isoforms.34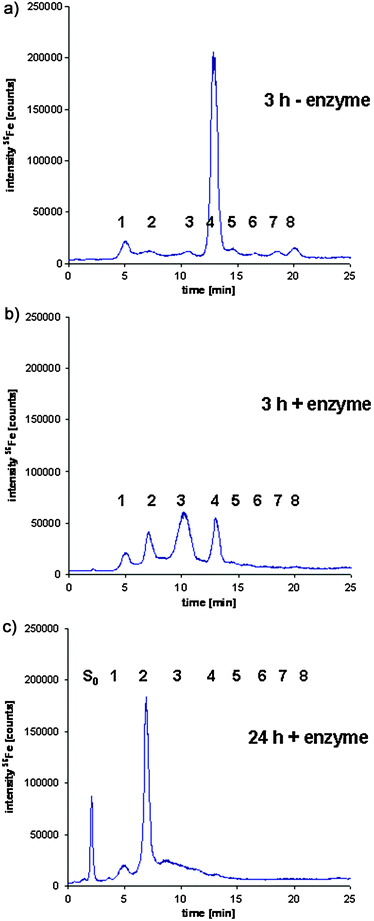 | ||
| Fig. 6 Enzymatic digestion of the Glycoproteins with α(2 → 3,6,8,9)Neuraminidase of serum of seal 1, prior to the sample preparation procedure and chromatographic separation of the Tf glycoforms using HPLC-ICP-MS detection: (a) incubation at 37 °C for 3 h without α(2 → 3,6,8,9)Neuraminidase; (b) incubation at 37 °C for 3 h with 5 U of α(2 → 3,6,8,9)Neuraminidase; (c) incubation at 37 °C for 24 h with 5 U of α(2 → 3,6,8,9)Neuraminidase. | ||
Cross species protein identification of the main protein fraction
PMF is a powerful tool for the rapid and accurate identification of single proteins isolated from complex sample mixtures. Cross species protein identification using such PMF data is particularly important for protein identification, when dealing with poorly or even un-sequenced species such as most marine mammals. Due to its conserved function with respect to iron transport, which represents one fundamental mechanism in all mammals, the common protein databases indicate Tf sequence homologies of more than 70%, when comparing different mammalian species. As a result of the sequence identity, there is a high statistical feasibility, that several tryptic peptides are absolutely conserved and therefore allow a reliable cross species protein identification even when working with poorly sequenced organisms.41 For the identification of seal Tf, database entries for related mammalian species were used. In Fig. S-3 of the supplementary material a typical PMF spectra obtained for spot 5 (see Fig. S-1), after digestion with PNGaseF and Trypsin is shown. For the verification of some of the peptide masses examined as well as their sequences, additional nano-HPLC-ESI-MS/MS experiments were performed. These experiments prove for a couple of peptides, that the mass is composed of exactly the same amino acids. Sequence coverage of about 17% was obtained with the serotransferrin precursor of Equus cabalus (Horse), which allows the final identification of the isolated protein species as seal Tf. All results are summarised in Table 3. In Fig. S-4 of the supplementary material MS/MS spectra of two peptides are shown, which were also observed during the nano-HPLC-ESI-MS/MS experiments. Additional MS Blast analysis of these two peptide sequences indicate that both were proteotypic peptides only present in proteins of the Tf family.| m/z | PMF Identified by Mascot | Verification by MS/MS data | MS Blast of peptide sequence | |
|---|---|---|---|---|
| Sequence | Organism | |||
| 887.4290 | SCHTGLGR | horse | ||
| 1757.8004 | AVSNFFAGSCVPCADR | horse | yes | Transferrin family only |
| 1994.8770 | CACSNHEPYFGYSGAFK | horse | yes | Transferrin family only |
| 2912.3328 | CLADGAGDDVAFVKHSTVLENLPQEADR | horse | ||
| 1486.7167 | SKDFHLFSSPHGK | horse | ||
| 1271.6701 | DFHLFSSPHGK | horse | ||
| 1097.5202 | YYGYTGAFR | horse | ||
| 2560.2551 | DDTQCLANLQPTTTYKTYLGEK | horse | ||
| 1146.5919 | LLEACTFHR | horse | ||
Conclusions and outlook
This metallomics approach demonstrates the potential of a complementary application of mass spectrometric detection techniques such as ICP-MS and MALDI/ESI-MS for the targeted isolation and identification of the diagnostically relevant iron containing acute phase protein transferrin (Tf). The isolated seal Tf showed similar characteristics in terms of its molecular weight compared to the known Tf molecular weight of other animal species, while the estimated pI values differ slightly from those reported e.g. for humans. Even though the seal proteome is only poorly described in the data bases commonly used, the fractionated protein could be clearly identified as Tf using either peptide mass fingerprinting via MALDI-TOF-MS or nanoLC-ESI-QTRAP-MS/MS based peptide sequencing followed by cross species protein identification. Some of the peptides were identified as proteotypic for proteins of the Tf family, which also indicates the conserved character of the amino acid sequence of this protein due to its general importance, particularly as the iron transporting protein in mammalian organisms. First qualitative screening results of smaller sample subsets has already shown differences in the observed isoform patterns, which may indicate the suitability of this parameter for describing the health status of these marine indicator organisms.17 Therefore, future work will be focused on the absolute quantitative analysis of the separated isoforms using HPLC-ICP-MS and iron specific detection, to allow the accurate investigation of differences in the Tf concentration as well as the individual isoform pattern, when comparing a larger set of samples derived from different animal groups (male vs. female, different age, origin). Within this background, a further focus will also lie on the investigation of micro-heterogeneities in the glycostructure of selected Tf isoforms.Acknowledgements
The authors would like to thank all colleagues at GKSS and at the Research and Technology Centre (FTZ) in Büsum, Germany, the Seal Station Friedrichskoog as well as the seal rangers along the North Sea coast for their cooperation and their support in obtaining the seal blood samples.References
- V. N. de Jonge, M. Elliott and V. S. Brauer, Marine monitoring: Its shortcomings and mismatch with the EU Water Framework Directive's objectives, Mar. Pollut. Bull., 2006, 53(1–4), 5–19 CrossRef CAS.
- A. T. Fisk, C. A. de Wit, M. Wayland, Z. Z. Kuzyk, N. Burgess, R. Letcher, B. Braune, R. Norstrom, S. Polischuk Blum, C. Sandau, E. Lie, H. J. S. Larsen, J. U. Skaare and D. C. G. Muir, An assessment of the toxicological significance of anthropogenic contaminants in canadian arctic wildlife, Sci. Total Environ., 2005, 351–352, 57–93 CrossRef CAS.
- R. J. Law, P. Bersuder, L. K. Mead and P. D. Jepson, PFOS and PFOA in the livers of harbour porpoises (phocoena phocoena) stranded or bycaught around the UK, Mar. Pollut. Bull., 2008, 56(4), 792–797 CrossRef CAS.
- J. Mi, A. Orbea, N. Syme and M. Ahmed, Peroxisomal proteomics, a new tool for risk assesment of peroxisome proliferating in the marine environment, Proteomics, 2005, 5(15), 3954–3965 CrossRef CAS.
- S. Mössner and K. Ballschmitter, Marine mammals as global pollution indicators for organochlorines, Chemosphere, 1997, 34(5–7), 1285–1296 CrossRef CAS.
- P. M. Bennett, P. D. Jepson, R. J. Law, B. R. Jones, T. Kuiken, J. R. Baker, E. Rogan and J. K. Kirkwood, Exposure to heavy metals and infectious disease mortality in harbour porpoises from england and wales, Environ. Pollut., 2001, 112(1), 33–40 CrossRef CAS.
- P. D. Jepson, P. M. Bennett, C. R. Allchin, R. J. Law, T. Kuiken, J. R. Baker, E. Rogan and J. K. Kirkwood, Investigating potential associations between chronic exposure to polychlorinated biphenyls and infectious disease mortality in harbour porpoises from england and wales, Sci. Total Environ., 1999, 243–244, 339–348 CrossRef CAS.
- U. Siebert, C. Joiris, L. Holsbeek, H. Benkes, K. Failing, K. Frese and E. Petzinger, Potential relation between mercury concentrations and necropsy findings in cetaceans from German waters of the north and baltic seas, Mar. Pollut. Bull., 1999, 38(4), 285–295 CrossRef CAS.
- M. P. Cajaraville, M. J. Bebianno, J. Blasco, C. Porte, C. Sarasquete and A. Viarengo, The use of biomarkers to assess the impact of pollution in coastal environments of the iberian peninsula: A practical approach, Sci. Total Environ., 2000, 247(2–3), 295–311 CrossRef CAS.
- I. Hasselmeier, S. Fonfara, J. Driver and U. Siebert, Differential hematology profiles of free-ranging, rehabilitated, and captive harbor seals (phoca vitulina) of the german north sea, Aquatic Mammals, 2008, 34, 149–156 CrossRef.
- A. Kakuschke, E. Valentine-Thon, S. Griesel, S. Fonfara, U. Siebert and A. Prange, Immunological impact of metals in harbor seals (phoca vitulina) of the north sea, Environ. Sci. Technol., 2005, 39(19), 7568–7575 CrossRef CAS.
- U. Siebert, P. Wohlsein, K. Lehnert and W. Baumgärtner, Pathological findings in harbour seals (phoca vitulina): 1996–2005, J. Comp. Pathol., 2007, 137(1), 47–58 CrossRef CAS.
- L. Ahrens, U. Siebert and R. Ebinghaus, Total body burden and tissue distribution of polyfluorinated compounds in harbor seals (phoca vitulina) from the german bight, Marine Pollution Bulletin, 2009, 76(2), 151–158 CAS.
- S. Griesel, A. Kakuschke, U. Siebert and A. Prange, Trace element concentrations in blood of harbor seals (phoca vitulina) from the wadden sea, Sci. Total Environ., 2008, 392(2–3), 313–323 CrossRef CAS.
- L. Weijs, K. Das, U. Siebert, N. van Elk, T. Jauniaux, H. Neels, R. Blust and A. Covaci, Concentrations of chlorinated and brominated contaminants and their metabolites in serum of harbour seals and harbour porpoises, Environ. Int., 2009, 35(6), 842–850 CrossRef CAS.
- L. Kuenstl, S. Griesel, A. Prange and W. Goessler, Arsenic speciation in bodily fluids of harbor seals (phoca vitulina) and harbor porpoises (phocoena phocoena), Environmental Chemistry, 2009, 6(4), 319–327 CAS.
- A. Kakuschke, E. Valentine-Thon, S. Griesel, J. Gandrass, O. Pérez Luzardo, L. Domínguez Boada, M. Zumbado Peña, M. Almeida González, M. Grebe, D. Pröfrock, H.-B. Erbsloeh, K. Kramer, S. Fonfara and A. Prange, First health and pollution study on harbor seals (phoca vitulina) living in the german elbe estuary, Marine Pollution Bulletin, 2010 DOI:10.1016/j.marpolbul.2010.07.011.
- A. Kakuschke, E. Valentine-Thon, S. Fonfara, S. Griesel, T. Rosenberger, U. Siebert and A. Prange, Metal-induced impairment of the cellular immunity of newborn harbor seals (phoca vitulina), Arch. Environ. Contam. Toxicol., 2008, 55(1), 129–136 CrossRef CAS.
- A. Kakuschke, H. B. Erbsloeh, S. Griesel and A. Prange, Acute phase protein haptoglobin in blood plasma samples of harbour seals (phoca vitulina) of the wadden sea and of the isle helgoland, Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol., 2010, 155(1), 67–71 CrossRef CAS.
- H. Rosenfeld, S. Lassen and A. Prange, Characterization of haptoglobin in the blood plasma of harbor seals (phoca vitulina), J. Proteome Res., 2009, 8(6), 2923–2932 CrossRef CAS.
- F. Ceciliani, A. Giordano and V. Spagnolo, The systemic reaction during inflammation: The acute-phase proteins, Protein Pept. Lett., 2002, 9(3), 211–223 CrossRef CAS.
- P. D. Eckersall, Recent advances and future prospects for the use of acute phase proteins as markers of disease in animals, Revue De Medecine Veterinaire, 2000, 151(7), 577–584 Search PubMed.
- H. Murata, N. Shimada and M. Yoshioka, Current research on acute phase proteins in veterinary diagnosis: An overview, Vet. J., 2004, 168(1), 28–40 CrossRef CAS.
- H. H. Petersen, J. P. Nielsen and P. M. H. Heegaard, Application of acute phase protein measurements in veterinary clinical chemistry, Vet. Res., 2004, 35(2), 163–187 Search PubMed.
- T. Arndt, Carbohydrate-deficient transferrin as a marker of chronic alcohol abuse: A critical review of preanalysis, analysis, and interpretation, Clinical Chemistry, 2001, 47(1), 13–27 Search PubMed.
- A. Helander, G. Eriksson, H. Stibler and J.-O. Jeppsson, Interference of transferrin isoform types with carbohydrate-deficient transferrin quantification in the identification of alcohol abuse, Clinical Chemistry, 2001, 47(7), 1225–1233 Search PubMed.
- Y. Murawaki, H. Sugisaki, I. Yuasa and H. Kawasaki, Serum carbohydrate-deficient transferrin in patients with nonalcoholic liver disease and with hepatocellular carcinoma, Clin. Chim. Acta, 1997, 259(1–2), 97–108 CrossRef CAS.
- E. Gruys, M. J. M. Toussaint, T. A. Niewold and S. J. Koopmans, Acute phase reaction and acute phase proteins, J. Zhejiang Univ., Sci., 2005, 6b(11), 1045–1056 CrossRef CAS.
- P. Leonhard, R. Pepelnik, A. Prange, N. Yamada and T. Yamada, Analysis of diluted sea-water at the ng l−1 level using an ICP-MS with an octopole reaction cell, J. Anal. At. Spectrom., 2002, 17(3), 189–196 RSC.
- S. D. Tanner, V. I. Baranov and D. R. Bandura, Reaction cells and collision cells for ICP-MS: A tutorial review, Spectrochim. Acta, Part B, 2002, 57(9), 1361–1452 CrossRef.
- N. Yamada, J. Takahashi and K. Sakata, The effects of cell-gas impurities and kinetic energy discrimination in an octopole collision cell ICP-MS under non-thermalized conditions, J. Anal. At. Spectrom., 2002, 17(10), 1213–1222 RSC.
- V. Schnaible, S. Wefing, A. Resemann, D. Suckau, A. Bucker, S. Wolf-Kummeth and D. Hoffmann, Screening for disulfide bonds in proteins by MALDI in-source decay and LIFT-TOF/TOF-MS, Anal. Chem., 2002, 74(19), 4980–4988 CrossRef CAS.
- M. E. del Castillo Busto, M. Montes-Bayon, E. Blanco-Gonzalez, J. Meija and A. Sanz-Medel, Strategies to study human serum transferrin isoforms using integrated liquid chromatography ICPMS, MALDI-TOF, and ESI-Q-TOF detection: Application to chronic alcohol abuse, Anal. Chem., 2005, 77(17), 5615–5621 CrossRef.
- M. E. del Castillo Busto, M. Montes-Bayon and A. Sanz-Medel, Accurate determination of human serum transferrin isoforms: Exploring metal-specific isotope dilution analysis as a quantitative proteomic tool, Anal. Chem., 2006, 78(24), 8218–8226 CrossRef CAS.
- J. O. Jeppsson, H. Kristensson and C. Fimiani, Carbohydrate-deficient transferrin quantified by HPLC to determine heavy consumption of alcohol, Clinical Chemistry, 1993, 39(10), 2115–2120 Search PubMed.
- H. Haraguchi, Metallomics as integrated biometal science, J. Anal. At. Spectrom., 2004, 19(1), 5–14 RSC.
- S. Mounicou, J. Szpunar and R. Lobinski, Metallomics: The concept and methodology, Chem. Soc. Rev., 2009, 38(4), 1119–1138 RSC.
- J. Szpunar, Advances in analytical methodology for bioinorganic speciation analysis: Metallomics, metalloproteomics and heteroatom-tagged proteomics and metabolomics, Analyst, 2005, 130(4), 442–465 RSC.
- M. M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal. Biochem., 1976, 72(1–2), 248–254 CrossRef CAS.
- R. P. Oda, R. Prasad, R. L. Stout, D. Coffin, W. P. Patton, D. L. Kraft, J. F. OBrien and J. P. Landers, Capillary electrophoresis-based separation of transferrin sialoforms in patients with carbohydrate-deficient glycoprotein syndrome, Electrophoresis, 1997, 18(10), 1819–1826 CAS.
- P. J. Lester and S. J. Hubbard, Comparative bioinformatic analysis of complete proteomes and protein parameters for cross-species identification in proteomics, Proteomics, 2002, 2(10), 1392–1405 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary information. See DOI: 10.1039/c0mt00009d |
| This journal is © The Royal Society of Chemistry 2010 |