Study of the Se-containing metabolomes in Se-rich yeast by size-exclusion—cation-exchange HPLC with the parallel ICP MS and electrospray orbital ion trap detection
Sandra Gil
Casal
,
Johann
Far
,
Katarzyna
Bierla
,
Laurent
Ouerdane
and
Joanna
Szpunar
CNRS/UPPA, Laboratoire de Chimie Analytique Bio-inorganique et Environnement, UMR5254, Hélioparc, 2, av., Pr. Angot, F-64053 Pau, France
First published on 29th July 2010
Abstract
Strong cation exchange HPLC with the parallel ICP MS and electrospray hybrid linear ion trap quadrupole orbital trap mass spectrometry (ESI Orbitrap MS) detection was developed for the study of the metabolomic pattern of selenium in selenium-rich yeast. The mobile phase composition (gradient of ammonium formate in 20% methanol) was optimized to obtain separation in conditions guaranteeing the identical ICP MS sensitivity during the entire chromatographic run and the compatibility with electrospray ionization. Twenty seven Se-containing metabolites observed in the HPLC-ICP MS chromatogram were identified by ESI Orbitrap MS based on the Se isotopic pattern, the accurate molecular mass, and the multistage fragmentation patterns. The method allowed for the first time the correlation of the differences observed in HPLC-ICP MS chromatography of water extracts of Se-rich yeast samples from different manufacturers with the identity of the eluted compounds determined by ESI MS.
Introduction
Selenium is known to be either essential or toxic for living organisms depending on the concentration and the chemical form.1,2 It has also been recognized as an antioxidant and chemo-preventive agent in cancer.3,4 The low levels of this element in the diet in many countries is responsible for the increasing interest in the supplementation of food and feed with this element.1,5 One of the most popular supplements is yeast grown in the presence of selenite (or selenate) which is able to accumulate up to 3000 mg Se kg−1.6–8 It is an attractive source of Se due to its low cost and high content of selenomethionine (SeMet) acting as a precursor for selenoprotein synthesis.2,7Se-rich yeast can be characterized by the selenium metabolic profile (selenometabolome) which is characteristic of yeast strain and fermentation parameters and can be a precious fingerprint of the origin of preparations available on the market and of the reproducibility of the production process.9 Despite recent advances in analytical methodology,8–10 methods allowing the rapid on-line identification of Se-metabolites and the acquisition of a Se-metabolomic profile are missing.
The fractionation pattern of selenocompounds in yeast was proposed by Ruiz Encinar et al.11 and Far et al.12 for the verification of the batch-to-batch reproducibility and the differentiation of Se-rich yeast origin. The fractionation was based on differential leaching followed by size-exclusion inductively coupled plasma (ICP) MS analyses of the extracted fraction of selenium. In addition to the problems related to the limited selectivity, recovery and reproducibility of the operationally defined procedure, the ICP MS-based fingerprinting protocols suffered from the lack of knowledge of the identity of the species used as markers of the yeast origin.
An insight into the identity of selenium compounds can be achieved by electrospray MS. A successful use of this technique, first demonstrated for the identification of an unknown selenium species in Se-rich yeast by Casiot et al.,13 has long been hampered by the insufficient sensitivity, largely related due to the impossibility of the on-line purification of Se-metabolites prior to electrospray MS. Consequently, the identified species were limited to molecules which could be separated from the sample salt fraction co-eluting with the rest of species.14–16 The methods were unsuitable for fingerprinting as all these species eluted as one peak in size-exclusion chromatography (SEC)-ICP MS after the total volume of the column.
A breakthrough in ESI MS identification of Se metabolites in yeast was made by Dernovics et al. who managed to purify (by 3D size-exclusion-anion-exchange-reversed-phase HPLC) a dozen Se-containing metabolites to a degree allowing their structural analysis by ESI MS/MS.17,18 The complexity of the purification procedure was alleviated later by using hydrophilic interaction chromatography (HILIC)-ESI Orbitrap MS19 and normal phase HPLC-ESI TOF MS12 to allow the identification of up to 10 compounds in a single run without, however, the separation quality allowing quantitative fingerprinting of selenometabolites by HPLC-ICP MS.
The objective of this work was to develop a straightforward analytical approach allowing the acquisition of an exhaustive metabolomic pattern of Se-compounds in yeast by HPLC-ESI MS. The adopted way was based on the isolation of a yeast extract fraction containing the largest possible set of selenium metabolites and optimisation of their high resolution chromatographic separation with the parallel detection by ESI MS and ICP MS.
Experimental
Samples and reagents
Samples of five leading manufactures worldwide [Alltech (KY), Angel (China), Biorigin (Brazil), Lallemand (Canada), Lesaffre (France)] were investigated. The samples were randomly coded with capital letters (A–E). One sample was analysed as two different production batches referred to as 1 and 2. A standard reference material SELM-1 (NRCC, Canada) was investigated along each analysis. The characteristics of the samples in terms of the total selenium, water-soluble selenium and selenomethionine concentrations are given in Table 1. Total selenium and selenomethionine by the methods described elsewhere20 and validated in an interlaboratory study.21 In brief, total selenium was determined upon digestion with HNO3–H2O2 by ICP MS; selenomethionine was determined after enzymatic digestion by HPLC–ICP MS. All the reagents were purchased from the Sigma group (Saint Quentin Fallavier, France). Deionised water (18.2 MΩ cm) obtained from a Milli-Q system (Millipore, Guyancourt, France) was used throughout the study. All samples were then prepared with the same method which allowed their relative comparison.| Sample | Se concentration, μg g−1 | Water extractable Se (%) | Selenomethionine concentration, μg g−1 (as Se) | Selenomethionine concentration (%)b |
|---|---|---|---|---|
| a Certified values are: 2059 ± 64 μg g−1 (selenium concentration) and 1381 ± 70 μg g−1 (selenomethionine concentration). b Determined selenomethionine concentration according Połatajko et al., ABC, 2005.20 | ||||
| A1 | 1857 ± 22 | 27 ± 9 | 1720 ± 95 | 93 ± 12 |
| A2 | 1870 ± 64 | 35 ± 2 | 1601 ± 177 | 86 ± 25 |
| B | 1819 ± 65 | 16 ± 1 | 795 ± 34 | 44 ± 7 |
| C | 1217 ± 40 | 21 ± 6 | 1035 ± 47 | 85 ± 13 |
| D | 2442 ± 41 | 23 ± 6 | 1901 ± 93 | 78 ± 10 |
| E | 1902 ± 117 | 18 ± 3 | 1560 ± 98 | 82 ± 5 |
| G (SELM-1)a | 2062 ± 63 | 23 ± 2 | 1187 ± 47 | 58 ± 8 |
Instrumentation
HPLC (size exclusion (SEC), cation-exchange (SCX) or anion-exchange (SAX))-ICP-MS coupling was achieved by using an Agilent 1200 Series HPLC system (Agilent Technologies, Palo Alto, CA) connected to an Agilent 7500ce Series ICP-MS equipped with a collision cell with hydrogen as the collision gas for element-specific detection of 77Se, 80Se, 82Se.For the SCX HPLC-ESI-MS experiments, an Ultimate 3000 pump (Dionex) was connected to an LTQ Orbitrap Velos (Thermo Scientific) linear ion trap quadrupole orbital trap mass analyzer used in the full-scan mode. The SCX-ESI-MS coupling was carried out via a Heated Electrospray Ion Source (HESI-II).
Water extraction was carried out using a Vibracell 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 115 (Bioblock Scientific, Illkirch, France) ultrasonic probe offering a nominal power of 500 W or a Branson 1210 ultrasonic bath. Size exclusion fractions were collected using an AKTA Prime (Amersham Pharmacia Biotech) fraction collector. Lyophilization was carried out using a Cryotec lyophilizer (Saint Gely du Fesc, France).
115 (Bioblock Scientific, Illkirch, France) ultrasonic probe offering a nominal power of 500 W or a Branson 1210 ultrasonic bath. Size exclusion fractions were collected using an AKTA Prime (Amersham Pharmacia Biotech) fraction collector. Lyophilization was carried out using a Cryotec lyophilizer (Saint Gely du Fesc, France).
Procedures
The ion source was operated in the positive ion mode. The optimum settings were: ion source voltage, 2.60 kV; capillary temperature, 280 °C; source heater temperature, 120 °C; sheath gas flow, 20; auxiliary gas flow, 5; S-lens RF level, 61%; resolution, 100000. Mass spectra were acquired in the 100–1000 m/z range, and were processed with Xcalibur 2.1 software (Thermo Scientific). The instrument was mass calibrated with a mixture of caffeine, n-butylamine, Met-Arg-Phe-Ala (MRFA), Ultramark 1621 and sodium dodecyl sulfonate (SDS), dissolved in 50% acetonitrile and 0.1% formic acid solution.
Results and discussion
Choice of the chromatographic fractionation technique
The chromatographic technique should assure the maximum separation of the Se compounds present in a yeast extract in conditions allowing their efficient electrospray ionization and reliable quantification by ICP MS. Size-exclusion LC has been the most exhaustively studied technique for the fractionation of the Se-containing metabolites in yeast.11,22 It allows the quantitative (100%) recovery of the injected selenium separated in relatively well resolved 6–7 peaks. As the elution is isocratic, the peak intensity is a measure of the concentration of selenium in the corresponding fraction. The chromatograms obtained for the set of analysed samples (Fig. 1) indicate a certain potential of SEC-ICP MS for the profiling of the yeast metabolome based on the intensity ratio of the peaks. The utility of this approach is, however, limited as the morphology of chromatograms for yeast samples of different manufacturers was quite frequently very similar.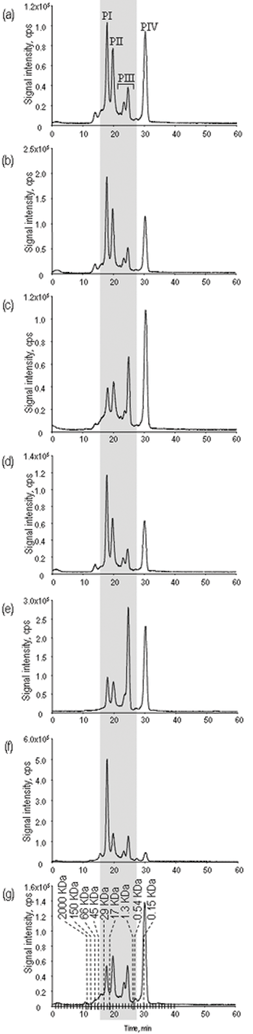 | ||
| Fig. 1 Chromatograms obtained by SEC-ICP MS (77Se) for the samples investigated. (a) A1, (b) A2, (c) B, (d) C, (e) D, (f) E, (g) SELM-1. Dash lines are retention times of molecular weight markers (blue dextran, globulin, bovine and chicken albumins, carbonic anhydrase, myoglobin, hydroxocobalamin, taurocholic acid and methionine, respectively). | ||
The peaks in SEC-ICP MS are not chromatographically pure and each of them is known to contain a number of compounds, some of them already published15,16,18,19,22,23 and found in the fractions eluting between 16–32 min in these chromatographic conditions. As the chromatographic resolution of the technique is low, selenium compounds co-elute with many others and they need to be extensively purified in order to be detectable by electrospray MS. Electrospray MS detection, at least in the full scan mode, does not allow the comprehensive metabolomics analysis, which raises a need for an orthogonal chromatographic separation mechanism prior to ESI MS.
The potentially most attractive, for a fine characterisation, was considered to be peak IV (see Fig. 1). It elutes after the total volume of the column, is well separated from the sample matrix and from other Se-peaks, and, according to literature, contains a number of selenium compounds. Attempts to separate these compounds by HILIC, NP (normal phase), RP (reverse phase), anion- and cation-exchange HPLC led to two conclusions: first, one compound, Se-adenosyl-Se-selenohomocysteine was strongly dominant, and second, the chromatograms resulted in 2–3 peaks only despite multiple Se-isotopic patterns detected in the ESI MS spectra, reducing the potential of the use of HPLC–ICP MS chromatograms of this peak for fingerprinting purposes. Hence, our attention was directed to the finer fraction of the other Se-containing fractions. As the baseline separation of peaks I–III was problematic and suggested the possibility of other compounds eluting in between, it was decided to use the entire 10 min elution range covering peaks I–III as the input sample for orthogonal chromatography (see Fig. 1).
Most of the compounds eluting in this range are hydrophilic which resulted in their elution in the void of reversed-phase HPLC. Attempts to use HILIC and normal phase HPLC proposed elsewhere12,19 resulted in a very limited number of fairly broad peaks. They allowed the identification of a dozen selenocompounds by ESI MS but the morphology of the chromatograms was not suitable for ICP MS fingerprinting. Therefore our attention was turned towards ion-exchange chromatography as the 2nd separation mechanism.
Strong anion-exchange HPLC was demonstrated to be an efficient technique allowing the baseline separation of a number of hydrophilic selenometabolites17,18 and appeared attractive for HPLC-ICP MS fingerprinting. The optimization of the flow rate, pH and gradient was carried out in this work with the purpose of obtaining the maximum number of peaks in the chromatograms. The optimized parameters included pH (in the range 4.5–7.5) and flow rate (in the range 1.2–1.5 ml min−1). Indeed, the chromatograms (Fig. 2) allow the separation of a dozen peaks and have a high added value in terms of the discrimination potential among the samples. The problem is the missed identity information because the mobile phase composition prevents sufficient ionization of the species for their detection by ESI MS.
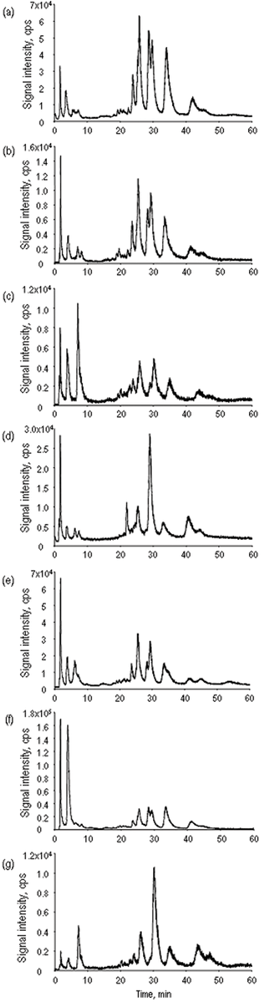 | ||
| Fig. 2 Chromatograms obtained by anion-exchange HPLC-ICP MS (77Se) for the samples investigated. (a) A1, (b) A2, (c) B, (d) C, (e) D, (f) E, (g) SELM-1. | ||
The highest power of resolution was found to be offered by cation-exchange HPLC which produced up to 30 peaks in chromatograms with ICP MS detection (Fig. 3). The mobile phase buffer (gradient of pyridine from 1 to 40 mM) is readily tolerated by ICP MS without affecting the sensitivity.23 However, the absence of an organic modifier and the high flow rate applied prevented the on-line analysis by ESI MS. Consequently, the SCX HPLC-ICP MS was downscaled and separation conditions optimized to be compatible with ESI MS.
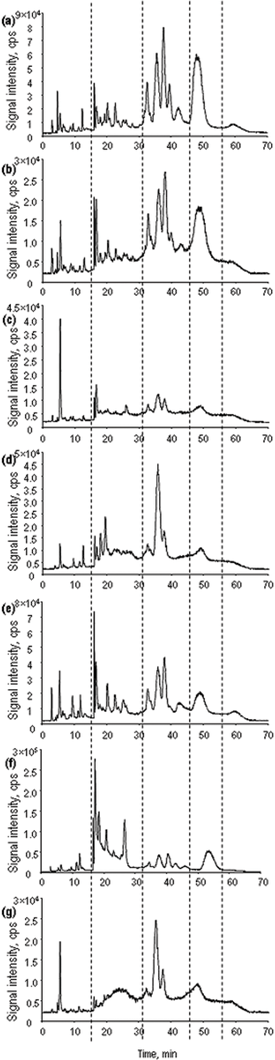 | ||
| Fig. 3 Chromatograms obtained by cation-exchange HPLC-ICP MS (77Se) for the samples investigated. (a) A1, (b) A2, (c) B, (d) C, (e) D, (f) E, (g) SELM-1. | ||
Optimization of the separation conditions for the parallel ICP MS and ESI MS detection
The same stationary phase was used in the narrowbore format which reduced the flow rate down to 500 μl min−1. The mobile phase was enriched with 20% methanol in order to facilitate the ionization in the electrospray source. Pyridine was replaced with more volatile ammonium formate. These modifications resulted in some loss in resolution (a 20% decrease of the number of peaks) but allowed the detection of 27 selenium isotopic patterns (adducts and reduced/oxidized forms not taken into account) throughout the chromatographic run. The comparison of the standard SCX HPLC-ICP MS chromatogram (pyridine buffer) and the narrowbore HPLC one obtained in the optimized conditions is given in Fig. 4, together with the retention times of the detected selenium compounds. The identification methodology, described elsewhere19 was extensively applied. The list of the empiric formula, selenium isotopic patterns recorded, and identified structures was given in Table 2. This is, to our knowledge, the largest set of identified metabolites in a Se-enriched yeast sample with an additional merit to be obtained on-line in a single chromatographic run.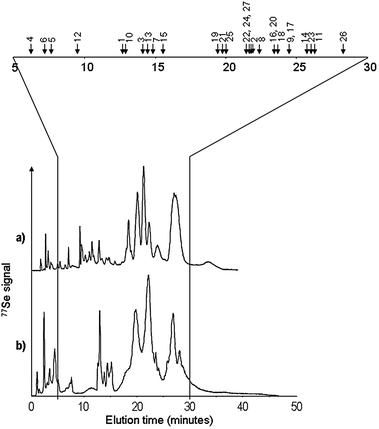 | ||
| Fig. 4 Comparison of cation-exchange HPLC-ICP MS (77Se) chromatograms obtained in (a) maximum resolution conditions, and (b) trade-off conditions allowing ESI MS detection of the maximum number of Se-species. The retention times (chromatogram b) of the species detected are marked with arrows and reported in Table 2. | ||
| No. | Th mass | Exp mass | Error ppm | Se isotopic pattern | Empiric formula | Proposed structures | Ref. |
|---|---|---|---|---|---|---|---|
| a The presence of free selenomethionine, 1, and new compounds related to selenocysteine and selenohomocysteine could be due in part to sample proteolysis between yeast harvest and the analysis | |||||||
| 1 | 198.00278 | 198.00293 | −0.74 |

|
C5H12N02Se+ |

|
—a |
| 195.98713 | 195.98706 (198-2H) | 0.37 | C5H10N02Se+ | ||||
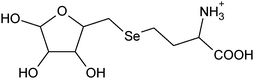
| 2 | 285.03481 | 285.03486 | −0.17 |
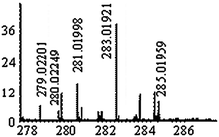
|
C8H17N204Se+ |
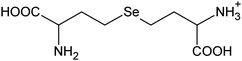
|
12, 17 |
| 283.01916 | 283.01921 (285-2H) | −0.16 | C8H15N204Se+ | ||||
| 3 | 316.02939 | 316.02932 | 0.21 |

|
C9H18N06Se+ |
|
24 |
| 4 | 345.01955 | 345.01965 | −0.30 |

|
C9H17N207Se+ |

|
12, 17 |
| 5 | 359.03520 | 359.03540 | −0.55 |

|
C10H19N207Se+ |
|
17, 24 |
| 357.01955 | 357.01946 (359-2H) | 0.24 | C10H17N207Se+ | ||||
| 6 | 376.99162 | 376.99146 | 0.46 |
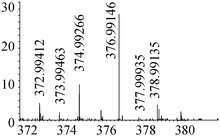
|
C9H17N207SSe+ |

|
—a |
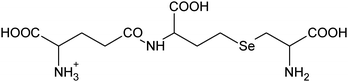
| 7 | 400.06175 | 400.06150 | 0.63 |
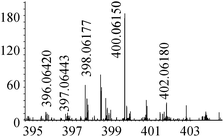
|
C12H22N307Se+ |
|
17, 24 |
| 8 | 402.02325 | 402.02309 | 0.39 |

|
C10H20N306SSe+ |
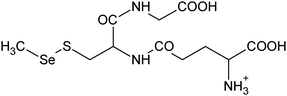
|
25 |
| 9 | 403.01850 | 403.01881 | −0.78 |

|
C11H20N306SSe+ |
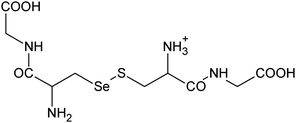
|
25 |
| 10 | 414.07740 |

|
C13H24N307Se+ |
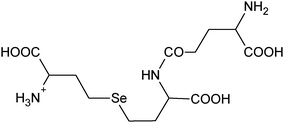
|
—a | ||
| 412.06175 | 412.06136 (414-2H) | 0.95 | C13H22N307Se+ | ||||
| 11 | 433.07332 | 433.07307 | 0.58 |

|
C14H21N605Se+ |

|
13, 26 |
| 431.05767 | 431.05780 (433-2H) | −0.30 | C14H19N605Se+ | ||||
| 12 | 434.01308 | 434.01225 | 1.90 |

|
C11H20N308SSe+ |
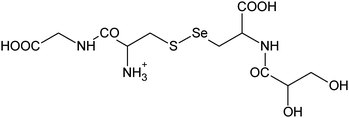
|
19 |
| 13 | 475.03963 | 475.03967 | −0.09 |

|
C13H23N408SSe+ |
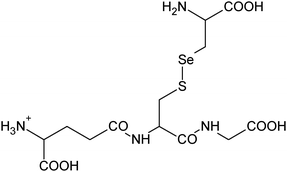
|
19 |
| 14 | 512.95211 | 512.95204 | 0.15 |

|
C12H21N2010Se2+ |
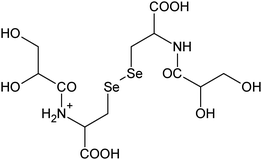
|
—a |
| 15 | 522.98408 | 522.98423 | −0.30 |
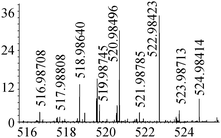
|
C13H23N408Se2+ |
|
—a |
| 16 | 532.06110 | 532.06125 | −0.28 |

|
C15H26N509SSe+ |

|
24 |
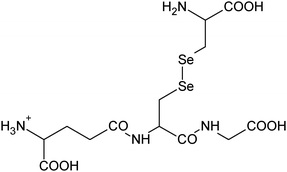
| 17 | 547.06076 | 547.06093 | −0.30 |
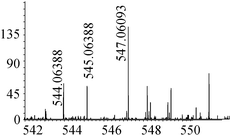
|
C16H27N4010SSe+ |
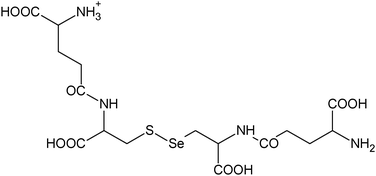
|
—a |
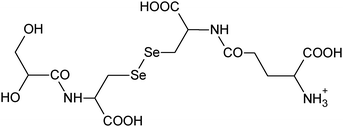
| 18 | 553.97866 | 533.97879 | −0.23 |

|
C14H24N3010Se2+ |
|
—a |
| 19 | 563.05568 | 563.05629 | −1.09 |
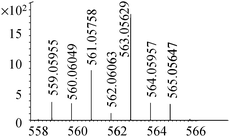
|
C16H27N4011SSe+ |
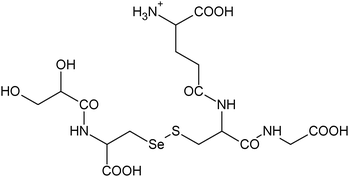
|
18, 19 |
| 585.03762 | 585.03786 (563+Na) | −0.41 | C16H26N4011SSeNa+ | ||||
| 601.01156 | 601.01174 (563+K) | −0.31 | C16H26N4011SSeK+ | ||||
| 20 | 577.07133 | 577.07156 | −0.41 |

|
C17H29N4011SSe+ |
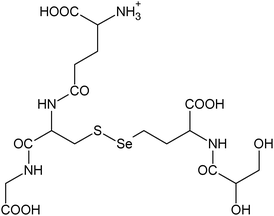
|
18, 19 |
| 21 | 604.08223 | 604.08207 | 0.26 |

|
C18H30N5011SSe+ |
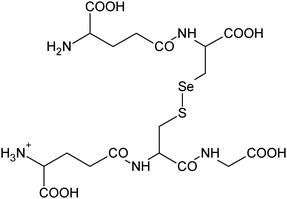
|
18, 19 |
| 22 | 611.00013 | 610.99976 | 0.60 |
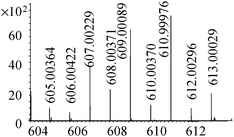
|
C16H27N4011Se2+ |
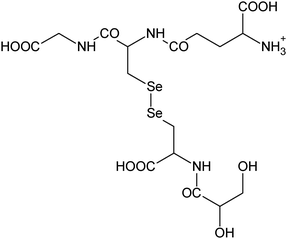
|
18, 19 |
| 632.98207 | 632.98226 (611+Na) | −0.30 | C16H26N4011Se2Na+ | ||||
| 23 | 625.01578 | 625.01546 | 0.50 |
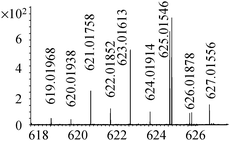
|
C17H29N4011Se2+ |

|
19 |
| 24 | 652.02668 | 652.02650 | 0.27 |

|
C18H30N5011Se2+ |
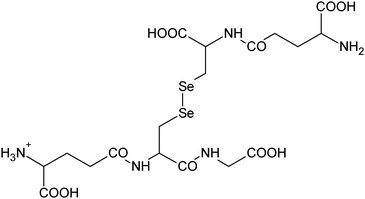
|
19 |
| 25 | 661.10369 | 661.10402 | −0.50 |

|
C20H33N6012SSe+ |
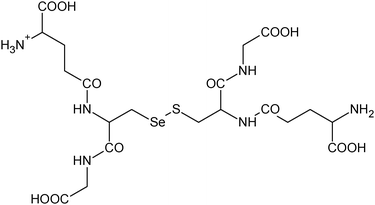
|
25 |
| 683.08563 | 683.08577 (661+Na) | −0.21 | C20H32N6012SSeNa+ | ||||
| 699.05957 | 699.05982 (661-H+K) | −0.36 | C20H32N6012SSeK+ | ||||
| 714.01516 | 714.01538 (661-3H+Fe) | −0.30 | C20H30N6012SSeFe+ | ||||
| 26 | 693.07576 | 693.07553 | 0.33 |
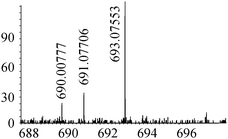
|
C20H33N6012S2Se+ |
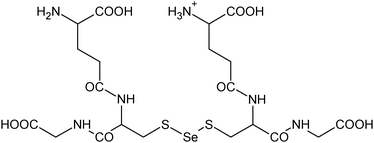
|
27 |
| 27 | 709.04814 | 709.04810 | 0.05 |

|
C20H33N6012Se2+ |

|
18, 19 |
| 731.03009 | 731.02986 (709-H+Na) | 0.32 | C20H32N6012Se2Na+ | ||||
| 747.00402 | 747.00339 (709-H+K) | 0.84 | C20H32N6012Se2K+ |
Fingerprinting of Se-enriched yeast by HPLC with the parallel ICP MS and ESI MS detection
Table 3 summarizes the species detected by ESI MS in the different samples. The samples appear very similar but some compounds are missing in some samples. Note that despite the large intrascan dynamic range the detection in ES MS is affected by other co-eluting compounds which may negatively affect the sensitivity for some species. Therefore, although the electrospray MS spectra are crucial for the interpretation and reporting of the data, the set of HPLC-anion and cation exchange ICP MS chromatograms is indispensable for the yeast samples discrimination.| Compound | A1 | A2 | B | C | D | E | SELM-1 |
|---|---|---|---|---|---|---|---|
| 1 | √ | √ | √ | √ | √ | √ | √ |
| 2 | √ | √ | √ | ||||
| 3 | √ | √ | √ | √ | √ | √ | √ |
| 4 | √ | √ | √ | √ | √ | √ | √ |
| 5 | √ | √ | √ | √ | √ | √ | √ |
| 6 | √ | √ | √ | √ | √ | √ | √ |
| 7 | √ | √ | √ | √ | |||
| 8 | √ | √ | √ | √ | √ | √ | |
| 9 | √ | √ | |||||
| 10 | √ | √ | √ | √ | √ | ||
| 11 | √ | √ | √ | √ | √ | ||
| 12 | √ | √ | √ | √ | √ | √ | |
| 13 | √ | √ | √ | √ | √ | √ | √ |
| 14 | √ | √ | √ | √ | √ | √ | √ |
| 15 | √ | √ | √ | √ | |||
| 16 | √ | √ | √ | √ | √ | ||
| 17 | √ | √ | |||||
| 18 | √ | √ | √ | √ | |||
| 19 | √ | √ | √ | √ | √ | √ | √ |
| 20 | √ | √ | √ | √ | √ | √ | √ |
| 21 | √ | √ | √ | √ | √ | ||
| 22 | √ | √ | √ | √ | √ | √ | √ |
| 23 | √ | √ | √ | ||||
| 24 | √ | √ | √ | ||||
| 25 | √ | √ | √ | √ | √ | √ | √ |
| 26 | √ | √ | |||||
| 27 | √ | √ | √ | √ |
Conclusions
A two dimensional size-exclusion–strong cation-exchange HPLC with the parallel ICP MS and ESI MS detection is an advanced tool for selenometabolomics studies of yeast, and potentially for other samples of plant origin. Information on the identity of the metabolites present can be acquired on-line in the high-throughput mode by ESI MS using an orbital trap. Although a larger number of yeast samples is necessary to be studied and dedicated chemometric tools need to be developed for data analysis, the variations in the morphologies of the chromatograms obtained make the developed method a potentially attractive tool for a fine batch-to-batch control of Se-rich yeast and for the discrimination of the products of the different manufacturers.Acknowledgements
S.G. acknowledges an Àngeles Alvariño post-doctoral fellowship of the Xunta de Galicia. The contribution of the Region of Aquitaine and the FEDER funds via CPER A2E (31486/08011464) project are acknowledged.References
- M. P. Rayman, British Journal of Nutrition, 2008, 100, 254 CAS.
- G. N. Schrauzer and P. F. Surai, Crit. Rev. Biotechnol., 2009, 29, 2 CrossRef CAS.
- M. I. Jackson and G. F. Combs Jr, Curr. Opin. Clin. Nutr. Metab. Care, 2008, 11, 718 CrossRef CAS.
- H. Zeng and G. F. Combs Jr, J. Nutr. Biochem., 2008, 19, 1 CrossRef CAS.
- G. N. Schrauzer, Journal of the American College of Nutrition, 2001, 20, 1 Search PubMed.
- M. P. Rayman, Br. J. Nutr., 2004, 92, 557 CrossRef CAS.
- G. N. Schrauzer, Pure Appl. Chem., 2006, 78, 105 CrossRef CAS.
- H. G. Infante, R. Hearn and T. Catterick, Anal. Bioanal. Chem., 2005, 382, 957 CrossRef.
- R. Lobinski, J. S. Edmonds, K. T. Suzuki and P. C. Uden, Pure Appl. Chem., 2000, 72, 447 CrossRef CAS.
- A. Polatajko, N. Jakubowski and J. Szpunar, J. Anal. At. Spectrom., 2006, 21, 639 RSC.
- J. Ruiz Encinar, M. Sliwka-Kaszynska, A. Polatajko, V. Vacchina and J. Szpunar, Anal. Chim. Acta, 2003, 500, 171 CrossRef CAS.
- J. Far, H. Preudhomme and R. Lobinski, Anal. Chim. Acta, 2010, 657, 175 CrossRef CAS.
- C. Casiot, V. Vacchina, H. Chassaigne, J. Szpunar, M. Potin-Gautier and R. Lobinski, Anal. Commun., 1999, 36, 77 RSC.
- J. F. Garcia-Reyes, M. Dernovics, P. Giusti and R. Lobinski, J. Anal. At. Spectrom., 2006, 21, 655 RSC.
- J. F. Garcia-Reyes, M. Dernovics, P. Ortega-Barrales, A. R. Fernandez-Alba and A. Molina-Diaz, J. Anal. At. Spectrom., 2007, 22, 947 RSC.
- S. McSheehy, J. Szpunar, V. Haldys and J. Tortajada, J. Anal. At. Spectrom., 2002, 17, 507 RSC.
- M. Dernovics, J. Far and R. Lobinski, Metallomics, 2009, 1, 317 RSC.
- M. Dernovics and R. Lobinski, J. Anal. At. Spectrom., 2007, 23, 72 Search PubMed.
- M. Dernovics and R. Lobinski, Anal. Chem., 2008, 80, 3975 CrossRef CAS.
- A. Połatajko, B. Banas, J. Ruiz Encinar and J. Szpunar, Anal. Bioanal. Chem., 2005, 381, 844 CrossRef CAS.
- Z. Mester, S. Willie, L. Yang, R. Sturgeon, J. A. Caruso, M. L. Ferná;ndez, P. Fodor, R. J. Goldschmidt, H. Goenaga-Infante, R. Lobinski, P. Maxwell, S. McSheehy, A. Polatajko, B. B. M. Sadi, A. Sanz-Medel, C. Scriver, J. Szpunar, R. Wahlen and W. Wolf, Anal. Bioanal. Chem., 2006, 385, 168 CrossRef CAS.
- A. Polatajko, M. Sliwka-Kaszynska, M. Dernovics, R. Ruzik, J. R. Encinar and J. Szpunar, J. Anal. At. Spectrom., 2004, 19, 114 RSC.
- M. Dernovics, T. Garcia-Barrera, K. Bierla, H. Preud'homme and R. Lobinski, Analyst, 2007, 132, 439 RSC.
- J. Far, S. Gil Casal, H. Preud'homme and R. Lobinski, submitted.
- H. G. Infante, G. O'Connor, M. Rayman, R. Hearn and K. Cook, J. Anal. At. Spectrom., 2006, 21, 1256 RSC.
- S. McSheehy, P. Pohl, J. Szpunar, M. Potin-Gautier and R. Lobinski, J. Anal. At. Spectrom., 2001, 16, 68 RSC.
- T. Lindemann and H. Hintelmann, Anal. Chem., 2002, 74, 4602 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |