Quantification of Phytochelatins in Chlamydomonas reinhardtii using ferrocene-based derivatization†
Anja
Bräutigam
a,
Susanne
Bomke
b,
Thorben
Pfeifer
b,
Uwe
Karst
b,
Gerd-Joachim
Krauss
a and
Dirk
Wesenberg
*a
aMartin-Luther-Universität Halle-Wittenberg, Institut für Biochemie und Biotechnologie, Ökologische und Pflanzen-Biochemie, Kurt-Mothes-Straße 3, 06120 Halle (Saale), Germany. E-mail: dirk.wesenberg@biochemtech.uni-halle.de; Fax: +49 3455527012
bWestfälische Wilhelms-Universität Münster, Institut für Anorganische und Analytische Chemie, Corrensstrasse 30, 48149, Münster, Germany
First published on 24th June 2010
Abstract
A method for the identification and quantification of canonic and isoforms of phytochelatins (PCs) from Chlamydomonas reinhardtii was developed. After disulfide reduction with tris(2-carboxyethyl)phosphine (TCEP) PCs were derivatized with ferrocenecarboxylic acid (2-maleimidoyl)ethylamide (FMEA) in order to avoid oxidation of the free thiol functions during analysis. Liquid chromatography (LC) coupled to electrospray mass spectrometry (ESI-MS) and inductively coupled plasma-mass spectrometry (ICP-MS) was used for rapid and quantitative analysis of the precolumn derivatized PCs. PC2−4, CysGSH, CysPC2−4, CysPC2desGly, CysPC2Glu and CysPC2Ala were determined in the algal samples depending on the exposure of the cells to cadmium ions.
Introduction
The green alga Chlamydomonas reinhardtii synthesizes phytochelatins (PCs) after cadmium exposure.1 PCs are peptides with the general structure (GluCys)nGly,2,3 which are synthesized from glutathione by the constitutive expressed enzyme phytochelatin synthase. Next to canonic PCs, the isoforms CysPCn (Cys(GluCys)nGly) and PCnAla ((GluCys)nAla) can be detected as well in Chlamydomonas reinhardtii. For physiological studies, identification and quantification of these peptides is essential.Not only for their biological function, but also for the analysis of PCs, the thiol function plays the key role and is of major interest. Due to their easy oxidation to disulfides, real samples will always contain a mixture of reduced and oxidized forms of PCs. Therefore, a reduction of the disulfides is required prior to their analysis. The sulfur content of PCs allows their quantification by element selective methods such as inductively coupled plasma-mass spectrometry (ICP-MS). As an example, Bluemlein et al. developed a liquid chromatography (LC)/ICP-MS procedure to quantify sulfur in arseno-PC-complexes.4 The detection of sulfur by ICP-MS is hampered by two major obstacles, the high first ionization potential of sulfur leading to an unfavorable ionization yield of this comparably electronegative element and interferences of 32S+ with 16O16O+ ions. Improved limits of detection and—depending on the particular derivatization technique—products, which are more stable towards oxidation may be obtained using one of the numerous derivatizing agents available for sulfhydryl groups.5 Thus, fluorophores like fluorescein (e.g. iodoacetamidofluorescein6 (IAF)), monobromobimane (MBB)7, benzofuranes (e.g. ammonium-7-fluorobenz-2-oxa-1,3-diazole-4-sulfonate8 (SBD-F)), ortho-phthalaldehyde9 (OPA) or chromophoric systems as Ellman’s reagent (5,5′-dithiobis-(2-nitrobenzoic acid)10,11 (DTNB)) are used for this purpose. If derivatizing agents with low polarity are used, an advantage of pre-column derivatization is the increased retention and therefore the improved separation of derivatized PCs on reversed-phase columns in comparison to the native, highly polar PCs. Moreover, pre-column derivatization prevents oxidation of thiol groups to disulfides during analysis. Selectivity of the applied reagents is another aspect to be considered, as some reagents as OPA react with primary amines as well, while others are highly selective towards thiols. Finally, phytochelatins as polythiols with at least two thiol functionalities are difficult to derivatize quantitatively, as bulky derivatizing agents will experience significant steric hindrance.10
The chemistry of ferrocene-based derivatizing agents is well established. Ferrocenes are iron(II) complexes of low polarity, which offer an extremely broad bandwidth with respect to available functional groups.12,13 After derivatization with a ferrocene-based reagent, the polarity of the derivatized analytes will be reduced compared to the native compounds, allowing simple reversed-phase high performance liquid chromatographic separation (RP-HPLC). Maleimides react selectively towards thiols at neutral pH to form the corresponding thioether bond. Ferrocenecarboxylic acid (2-maleimidoyl)ethylamide (FMEA) was originally developed by Seiwert and Karst to quantify oxidized thiols simultaneously with reduced ones in a two-step derivatization procedure. This consists of a derivatization of free thiols with a first reagent, removal of the reagent excess, reduction of the disulfide group and derivatization of the generated thiols with the second reagent.14 The derivatizing agent FMEA contains a N-substituted maleimide group, which reacts selectively towards thiol functionalities. Moreover, the Fe atom is applicable to ICP-MS for quantification, although ferrocene derivatives suffer from the inherent drawback of only moderate limits of detection in unit resolution ICP-MS due to the formation of isobaric [40ArO]+ ions (m/z 56 cannot be discriminated from the respective 56Fe isotope at low resolution). However, this problem can be overcome by either high resolution MS or the use of reaction/collision cell technology.15,16 Seiwert et al. also showed that even cysteines in sterically demanding positions are derivatized quantitatively using FMEA under appropriate reaction conditions.17 Therefore, FMEA derivatization appears to be a promising tool for the modification of thiol-rich PCs.
Aim of this work was the development of a FMEA derivatization procedure for PC identification and quantification in C. reinhardtii. For this purpose, pre-column derivatization with FMEA and subsequent liquid chromatography with electrospray mass spectrometry (LC/ESI-MS) was used for quantification of the resulting PCn-(FMEA)n-adducts and to verify PC isoforms, which were only suspected to exist according to earlier work by Bräutigam et al.1
Experimental
Chemicals
Cysteine (99%), formic acid (98%) and GSH (reduced, 98%) were purchased from Acros Organics (Geel, Belgium). Tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 99%) was ordered from Sigma Aldrich (Steinheim, Germany), ammonium bicarbonate from Fluka Chemie GmbH (Buchs, Switzerland). Acetonitrile (ACN) for HPLC was obtained in gradient-grade quality from Merck KGaA (Darmstadt, Germany). Water for HPLC was purified using a Milli-Q Gradient A 10 system and was filtered through a 0.22 μm Millipak 40 filter unit (Millipore, Billerica, MA, USA). FMEA was synthesized from ferrocenecarboxylic acid chloride with N-(2-aminoethyl) maleimide according to a method described earlier.14 Hydrochloric acid was obtained from KMF Laborchemie Handels GmbH (Lohmar, Germany). Phytochelatins were purchased from GeneCust (Dudelange, Luxemburg) with >98% purity.Instrumentation
LC/ESI-MS
The LC/ESI-MS setup comprised a Shimadzu (Duisburg, Germany) LC system. The LC system consisted of two LC-10ADVP pumps, a DGC-14A degasser, a SIL-HTA autosampler, a CTO-10AVP column oven and a SPD-10AVVP UV/vis detector. A QTRAP mass spectrometer (Applied Biosystems, Darmstadt, Germany), equipped with a Turbo IonSpray (pneumatically assisted ESI source) source was used for detection in the positive ion mode with an integration time of 100 ms per analyte. The analytes were ionized with an ionspray voltage of 5000 V, using 20 psi nebulizer gas and 50 psi dry gas with a temperature of 450 °C. The curtain gas was set at 25 psi, declustering potential at 50 V and entrance potential at 8 V.Separation of derivatized phytochelatins was carried out using a Discovery® C8 (Supelco, Taufkirchen, Germany) column with the following dimensions: 150 mm length x 2.1 mm i.d., 5 μm particle size 180 Å. The column was operated at ambient temperature. The flow rate of the mobile phase was 0.3 mL min−1. For all separations, eluent A of the mobile phase was 0.1% formic acid in 1 L of deionized water (pH 2.4). Eluent B was acetonitrile. The injection volume was 10 μL. The derivatives were eluted with the following gradient profile: Start with 5% B, immediately followed by a 60 min gradient to 90% B, 90% B were held for 3 min. The column was reequilibrated to initial conditions with a 1 min linear gradient to 5% B and an isocratic period at 5% B of 6 min. Additional data were acquired by UV-detection at 254 nm.
ESI-ToF-MS
ESI-ToF-MS measurements were performed on a Bruker MicrOToF mass spectrometer (Bruker Daltonics, Bremen, Germany), which was controlled by MicrOTOFcontrol v.1.1. Peaks were integrated using the DataAnalysis software v.3.3 (Bruker Daltonics).Full-scan spectra (m/z 300–2000) were recorded after HPLC separation using the ESI(+) mode under the following conditions: end plate offset, −400 V; capillary, 4200 V; nebulizer gas (N2), 0.8 bar; drying gas (N2), 6.0 mL min−1; drying temperature 200 °C; capillary exit, 180 V; skimmer 1, 60.0 V; skimmer 2, 26.5 V; hexapole 1, 23.0 V; hexapole 2, 21.4 V; hexapole rf, 350 V; transfer time, 75.0 μs; prepulse storage, 22.0 μs; detector, −1000 V. Internal calibration was performed by using sodium formate clusters at the beginning of each HPLC run.
ICP-MS
ICP-MS data were recorded using an Agilent 7500ce instrument equipped with an octopole reaction system (ORS) (Agilent Technologies Inc., Santa Clara, United States). All gases were used according to the requirements of the manufacturer. The determination of the nebulizer gas flow rate and the ion lens voltage was determined applying a daily standard optimization procedure. The detector was operated in both pulse count and analog mode (dual mode). Data treatment and integration was done with OriginPro 8G. The ICP-MS conditions were optimized and are compiled in Table 1.| Sample introduction and plasma | |
| Nebulizer | MicroFlow PFA-ST |
| Spray Chamber | SCOTT Type, double pass |
| Torch | Fassel |
| Sampler/skimmer cones | Platin, 1.0 mm ID (sampler) |
| 0.6 mm ID (skimmer) | |
| RF Power | 1500 W |
| Plasma gas flow rate | 15.00 L min−1 |
| Auxiliary gas flow rate | 0.90 L min−1 |
| Makeup gas flow rate | 0.10 L min−1 |
| Carrier Gas | 0.65 L min−1 |
| O2 flow rate | 50 mL min−1 |
| Data acquisition | |
| Acquisition mode | Spectrum (time resolved) |
| Acquired masses | 56 (Fe), 111 (Cd) |
| Integration time | 0.2 s/points |
| Number of points per mass | 1 |
| Reaction cell gases and run lens voltages | |
| Cell gas flow rate (H2) | 2.3 mL min−1 |
| QP Focus | −9 V |
| Octopole Bias | −18 V |
| Quadruple Bias | −15 V |
For LC/ESI-ToF-MS and LC/ICP-MS, an Agilent Technologies (Waldbronn, Germany) HP1200 liquid chromatograph consisting of a binary gradient pump model G1312A and an autosampler model G1313A was used.
ICP-OES
For ICP-optical emission spectroscopy (OES) measurements, a Spectro CIROSCCD ICP optical emission spectrometer (Spectro Analytical Instruments, Kleve, Germany) with axial plasma viewing was used. All ICP operating parameters such as gas flows and positioning of the discharge container in front of the optical interface were adjusted by the Smart Analyzer CIROSCCD software (version 3.2, Spectro).Algae culture conditions
Chlamydomonas reinhardtii DANGEARD (wt −11/32B) was maintained mixotrophically in 50 mL tris acetate phosphate medium (TAP)18 in 100 mL Erlenmeyer flasks at 25 °C under long day conditions (16 h, 40 μE, 120 rpm). For experiments, the medium was inoculated with 1.5 mL of a three days old culture (optical density at 730 nm (OD730nm) 1.3). After 24 h, CdCl2 was added to a final concentration of 70 μM. Samples were taken 1, 4, 24 and 48 h after cadmium addition. All glassware was rinsed with 1% HNO3 to remove metal residues before use. The OD730nm was measured for growth mapping. To check the vitality of the cells, the chlorophyll content was determined photometrically at 652 nm in 80% acetone.19FMEA derivatisation of phytochelatins
Prior to FMEA derivatization, 100 mg of biomass were suspended in 300 μL 0.1 N HCl, containing 10 mM Na2EDTA. The extract was centrifuged (15 min, 1500 g) and the supernatant filtrated through a Whatman filter (Spartan, diameter 13 mm, pore diameter 0.2 μm). A volume of 50 μL of the flow through was incubated with NH4HCO3 (150 μL, 100 mM) and tris(2-carboxyethyl)phosphine hydrochloride (TCEP, 50 μL, 100 mM in 100 mM NH4HCO3) at room temperature (RT, 1 h) in order to reduce the disulfides in the sample to thiols. Afterwards, 150 μL (FMEA, 20 mM in acetonitrile) were added. After 5 min reaction time, 5 μL HCOOH were added.Quantification of phytochelatins
For external calibration, cysteine, glutathione, PC2, PC3, CysPC2 and CysPC3 were used as standards. A 1 mM stock solution of each analyte was prepared using 10 mM Na2-EDTA in 0.1 HCl. The diluted solutions (0.5, 0.2, 0.1, 0.075, 0.05 and 0.025 mM) were derivatized according to the procedure described above.Results and discussion
C. reinhardtii physiological parameters
C. reinhardtii was stressed with 70 μM CdCl2, which is the highest Cd concentration found in soil solution.20 During the experiment, no significant change in optical density or chlorophyll content was observed. OD730nm varied between 101% and 106% and chlorophyll content from 85% to 104% of the control level. Thus, it is demonstrated that the Cd-treated cells were vital.Derivatization procedure
For PC derivatization, an extraction procedure had to be developed. For PC extraction of Cd treated samples, usually an acidic solvent is applied to not only precipitate proteins, but also to demetallate thiol groups. The demetallation is necessary for further derivatization. Therefore, a solution of 10 mM Na2EDTA in 0.1 N HCl was added. The signal intensity of derivatized PCs in ICP-MS was 60% lower if only 0.1 N HCl without Na2EDTA was used for extraction. As a second step prior to derivatization, the quantitative reduction of disulfide bridges had to be accomplished. Therefore, TCEP, as the most effective agent, was used.1 After the reduction, no inter- or intramolecular oxidation products could be observed by ESI-MS. Hence, the free SH groups were accessible for FMEA derivatization. The derivatizing agent FMEA contains a maleimide ring as reactive group. With their activated double bond, N-substituted maleimides undergo a Michael-type electrophilic addition reaction by forming a stable thioether bond with sulfhydryl groups. The reaction scheme is shown in Fig. 1. The reaction preferentially takes place at neutral pH, at which the thiolate anion is formed. At a pH of 7, other functional groups, in particular amino or hydroxyl groups do not react with maleimide rings.21 The final FMEA concentration of 11.3 mM was higher than in earlier work of Seiwert et al.12, because 4 mM were not sufficient for derivatization of all PCs. The employed concentration of FMEA was adequate for quantitative derivatization, incomplete derivatized PCs were only of low abundance. After a reaction time of five minutes, formic acid was added to decrease the pH value to acidic conditions to ensure the stability of the derivatized PCs. Furthermore, under alkaline conditions, a hydrolytic cleavage of the maleimide ring (N1–C2 or N1–C5) may take place, which may lead to the formation of a succinamic acid derivative.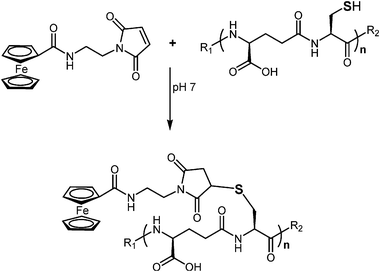 | ||
| Fig. 1 Reaction scheme of PC and FMEA, n = 2–4, R1 = H, Cys, R2 = OH, Glu, Ala. | ||
Identification of FMEA derivatised PCs
The hyphenation of LC/ESI-MS was used for identification of PC-FMEA compounds. This technique offers not only the possibility to prove whether the derivatization is complete, moreover, it is also a prerequisite for later peak identification in LC/ICP-MS.The derivatized PCs were separated on a reversed-phase HPLC column. The spectra were recorded in the full-scan mode in the range of m/z 300–1500. Further measurements were recorded in the selected ion monitoring (SIM) mode, which provides higher selectivity and, therefore, lower limits of detection (LOD). The separation of several derivatized PCs, which could be identified in a 48 h Cd treated C. reinhardtii sample, is shown in Fig. 2. Next to canonic PC2–4 also CysGSH, CysPC2–4 and PC2-Ala could be identified, which is in accordance to earlier work by Bräutigam et al.1 PC5 and CysPC5 were not detected in this work, possibly due to only moderate sample preconcentration.
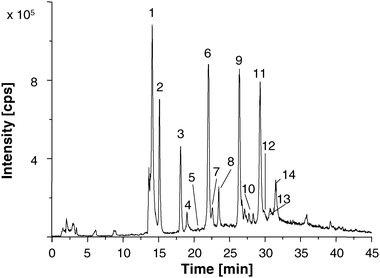 | ||
| Fig. 2 Extracted ion LC/ESI-MS chromatogram recorded in the SIM mode of an 48 h Cd sample: 1: Cysteine-FMEA, 2: GSH-FMEA, 3: CysPC2Glu-(FMEA)3/PC3desGly-(FMEA)3, 4: CysPC2desGly-(FMEA)3, 5: CysGSHGlu-(FMEA)2/PC2desGly-(FMEA)2, 6: CysGSH-(FMEA)2, 7: CysGSHdesGly-(FMEA)2, 8: PC2-(FMEA)2, 9: CysPC2-(FMEA)2, 10: PC3-(FMEA)4, 11: CysPC3-(FMEA)2, 12: PC2Ala-(FMEA)2, 13: PC4-(FMEA)4, 14: CysPC4-(FMEA). Monitored ions are listed in Table 2. | ||
PC2–3desGly, CysGSHGlu and CysPC2–3Glu were identified in the ESI-MS spectra as well. The difficulty in identifying these PCs is the mass similarity of PCndesGly and CysPCn−1Glu. These isoforms can only be distinguished via ESI-MS/MS experiments. For a better overview, Table 2 shows all PCn-(FMEA)n ions, which were identified in a C. reinhardtii sample treated with Cd for 48 h. Unlabelled peaks in Fig. 2, result from the use of algal raw extracts for derivatization and could not be identified.
| Derivatized thiol | m/z measured | m/z calculated | Number of FMEA-residues | Charge |
|---|---|---|---|---|
| Cysteine | 474.2 | 473.2 | 1 | 1 |
| GSH | 660.5 | 659.1 | 1 | 1 |
| CysPC2Glu/PC3desGly | 886.7 | 885.6 | 3 | 2 |
| CysPC2desGly | 821.6 | 821.1 | 3 | 2 |
| CysGSHGlu/ PC2desGly | 1187.5 | 1186.1 | 2 | 1 |
| CysGSH | 1115.5 | 1114.1 | 2 | 1 |
| CysGSHdesGly | 1058.8 | 1057.1 | 2 | 1 |
| PC2 | 1244.8 | 1243.1 | 2 | 1 |
| CysPC2 | 849.6 | 849.6 | 3 | 2 |
| PC3 | 915.6 | 914.1 | 3 | 2 |
| CysPC3 | 1143.1 | 1141.6 | 4 | 2 |
| PC2Ala | 1258.7 | 1257.6 | 3 | 2 |
| PC4 | 1207.7 | 1206.1 | 4 | 2 |
| CysPC4 | 1434.8 | 1433.6 | 5 | 2 |
An additional complexity factor results from FMEA derivatization. Due to the oxidation of ferrocene (Fe2+) to the corresponding ferrocinium cation (Fe3+) in the ESI interface, every PC-FMEA can form two singly charged ions, namely [M]+ additional to [M+H]+, which have a m/z difference of 1. PCs, derivatised with nFMEA molecules, can be oxidised n times. For example, the resulting three different doubly charged ions for n ≥ 2 are [M]2+, [M+H]2+ and [M+2H]2+, differing in only 0.5 m/z units. In the case of the derivatized phytochelatins CysPC2Glu-(FMEA)3 and PC3desGly-(FMEA)3 (peak 3 in Fig. 2) and PC2desGly-(FMEA)2 and CysGSHGlu-(FMEA)2 (peak 5 in Fig. 2), the analytes cannot be distinguished by their mass to charge ratios. An unambiguous identification can only be performed with the help of ESI-MS/MS. However, the isoforms CysGSHdesGly as well as CysPC2desGly were allocated clearly.
In summary, the LC/ESI-MS coupling enabled the confirmation of PC isoforms, which cannot be distinguished by ESI-MS alone, because collision induced fragmentation may result in misleading data.1 For further unambiguous identification of the PCn-FMEAn complexes, high resolution ESI/ToF-MS was applied, which could, in most cases, confirm the identification of the analytes due to small mass deviations of calculated and detected m/z values (Table S-1, ESI†).
LC/ICP-MS of PC standards
The FMEA labelled PCs shall be quantified via the Fe56-trace using the hyphenation of LC/ICP-MS. Unfortunately, no constant Fe signal is observed in ICP-MS caused by the gradient elution in LC (Fig. S-1, ESI†). However, with an isocratic elution, no acceptable baseline separation of the derivatized PCs could be achieved. Thus, calculation of PC concentration was not possible by species independent Fe determination and had to be performed with the help of standard compounds.First, the standards Cys, GSH, PC2, CysPC2, PC3, and CysPC3 were derivatized and analyzed via LC/ICP-MS. The observed chromatogram is shown in Fig. 3. A retention time shift of approximately +1 min in comparison to the LC/ESI-MS system was observable due to different volumes of the mixing chamber and length of the used capillaries of the two LC systems. Cys-FMEA eluted at the same retention time as the TCEP-FMEA adduct (14.6 min) and GSH-FMEA (16.0 min) shortly thereafter. Another unknown Fe-containing substance overlapped with PC3. Because of this limited chromatographic resolution, only PC2, CysPC2, and CysPC3 were left for further quantification. The other peaks observed in the chromatogram could not be assigned. Calibration was carried out in a range of 0.025–0.2 mM. The linearity of calibration shows the applicability of FMEA derivatization for PC quantification. Instrumental detection limits were estimated according to the 3σ criterion for CysPC2 and PC2 as 0.69 μM and 0.59 μM respectively.
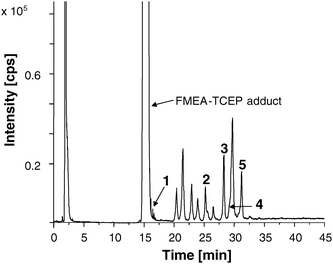 | ||
| Fig. 3 LC/ICP-MS chromatogram showing the 56Fe-trace of a standard mix with 1: GSH-FMEA, 2: PC2-(FMEA)2, 3: CysPC2-(FMEA)3 and 4: PC3-(FMEA)3, 5: CysPC3-(FMEA)4. The concentration for each standard is 0.125 mM. The other peaks could not be assigned. | ||
LC/ICP-MS of C. reinhardtii samples
To test the application of the method in biological samples, Cd exposed C. reinhardtii cells were used for further investigations. A LC/ICP-MS chromatogram showing the 56Fe-trace of a real sample after Cd exposure for 48 h is presented in Fig. 4. Cd was complexed via EDTA and could be detected by ICP-MS only within the void volume of the chromatographic run. The Cd-EDTA complex could not be detected in ESI-MS, possibly due to the unsatisfactory ionization efficiency in the ESI(+) mode. Neither in blanks nor in standards or control cultures, could Cd be detected.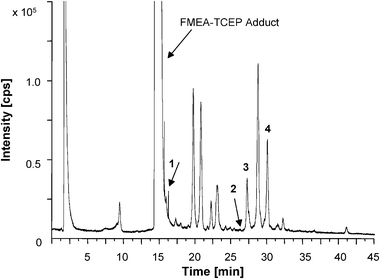 | ||
| Fig. 4 LC/ICP-MS chromatogram showing the 56Fe-trace of a real sample (exposition with Cd for 48 h) with 1: GSH-FMEA, 2: PC2-(FMEA)2, 3: CysPC2-(FMEA)3 and 4: CysPC3-(FMEA)4. | ||
56Fe-containing substances even elute within the void volume (1.8 min). In comparison to the measurement of blank and standard samples, the intensity of the 56Fe peak in the void volume of the algae samples was approximately two times higher. This is probably due to metal ion concentrations in the TAP medium, which was used as culture medium during sample preparation. With the help of ICP-OES measurements, the concentration of several metals in the culture medium was investigated (Table S-2, ESI†). Thereby, the culture medium contains 264.4 ppb Fe as micronutrient, which could explain the high intensity of the 56Fe-peak in the form of the 56Fe-EDTA complex at the beginning of the chromatogram.
The quantification results for PC2, CysPC2 and CysPC3 in real samples are summarized in Table 3. Several algae samples of C. reinhardtii were treated with Cd from 1 to 48 h and analyzed via LC/ICP-MS.
| 1 h | 4 h | 24 h | 48 h | ||
|---|---|---|---|---|---|
| PC2 | 70 μM Cd/nmol g−1 FW | 26.3 ± 1.4 | 24.8 ± 1.7 | 13.4 ± 1.2 | 12.5 ± 2.0 |
| control/nmol g−1 FW | 0.9 | n.d. | 2.0 ± 2.9 | n.d. | |
| CysPC2 | 70 μM Cd/nmol g−1 FW | n.d. | 73.8 ± 2.3 | 151.1 ± 6.1 | 189.3 ± 22.6 |
| control/nmol g−1 FW | n.d. | n.d. | 100.6 ± 51.1 | 63.8 ± 0.4 | |
| CysPC3 | 70 μM Cd/nmol g−1 FW | 73.0 ± 2.1 | 72.2 ± 6.8 | 138.1±24.6 | 251.7 ± 44.0 |
| control/nmol g−1 FW | 70.7 | 64.3 | 88.6 ± 55.1 | 58.7 ± 50.9 |
In the control cultures the content of analyzed PCs was lower than in the Cd-treated ones. The PC synthesis in control samples can be explained by the used TAP culture medium, containing essential metals (Table S-2, ESI†), which can activate the enzyme phytochelatin synthase as well.
The large standard deviation (SD) may be traced back to the biological deviation, because only biologically independent algae cultures were investigated. The analysis of the same algae samples in triplicate led to RSDs of the peak area of less than 2%. CysPC2, whose m/z could not be detected in ESI-MS of the control cultures, possibly due to signal suppression, was detectable in ICP-MS.
The CysPC3-(FMEA)3 peak showed peak fronting in both the control and the cadmium exposed cultures, which made the integration difficult and may explain the relatively high concentrations in control and Cd-treated samples. Most likely, another unidentified thiol-containing substance is synthesized by C. reinhardtii, which coelutes with CysPC3-(FMEA)3 and disturbs the quantification.
PC2 and CysPC2 showed distinct peaks. The concentration of PC2 could already be analyzed after 1 h Cd incubation and showed a decrease up to 50% till 48 h. CysPC2 contents of Cd-exposed cultures increased during Cd exposition. CysPC2 is concentrated higher than PC2, which is in good agreement with the earlier estimation.1
Conclusions
This work describes the general suitability of ferrocene derivatization in qualitative as well as quantitative phytochelatin analysis. The use of LC/ESI-MS enabled the identification of canonic PC2–4, CysGSH, CysGSH, CysPC2–4, CysPC2desGly, CysPCGlu and CysPCAla in Cd exposed C. reinhardtii cells. Thus, among others, PC3desGly was confirmed, which was only proposed before.1 Other phytochelatins like PC5 could not be detected, probably due to lacking preconcentration. For quantification, the hyphenation of LC/ICP-MS was exploited. Because a baseline separation of the derivatized PC could only be achieved by gradient elution, a species independent Fe determination by LC/ICP-MS was not possible. Thus, only PC2, CysPC2 and CyPC3 were unambiguously identified due to the availability of standards. However, isocratic elution might offer improved possibilities for quantification without PC standards in the future.Acknowledgements
Financial support by the Fonds der Chemischen Industrie (Frankfurt/Main, Germany) is gratefully acknowledged.References
- A. Bräutigam, D. Schaumlöffel, G. J. Krauss and D. Wesenberg, Anal. Bioanal. Chem., 2009, 395, 1737–1747 CrossRef.
- W. H. O. Ernst, G.-J. Krauss, J. A. C. Verkleij and D. Wesenberg, Plant Cell Environ., 2008, 31, 123–143 CAS.
- E. Grill, E. L. Winnacker and M. H. Zenk, Science, 1985, 230, 674–676 CrossRef CAS.
- K. Bluemlein, E. M. Krupp and J. Feldmann, J. Anal. At. Spectrom., 2009, 24, 108–113 RSC.
- K. Shimada and K. Mitamura, J. Chromatogr., B: Biomed. Sci. Appl., 1994, 659, 227–241 CrossRef CAS.
- E. Caussé, C. Issac, P. Malatray, C. Bayle, P. Valdiguie, R. Salvayre and F. Couderc, J. Chromatogr., A, 2000, 895, 173–178 CrossRef CAS.
- G. L. Newton and R. C. Fahey, Methods Enzymol., 1995, 251, 148–166 CAS.
- T. Santa, C. Aoyama, T. Fukushima, K. Imai and T. Funatsu, Biomed. Chromatogr., 2006, 20, 656–661 CrossRef CAS.
- I. Molnar-Perl, J. Chromatogr., A, 2001, 913, 283–302 CrossRef CAS.
- F. E. C. Sneller, L. M. v. Heerwaarden, P. L. M. Koevoets, R. Vooijs, H. Schat and J. A. C. Verkleij, J. Agric. Food Chem., 2000, 48, 4014–4019 CrossRef CAS.
- M. Berlich, S. Menge, I. Bruns, J. Schmidt, B. Schneider and G.-J. Krauss, Analyst, 2002, 127, 333–336 RSC.
- B. Seiwert and U. Karst, Anal. Bioanal. Chem., 2008, 390, 181–200 CrossRef CAS.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5985 CrossRef CAS.
- B. Seiwert and U. Karst, Anal. Chem., 2007, 79, 7131–7138 CrossRef CAS.
- C. Ingle, N. Langford, L. Harvey, J. R. Dainty, C. Armah, S. Fairweather-Tait, B. Sharp, H. Crews, M. Rose and J. Lewis, J. Anal. At. Spectrom., 2002, 17, 1498–1501 RSC.
- K. J. R. Rosman and P. D. P. Taylor, J. Anal. Atom. Spectrom., 1999, 14, 5n–24n Search PubMed.
- B. Seiwert, H. Hayen and U. Karst, J. Am. Soc. Mass Spectrom., 2008, 19, 1–7 CrossRef CAS.
- E. H. Harris, The Chlamydomonas Sourcebook: A Comprehensive Guide to Biology and Laboratory Use, Academic Press, San Diego, 1989 Search PubMed.
- D. I. Arnon, Plant Physiol., 1949, 24, 1–4 CrossRef CAS.
- T. M. Lee, H. Y. Lai and Z. S. Chen, Chemosphere, 2004, 57, 1459–1471 CrossRef CAS.
- D. G. Smyth, A. Nagamatsu and J. S. Fruton, J. Am. Chem. Soc., 1960, 82, 4600–4604 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: ICP-MS signal of 56Fe, LC gradient course, measured and calculated exact masses of phytochelatins and concentrations of Fe, Cu and Zn in the TAP culture medium determined by ICP-OES. See DOI: 10.1039/c005014h |
| This journal is © The Royal Society of Chemistry 2010 |