Synthesis, characterization and biological evaluation of In(III) complexes anchored by DOTA-like chelators bearing a quinazoline moiety†
Raquel
Garcia
a,
Vojtěch
Kubíček
b,
Bohuslav
Drahoš
b,
Lurdes
Gano
a,
Isabel C.
Santos
a,
Paula
Campello
a,
António
Paulo
a,
Éva
Tóth
b and
Isabel
Santos
*a
aUnidade de Ciências Químicas e Radiofarmacêuticas, Instituto Tecnológico e Nuclear, Estrada Nacional 10, Apartado 21, 2686-953 Sacavém, Portugal
bCentre de Biophysique Moléculaire CNRS, Rue Charles Sadron 45071 Orléans Cedex 2, France
First published on 1st July 2010
Abstract
Following previous studies with a DOTA-like bifunctional chelator (H3L1) containing an ethylenic linker between the macrocycle backbone and a quinazoline pharmacophore, we synthesized and fully characterized a congener macrocyclic ligand (H3L2) having a longer, five-carbon spacer for the linkage of the quinazoline moiety. Both H3L1 and H3L2 were used to prepare indium(III) complexes aiming at their evaluation as radioactive probes for in vivo targeting of EGFR-TK. The protonation constants (log KHi) of H3L2 were determined by potentiometry and UV-Vis spectrophotometry and the values found are 12.18, 9.74, 4.99, 3.91 and 2.53. The stability and protonation constants of InL (L = L1, L2) were also obtained from a combined potentiometry and UV-VIS spectrophotometry study. The reaction of InCl3 with H3L1 and H3L2 led to the formation of the well-defined complexes InL1 and InL2, containing In(III) ions coordinated by a seven (N4,O3) donor atom set. These new complexes were fully characterized by spectroscopic methods (IR, NMR, ESI-MS), HPLC and by X-ray diffraction analysis in the case of InL1. The radioactive congener 111InL2 was prepared from the reaction of 111In–chloride with H3L2, in high yield and high radiochemical purity. 111InL2 is a neutral complex that presents a hydrophilic character and exhibits a high in vitro and in vivo stability. H3L2 and InL2 do not inhibit the cell growth of A431 cervical carcinoma cells. In this EGFR-expressing cell line, 111InL2 has shown very low cell internalization. These findings indicate that these DOTA-like chelators are not the best suited bifunctional ligands to obtain In(III) complexes with adequate biological properties for targeting the EGFR-TK.
1. Introduction
Overexpression of the epidermal growth factor receptor tyrosine kinase (EGFR-TK) occurs in several human cancers of epithelial origin, being correlated with resistance to treatment and poor prognosis.1 The EGFR belongs to the human epidermal receptor (HER) family, which consists of four closely related receptors: EGFR (HER1, erbB1), HER2 (erbB2), HER3 (erbB3) and HER4 (erbB4). The role that ErbB receptors and their ligands play in proliferation, migration and cell survival has made them an attractive target in the development of cancer therapies.2 Several approaches have been developed to target EGFR and to interfere with EGFR-mediated cellular effects. Two of these approaches are based on monoclonal antibodies that competitively inhibit ligand binding at the external domain of EGFR and on small-molecule tyrosine kinase inhibitors which exert their effects at the intracellular portion of the receptor, preventing tyrosine kinase phosphorylation and subsequent activation of signal transduction pathways.3 Recently, erlotinib (Tarceva®) and gefitinib (Iressa®), which selectively and reversibly inhibit phosphorylation of the EGFR tyrosine kinase, have received approval for treatment of Non Small Cell Lung Carcinoma (NSCLC). These agents are anilinoquinazoline derivatives that exert their antitumor activity through inhibition of proliferation and tumor angiogenesis and through induction of apoptosis.4The finding of radioactive probes for in vivo imaging of EGFR expression should allow the selection of patients to anti-EGFR therapeutic approaches. In recent years, radiolabelled quinazoline derivatives emerged as a promising class of radiotracers for molecular imaging of EGFR overexpressing tumors by Positron Emission Tomography (PET).1 Since tyrosine kinase inhibitors are small molecules, they were expected to be labeled with PET radionuclides (e.g.11C, 18F and 124I) without significantly affecting EGFR binding affinity.5–16 In contrast, the development of Single Photon Emission Computed Tomography (SPECT) imaging probes for EGFR-TK imaging is a more demanding task. The most useful SPECT radionuclides are isotopes from metallic elements and, therefore, it is necessary the design of bifunctional chelating agents that support sophisticated receptor targeting, while still being able to stabilize the radiometal. The choice of the appropriate bifunctional chelate is radiometal-dependent, as the coordination requirements of the metal, such as denticity and donor atoms, must be considered to achieve a kinetically inert and thermodynamically stable complex.
Recently, our research group has been studying and evaluating in vitro and in vivo125I-, 99mTc- and 67Ga-labelled quinazoline derivatives as radioactive probes for a possible detection and staging of EGFR-positive tumors.17–19 In the case of 67Ga, we have evaluated the complex 67GaL1, anchored on the DOTA-like chelator 1,4,7,10-tetraazacyclododecane-1-{4-[(3-chloro-4-fluorophenyl)amino]quinazoline-6-yl}propionamide-4,7,10-triacetic acid (H3L1) containing an ethylenic linker between the macrocycle backbone and the quinazoline pharmacophore. The cold congener, GaL1, was unable to inhibit the cell growth of EGFR-expressing A431 cervical carcinoma cell line, in agreement with the negligible cell internalisation found for 67GaL1. These findings could be due to the highly hydrophilic character (log D = −1.02 ± 0.03) of 67GaL1, which justifies its poor ability to cross the cellular membrane by passive diffusion and reach the putative intracellular target. In part, the high hydrophilicity of GaL1 is a consequence of the presence of a non-coordinated and ionizable carboxylate pendant arm, as Ga(III) complexes with DOTA-like chelators are usually hexacoordinated by the four nitrogen atoms of the macrocycle backbone and by two oxygen atoms of the carboxylate pendant arms. We hypothesized that the use of indium instead of gallium should lead to more lipophilic complexes with DOTA-like chelators bearing a quinazoline-containing pendant arm, because all the three non-functionalised carboxylate pendant arms should coordinate the In(III) ion, forming a heptacoordinated complex.20
Searching for more lipophilic complexes with an enhanced ability to enter the cells and best suited to design radioactive probes for the targeting of EGFR-TK, we have studied the possibility of preparing In(III) complexes with the novel macrocyclic ligand 1,4,7,10-tetraazacyclododecane-1-{4-[(3-chloro-4-fluorophenyl)amino]quinazoline-6-yl}hexanamide- 4,7,10-triacetic acid (H3L2), both at macroscopic level and using carrier-free 111In. The studies at the macroscopic level were also extended to the previously reported ligand H3L1.19 The resulting complexes, InL1 and InL2, were fully characterized by common analytical techniques, which also included X-ray diffraction analysis in the case of InL1. The studies reported herein comprise the assessment of the protonation constants of H3L2 by potentiometric titration and UV-Vis spectrophotometry, as well and the determination of the stability constants of the corresponding indium complexes, InL1 and InL2. To have a first insight into the potential relevance of these complexes in the design of SPECT probes for the targeting of epidermal growth factor receptors-tyrosine kinase, cellular uptake studies with 111InL2 and inhibition studies with InL2 in A431 cervical carcinoma cells have been performed, and are also reported.
2. Experimental
All chemicals and solvents were of reagent grade and were used without purification. NMR experiments were performed in a Varian Unity 300 MHz spectrometer. 1H and 13C chemical shifts are reported in parts per million and were referenced relative to the residual solvent signals or to tetramethylsilane. IR spectra were recorded as KBr pellets with a Bruker Tensor 27 Fourier transform IR spectrometer. C, H, N analyses were performed using a CE Instruments EA 110 automatic analyser. Electrospray ionisation mass spectrometry (ESI-MS) was performed using a Bruker HCT electrospray ionization quadrupole ion trap mass spectrometer. 1,4,7,10- Tetraazacyclododecane-1,4,7-tris(t-butyl acetate) (DO3AtBu), 1,4,7,10-tetraazacyclododecane-1-{4-[(3-chloro-4-fluorophenyl)amino]quinazoline-6-yl}propionamide- 4,7,10-triacetic acid (H3L1) and 6-Amino-4-[(3-chloro-4-fluorophenyl) amino] quinazoline were prepared according to published methods17,19,21 Indium-111 chloride was purchased from Mallinckrodt Medical (The Netherlands). High-performance liquid chromatography (HPLC) analysis of 1,4,7,10-tetraazacyclododecane-1-{4-[(3-chloro-4-fluorophenyl)amino]quinazoline-6-yl}hexanamide- 4,7,10-triacetic acid (H3L2), InL1, InL2 and 111InL2 were performed with a PerkinElmer liquid chromatography (LC) pump 200 coupled to an LC 290 tunable UV-Vis detector and to a Berthold LB-507A radiometric detector. Analysis and purification were achieved on Nucleosil columns (10 μm, 250 mm × 4 mm and 7 μm, 250 mm × 8 mm) using flow rates of 1 and 2 mL min−1, respectively; UV detection was at 254 nm. For H3L2, eluent A was aqueous 0.05% CF3COOH solution and eluent B was acetonitrile with 0.05% CF3COOH. The gradient was as follows: 0–1 min, 85% eluent A, 15% eluent B; 1–36 min, 15–100% eluent B; 36–43 min, 100% eluent B; 43–45 min, 0–85% eluent A; 45–50 min, 85% eluent A, 15% eluent B. For InL1, InL2, and 111InL2, eluent A was aqueous NEt3/CH3COOH (2.1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8 (v/v)) solution and eluent B was acetonitrile. The gradient was as follows: 0–15 min, 80% eluent A, 20% eluent B; 15–25 min, 20–30% eluent B; 25–45 min, 70% eluent A, 30% eluent B. Labelling efficiency was assessed using ascending silica gel thin-layer chromatography (ITLC-SG) strips (Polygram, Macherey Nagel) developed with the mobile phase aqueous 0.9% NaCl solution/CH3COOH (9:1 (v/v)). The radioactive distribution on the thin-layer chromatography strips was detected using a Berthold LB 505 detector coupled to a radiochromatogram scanner.
:2.8 (v/v)) solution and eluent B was acetonitrile. The gradient was as follows: 0–15 min, 80% eluent A, 20% eluent B; 15–25 min, 20–30% eluent B; 25–45 min, 70% eluent A, 30% eluent B. Labelling efficiency was assessed using ascending silica gel thin-layer chromatography (ITLC-SG) strips (Polygram, Macherey Nagel) developed with the mobile phase aqueous 0.9% NaCl solution/CH3COOH (9:1 (v/v)). The radioactive distribution on the thin-layer chromatography strips was detected using a Berthold LB 505 detector coupled to a radiochromatogram scanner.
2.1 Synthesis
1H NMR (300 MHz) δ (ppm) [(CD3)2SO]: 11.64 (1H, s, CON![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 10.75 (1H, s, N), 9.06 [1H, s, CH, H(5)], 8.91 [1H, s, CH, H(2)], 8.08–7.69 [3H, m, CH, H(8), H(2′), H(7)], 7.67–7.66 [1H, m, CH, H(6′)], 7.59 [1H, t, CH, H(5′)], 3.54 (2H, t, CH2), 2.43 (2H, t, CH2), 1.89–1.80 (2H, m, CH2), 1.72–1.65 (2H, m, CH2), 1.51–1.44 (2H, m, CH2). 13C NMR δ (ppm) [(CD3)2SO]: 171.7 (
), 10.75 (1H, s, N), 9.06 [1H, s, CH, H(5)], 8.91 [1H, s, CH, H(2)], 8.08–7.69 [3H, m, CH, H(8), H(2′), H(7)], 7.67–7.66 [1H, m, CH, H(6′)], 7.59 [1H, t, CH, H(5′)], 3.54 (2H, t, CH2), 2.43 (2H, t, CH2), 1.89–1.80 (2H, m, CH2), 1.72–1.65 (2H, m, CH2), 1.51–1.44 (2H, m, CH2). 13C NMR δ (ppm) [(CD3)2SO]: 171.7 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ONH), 159.6, 155.3, 149.7, 139.1, 135.3, 134.2, 129.4, 127.0, 125.6, 121.2, 117.0, 114.1, 112.0, 45.3, 36.0, 31.8, 25.9, 24.2. IR: νmax/cm−1 (KBr): 1703s, 1616vs, 1572vs, 1498vs, 1443vs. ESI-MS (+): m/z calcd. for C20H20ON4FClBr: 466.76; found: 466.9.
ONH), 159.6, 155.3, 149.7, 139.1, 135.3, 134.2, 129.4, 127.0, 125.6, 121.2, 117.0, 114.1, 112.0, 45.3, 36.0, 31.8, 25.9, 24.2. IR: νmax/cm−1 (KBr): 1703s, 1616vs, 1572vs, 1498vs, 1443vs. ESI-MS (+): m/z calcd. for C20H20ON4FClBr: 466.76; found: 466.9.
RP-HPLC: Rt = 12.6 min. 1H NMR (300 MHz) δ (ppm) (CD3CN): 9.79 (1H, s, CON), 8.70 [1H, s, CH, H(2)], 8.61 [1H, s, CH, H(5)], 8.08 [1H, d, CH, H(8)], 7.87 [2H, dd, CH, H(7), H(2′)], 7.66 [1H, m, CH, H(6′)], 7.27 [1H, t, CH, H(5′)], 3.57–2.94 (24H, m, CH2, CH2N, CH2COOH), 2.38 (2H, t, CH2), 1.75–1.64 (4H, m, CH2), 1.39–1.35 (2H, m, CH2). 13C NMR δ (ppm) (CD3CN): 174.4 (COOH), 174.1 (CONH), 168.9 (COOH), 160.6, 157.0, 150.2, 140.6, 135.5, 134.4, 130.0, 127.4, 125.7, 123.0, 119.1, 117.8, 115.1, 56.3, 55.4, 54.0, 52.8, 51.2, 49.3, 36.6, 28.6, 26.0, 24.8, 23.8. IR: νmax/cm−1 (KBr): 1677s (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 3470b (νO–H). ESI-MS (+): m/z calcd. for C34H44O7N8FCl: 731.22; found: 731.0.
O), 3470b (νO–H). ESI-MS (+): m/z calcd. for C34H44O7N8FCl: 731.22; found: 731.0.
:10 mg, 67%; InL2:8 mg, 53%).
InL1. RP-HPLC: Rt = 20.1 min. 1H NMR (300 MHz) δ (ppm) [(CD3)2SO]: 10.40 (1H, s, NH), 10.14 (1H, br, NH), 8.77 (1H, s, CH), 8.54 (1H, s, CH), 8.10 (1H, dd, CH), 7.74 (3H, m, CH), 7.39 (1H, t, CH), 3.48–3.02 (28H, m, CH2COO, CH2N, CH2), 2.90 (2H, m, CH2), 2.73 (2H, m, CH2), 2.60 (2H, m, CH2). 13C NMR δ (ppm) [(CD3)2SO]: 170.9, 170.6, 169.8, 160.3, 157.3, 153.0, 151.6, 136.6, 128.5, 127.0, 123.7, 123.0, 118.8, 116.6, 116.4, 115.3, 111.9, 59.8, 58.2, 52.1, 50.4, 49.4, 46.4, 29.2, one CH2 of the linker occasionally overlaps with the solvent resonance. IR: νmax/cm−1 (KBr): 1664s (νC
O), 1622s (νCO), 1388s (νC–N), 1370s (νC–N) cm−1. ESI-MS (+): m/z calcd. for C31H35O7N8FClIn: 800.94; found: 801.1.
InL2. RP-HPLC: Rt = 26.6 min. 1H NMR (300 MHz) δ (ppm) [(CD3)2SO]: 10.37 (1H, s, NH), 10.13 (1H, br, NH), 8.74 (1H, s, CH), 8.56 (1H, s, CH), 8.29 (1H, s, CH), 8.14 (1H, dd, CH), 7.89–7.75 (2H, m, CH), 7.47 (1H, t, CH), 3.48–2.63 (26H, m, C
2N, CH2, C2COO), 1.62 (2H, m, CH2), 1.50 (2H, m, CH2), 1.27 (2H, m, CH2). 13C NMR δ (ppm) [(CD3)2SO]: 171.5, 171.0, 170.7, 157.3, 152.9, 146.5, 137.1, 136.8, 128.4, 127.2, 123.8, 122.7, 118.6, 116.7, 115.4, 111.6, 59.8, 58.2, 53.0, 52.1, 50.4, 46.4, 36.2, 26.0, 24.9, 20.1, one CH2 of the linker occasionally overlaps with the solvent resonance). IR: νmax/cm−1 (KBr): 1626s (νCO), 1658s (νCO), 1390 m (νC–N), 1375 m (νC–N). ESI-MS (+): m/z calcd. for C34H41O7N8FClIn: 843.02; found: 843.2.
2.2 Potentiometric and UV-Vis studies of H3L2 and the indium(III) complexes of L1, L2 and DOTA
To determine the ligand protonation constants and the stability constants of InL complexes (L = L1, L2), potentiometric titrations were carried out. For comparison, we also determined the stability constant of the InDOTA complex. H4DOTA was synthesized according to the published procedure.22 The measurements were performed in a cell thermostated at 25 °C, at an ionic strength I(KCl) = 0.1 M and in the presence of extra HCl or KOH in the −log[H+] range 3–12, using a combined glass electrode (LL Biotrode, Metrohm) connected to a Metrohm 827 pH/ion-meter, equipped with a Metrohm Dosimat 765 automatic burette. The initial volume was 3 mL, the concentration of the ligand was 0.001, 0.002 or 0.004 mol L−1 and the In(III)-to-ligand ratio was 1:1. Variable equilibration times (3–10 min per point ∼3–11 h per titration) were applied to confirm the equilibrium conditions. An inert atmosphere was ensured by constant passage of N2 through the solution. The protonation constants βn are defined by βn = [HnL]/([H]n×[L]), therefore log KH1 = log β1, log KH2 = log β2 − log β1, etc. The stability constant is defined by β = [MpHqLr]/([M]p × [H]q × [L]r). The constants (with standard deviations) were calculated with the program OPIUM.23 The program minimizes the criterion of the generalized least-squares method using the calibration function E = E0 + S × log[H+] + jA × [H+] + jB × Kw/[H+], where the additive term E0 contains the standard potentials of the electrodes used and contributions of inert ions to the liquid-junction potential, S corresponds to the Nernstian slope and jA × [H+] and jB × [OH−] terms are contributions of the H+ and OH− ions to the liquid-junction potential. It is clear that jA and jB cause deviation from a linear dependence between E and −log[H+] only in strong acid and strong alkaline solutions. The calibration parameters were determined from titration of standard HCl with standard KOH before any titration of the ligand to give a pair of calibration titrations used for calculations of the constants. The errors given correspond to one standard deviation.
The protonation constants of the ligand H3L2 and of the InL1 and InL2 complexes have been also assessed by UV-Vis measurements carried out on a PERKIN ELMER Lambda 19 spectrometer in the region 200–400 nm with data steps of 1 nm. The sample concentrations were ∼15 μM. The ligand to metal ratio was 1:1. A constant temperature of 25 °C was maintained by using thermostated cells with a 1 cm optical length. The value of the protonation constant was calculated with program OPIUM by simultaneous treatment of the data in range 320–400 nm.
2.3 X-ray crystallographic analysis
Monocrystals suitable for X-ray diffraction analysis were obtained by slow concentration of a solution of InL1 in H2O–CH3CN. The X-ray diffraction analysis of InL1 has been performed on a Bruker AXS APEX CCD area detector diffractometer, using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Empirical absorption correction was carried out using SADABS.24 Data collection and data reduction were done with the SMART and SAINT programs.25 The structure was solved by direct methods with SIR9726 and refined by full-matrix least-squares analysis with SHELXL-9727 using the WINGX28 suite of programmes. All non-hydrogen atoms were refined anisotropically. The remaining hydrogen atoms were placed in calculated positions. Molecular graphics were prepared using ORTEP3.29 A summary of the crystal data, structure solution and refinement parameters are given in Table 3. Selected bond distances and angles are listed in Table 4.2.4 Preparation of 111InL2
To a 5 mL glass vial was added 200 μL (3.7–15.91 MBq/100–430 μCi) of indium chloride and 100 μL of a 0.3 mM aqueous solution of H3L2 and the pH was adjusted to approximately 6 with an aqueous solution of NaOH. The vial was heated at 100 °C for 15 min, cooled to room temperature and then analysed by RP-HPLC. The presence of hydrolysed indium(III) species was checked by ITLC-SG.RP-HPLC: Rt = 27.10 min; ITLC-SG: Rf 0.25.
2.5 Determination of the overall charge 111InL2
The overall charge of 111InL2 was determined by electrophoresis, using paper strips (Whatman no. 1, 3 MM) and a tris(hydroxymethyl)aminomethane hydrochloride buffer (0.1 mol L−1, pH 7.4). A constant voltage of 250 V was applied for 1 h. The radioactive distribution on the strips was analysed using a Berthold LB 505 detector coupled to a radiochromatogram scanner.2.6 Octanol-water partition coefficient of 111InL2
The log Po/w value of 111InL2, under physiological conditions [n-octanol/0.1mol L−1 phosphate-buffered saline (PBS), pH 7.4], was determined by the multiple back extraction method.302.7 In vitro evaluation: cell line studies
The EGFR-expressing cell lines, A431 cervical carcinoma cells, kindly provided by J. Pirmettis and M. Paravatou (Demokritos Center, Greece), were maintained in Dulbecco’s modified Eagle’s medium containing Glutamax I and supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin (Gibco, Invitrogen, UK), 100 IU per 100 μg mL−1, in a 5% humidified CO2 atmosphere at 37 °C. Cells were subcultured every 2 or 3 days.000 cells per 200 μL per well and incubated for 24 h for attachment. The day after seeding, exponentially growing cells were incubated with various concentrations of H3L2 or 111InL2 (ranging from 1 nM to 100 μM in 4 replicates) during 24 h. Controls consisted of wells without drugs. The medium was removed and the cells were incubated for 3 h in the presence of 0.5 mg mL−1 MTT (Sigma) in PBS (Gibco, Invitrogen, UK) at 37 °C. The MTT solution was removed and 200 mL well−1 of DMSO was added. After thorough mixing, absorbance of the wells was read in an ELISA reader at test and reference wavelengths of 540 and 620 nm, respectively. The mean of the optical density of different replicates of the same sample and the percentage of each value was calculated (mean of the OD of various replicates/OD of the control).
2.8 Biodistribution studies
The in vivo behavior of 111InL2 complex was evaluated in groups of 3 female CD-1 mice (randomly bred, Charles River) weighting approximately 25 g each. Animals were injected intravenously with 100 μL (1.7–2.5 MBq) of each preparation via the tail vein and were maintained on normal diet ad libitum. All animal studies were conducted in accordance with the highest standards of care, as outlined in the European Law. Mice were sacrificed by cervical dislocation at 15 min, 1 h, 4 h and 24 h post-injection. The injected radioactive dose and the radioactivity remaining in the animal after sacrifice were measured in a dose calibrator (Aloka, Curiemeter IGC-3, Tokyo, Japan). The difference between the radioactivity in the injected and sacrificed animal was assumed to be due to total excretion from whole animal body. Blood samples were taken by cardiac puncture at sacrifice. Tissue samples of the main organs were then removed, weighted and counted in a gamma counter (Berthold). Biodistribution results were expressed as percentage of the injected dose per gram tissue (%ID/g). Blood samples, collected at sacrifice time, were centrifuged, the serum separated and treated with ethanol to precipitate the proteins and the supernatant was analysed by HPLC for stability evaluation. The urine samples were filtered trough a Millipore filter (0.22 μm) and also analyzed by RP-HPLC.3. Results and discussion
3.1 Synthesis, characterization and acid–base behavior of the quinazoline-containing DOTA-like chelator
The functionalization of the tetraazamacrocycle derivative with a quinazoline moiety, using a five-carbon spacer, has been performed following the same procedure previously described for the congener ligand (H3L1) with a shorter ethylenic linker.19 As depicted in Scheme 1, the resulting novel DOTA-like chelator (H3L2) was successfully synthesized by alkylation of DO3tBu3 with a bromine quinazoline derivative (1), in the presence of potassium carbonate, followed by removal of the tert-butyl protecting groups with trifluoroacetic acid. After HPLC purification, H3L2 was obtained in a pure form with an overall yield of 57%.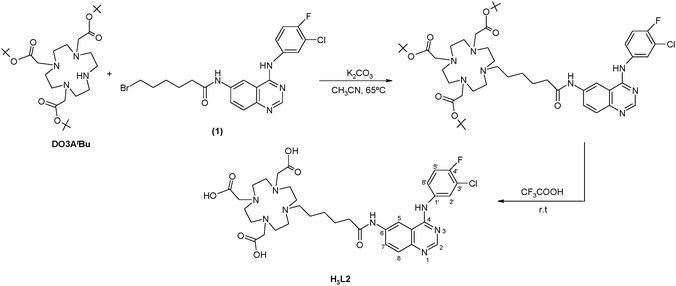 | ||
| Scheme 1 Synthesis of H3L2. | ||
H3L2 was recovered as a pale yellow solid, which is soluble in most common organic solvents and in water. Its characterization has been done by IR, 1H and 13C NMR spectroscopy, RP-HPLC and mass spectrometry. The spectroscopic data obtained for H3L2 are consistent with the presence of one pendant arm bearing the quinazoline moiety and the five-carbon linker. The positive-ion mass spectra showed the expected molecular-ion peak ([M]+, m/z: 731.0), which confirmed the formulation proposed for H3L2.
Potentiometric studies of the ligand H3L2 were performed to determine its protonation constants. The five log KHi values are listed in Table 1 (KHi = [HiL]/[Hi−1L][H]). As proposed previously for the congener H3L1 and reported by other authors for H4DOTA and related tetraazamacrocycles,19 the first two protonation constants have been assigned to the protonation of macrocyclic amines, while the last two protonation constants were attributed to the carboxylates. The third protonation constant (log KH3 = 4.99) was ascribed to the secondary amine of the quinazoline pharmacophore. A slightly higher value, log KH3 = 5.24, was previously found for H3L1, where this protonation constant, intermediate between the higher values of the macrocyclic amines and the lower values of the carboxylates, has been unambiguously assigned to the quinazoline moiety on the basis of the pH-dependent UV-Vis absorption spectra.19 We should also note the relatively high value of the first protonation constant of H3L2, log KH1 = 12.18 in comparison to log KH1 = 10.47 for H3L1. This large difference can be rationalized with the electron donating effect of the longer alkyl spacer in H3L2 (pentyl vs. ethyl in H3L1).
3.2 Synthesis, characterization and stability constants of the indium(III) complexes
The stability and the protonation constant of the InL (L = L1, L2 and DOTA) complexes were determined in aqueous solution by using direct pH-potentiometric titrations and UV-Vis spectrophotometry (for L1 and L2). In order to find the equilibrium conditions, variable equilibration times were applied for potentiometric measurements. The identical titration curves confirmed that the complexation rate is sufficient for determining the stability constants by the in-cell method.For the L1 and L2 systems, from the potentiometric titration only the stability constant of the protonated complexes, HInL1+ and HInL2+ could be determined (logβHInL1 = 23.76 ± 0.02 and logβHInL2 = 26.67 ± 0.02). These complexes are protonated on the secondary amine of the quinazoline. Above pH 3, a slight precipitation (likely indium-hydroxide) was observed in the solution which prevented the determination of the protonation constants of the complexes. They have been therefore assessed by UV-Vis spectrophotometry, at a much lower concentration (∼15 μM). UV-Vis spectra have been recorded in the pH range 3.6–6.8 (see Fig. 1 for the In-L1 system). In the region from 220 to 400 nm, the spectral changes occur between pH 4 and 6.5 and correspond to the protonation of the amine nitrogen on the noncoordinated aromatic pendant arm which does not participate in the complex formation. The pH-dependent spectra allowed us to calculate a protonation constant of log KHInL = 5.23 ± 0.01 and 5.26 ± 0.03 for InL1 and InL2, respectively. These constants were then used to calculate the stability constant of the non-protonated complexes, log KInL (Table 2). For InDOTA, both the stability constant and the protonation constant of the complex were obtained from the potentiometric titration curve. The stability constant is very similar to that obtained previously by Clarke and Martell from spectrophotometric measurements using a ligand competition method.32
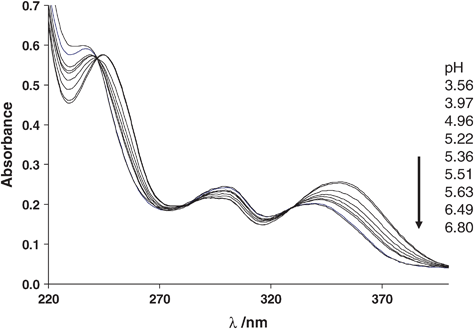 | ||
| Fig. 1 Representative UV–Vis spectra of the In-L1 system at 25 °C, I = 0.1 M (KCl) and variable pH. cIn = cL = 15 μM. | ||
Table 2 presents the stability and protonation constants, as well as pM values for these InL systems and for other indium complexes with related macrocycles. According to the pM values calculated for InL1 and InL2 in comparison to the other macrocycles (Table 2), we can conclude that L1 and L2 are slightly less good chelators for In3+ than DOTA and they form also less stable complexes than other tetraazamacrocycles with a larger cavity size, such as TETA and TRITA.
As shown in Scheme 2, the synthesis of complexes InL1 and InL2 has been done by reacting H3L (L = L1 and L2) with 3 equivalents of indium trichloride, at pH between 5 and 6. The formation of InL1 and InL2, after 1 h of heating at 95 °C, was reliably observed by reversed-phase HPLC. Both compounds were purified by semi-preparative HPLC, being recovered with moderate yields of 67 and 53%, respectively.
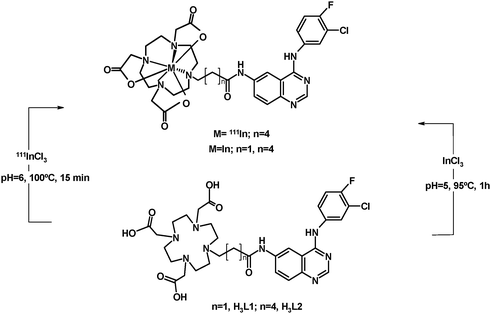 | ||
| Scheme 2 Synthesis of the indium(III) complexes. | ||
The characterization of InL (L = L1, L2) by IR, NMR and mass spectrometry has provided spectroscopic data consistent with the formation of well-defined complexes, anchored by quinazoline-containing macrocycles. The IR spectra of InL1 and InL2 present strong bands, between 1679 and 1622 cm−1, due to the coordinated carboxylate and to the carbonyl groups. Their ESI-MS spectra have shown molecular-ions ([M]+) at m/z 801.1 and 843.2, respectively, in agreement with the proposed formulations.
The molecular structure of InL1 was determined by X-ray crystallography (see Fig. 2). Crystals suitable for X-ray diffraction analysis were obtained by slow concentration of a solution of the complex in CH3CN–H2O. The crystallographic data are presented in Table 3. Selected bond lengths (Å) and angles (°) are listed in Table 4.
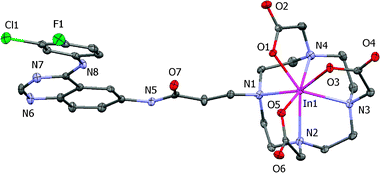 | ||
| Fig. 2 ORTEP diagram of InL1 with ellipsoids drawn at the 40% probability level. | ||
| Empirical formula | C31H41ClFInN8O10 |
| M g−1 mol−1 | 854.99 |
| T/K | 150(2) |
| Wavelength/Å | 0.71073 |
| Cryst. Syst., space group | Monoclinic, P21/c |
| a/Å | 9.3886(3) |
| b/Å | 26.4945(8) |
| c/Å | 13.4424(4) |
| α (°) | 90 |
| β (°) | 93.837 |
| γ (°) | 90 |
| Volume/Å3 | 3336.25(18) |
| Z | 4 |
| D c/Mg m−3 | 1.702 |
| μ/mm−1 | 0.866 |
| F(000) | 1752 |
| cryst size/mm | 0.15 × 0.12 × 0.10 |
| Theta range/° | 3.08–25.02 |
| Limiting indices, hkl | −11/11, −29/31, −15/15 |
| Reflec. Collected/unique | 20302/5668 [R(int) = 0.0827] |
| completeness to θ [%] | 96.3 (θ = 25.02°) |
| absorption correction | semi-empirical |
| max. and min. | 0.9184 and 0.8811 |
| T max and Tmin | 0.9184 and 0.8811 |
| refinement method | Full-matrix least-squares on F2 |
| data/restraints/params | 5668/0/477 |
| S on F2 | 1.019 |
| R 1, wR2 [I > 2σ(I)]] | 0.0395, 0.0894 |
| R 1, wR2 (all data) | 0.0558, 0.0943 |
| Δρ max, min/e Å−3 | 0.836 and −0.867 |
| In(1)–O(5) | 2.156(3) |
| In(1)–O(3) | 2.173(3) |
| In(1)–O(1) | 2.201(2) |
| In(1)–N(4) | 2.315(3) |
| In(1)–N(2) | 2.364(3) |
| In(1)–N(1) | 2.394(3) |
| In(1)–N(3) | 2.425(3) |
| O(5)–In(1)–O(3) | 88.91(11) |
| O(5)–In(1)–O(1) | 85.87(10) |
| O(3)–In(1)– O(1) | 78.83(10) |
| O(5)–In(1)–N(4) | 162.00(10) |
| O(3)–In(1)–N(4) | 88.12(11) |
| O(1)–In(1)–N(4) | 76.14(10) |
| O(5)–In(1)–N(2) | 74.44(10) |
| O(3)–In(1)–N(2) | 128.15(11) |
| O(1)–In(1)–N(2) | 145.05(10) |
| N(4)–In(1)–N(2) | 120.71(11) |
| O(5)–In(1)–N(1) | 99.07(11) |
| O(3)–In(1)–N(1) | 155.47(10) |
| O(1)–In(1)–N(1) | 78.66(10) |
InL1 crystallized in the P21/c space group with four molecules in the unit cell, along with three crystallization water molecules surrounding each molecule of InL1. The In3+ ion is heptacoordinated by the four nitrogens of the macrocycle backbone and by the three carboxylate pendant arms. As shown in Fig. 3, the coordination geometry around the metal can be described as capped trigonal prismatic. The trigonal faces are defined by two of the nitrogen atoms and one oxygen from the carboxylate groups: N(1)N(2)O(5) and N(3)N(4)O(3), respectively. The oxygen atom O(1) caps the quadrangular face formed by N(1), N(4), O(3) and O(5). The same type of coordination geometry has been previously reported for [In(DO3A)]·2H2O·MeOH.33 By contrast, the structure of In(DO3A-xy-TPP)+ (DO3A-xy-TPP = triphenyl(4-((4,7,10-tris(carboxymethyl)-1,4,7,10-tetraazacyclododecan-1-yl)methyl)benzyl)phosphonium), containing also one functionalized pendant arm, is best described as a monocapped octahedron.34 In the structure of InL1, the quinazoline moiety is kept away from the metal center, and therefore no direct or indirect interaction with the indium chelate has been established, minimizing possible interference with the receptor binding.
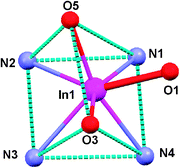 | ||
| Fig. 3 Diagram of the coordination polyhedron of InL1. | ||
The In–N bond distances average 2.374 Å, being In–N(4) the shortest one with a value of 2.315 Å. The average In–N bond distances in InL1 is the same as in In(DO3A-xy-TPP)+ (average In–N distance = 2.374 Å),34 and is also comparable to that (average In–N distance = 2.356 Å) of In(DO3A)·2H2O·MeOH.33 For InL1, the average In–O bond length of 2.177 Å is also comparable to that reported for In(DO3A-xy-TPP)+ (average In–O distance = 2.174 Å)34 and for In(DO3A)·2H2O·MeOH (average In–O distance = 2.181 Å).33
3.3 Synthesis, characterization and in vitro evaluation of 111InL2
The radioactive complex 111InL2 was prepared by reacting H3L2 (1 × 10−4 mol L−1) with 111InCl3, at pH = 6 and with heating at 95 °C for 15 min (Scheme 2). The chemical identity of 111InL2 was assessed by comparison of its HPLC profile with that of the corresponding non-radioactive indium complex, as shown in Fig. 4. During the synthesis of 111InL2, the presence of 111In hydroxide species was checked by ITLC-SG with 0.9%NaCl/CH3COOH (9:1 (v/v)) as eluent. In these conditions the hydrolysed species are retained in the origin (Rt = 0), whereas 111InCl3 migrates with Rt = 1. The ITLC-SG chromatograms obtained for 111InL2 have shown only one single peak (Rf 0.25) corresponding to the desired radioactive complex. Together with the HPLC results, these data have shown that InL2 was obtained with a high radiochemical purity (>97%). The electrophoretic behavior of 111InL2 was studied under physiological conditions (pH = 7.4) in order to evaluate the overall charge of the complex. 111InL2 did not migrate, confirming its neutral character as found by the X-ray structural analysis of the non-radioactive congener.
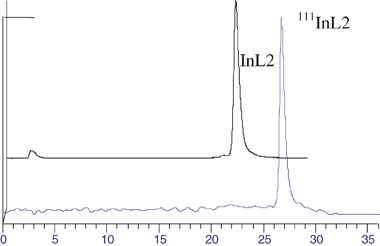 | ||
| Fig. 4 HPLC chromatograms of InL2 and 111InL2. | ||
111InL2 displays a great stability in vitro under physiological conditions. In fact, the complex remains almost intact (>95%) after incubation in PBS (pH 7.4, 37 °C) for 5 days and in serum for 3 h (Fig. 6). The lypophilicity (log Po/w) of 111InL2 was assessed by the multiple back extraction method, under physiological conditions, and the value found was −0.57 ± 0.14. This value is considerably higher than the one that we have reported for a related gallium complex, 67GaL1 (log D = −1.02 ± 00.03). This difference is certainly due to the presence of a five-carbon spacer in L2, instead of the ethylenic linker in L1, and to the different coordination spheres of In(III) and Ga(III) in the respective complexes. Differences in the coordination sphere have also been invoked to explain changes on the lipophilicity of other trivalent metal complexes (e.g.111In vs.90Y) anchored by DOTA-like chelators functionalised with small biomolecules.20
3.4 In vitro cellular uptake, inhibition studies and biodistribution
To evaluate the potential utility of 111InL2 as a biomarker for EGFR-TK imaging we have performed in vitro cellular studies to assess the degree of internalization of the complex in intact A431 cells, as described in the experimental section. The uptake kinetics of 111InL2 in A431 cells is shown in Fig. 5.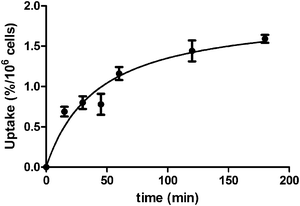 | ||
| Fig. 5 Uptake kinetics of 111InL2 into A431 cells at 37 °C. | ||
The results indicate that there is a slight increase of internalization with time, but the overall value is relatively low, even after 3 h of incubation. The inhibition of intact A431 cell growth by H3L2 and InL2 was also evaluated by a MTT assay. It was found that both compounds do not have any effect on the cell growth, for concentrations in the range 1 nM–100 μM. Taken all together these data indicate a low level of internalization and/or potency to inhibit EGFR autophosphorylation. Such behavior indicates that the chemical modifications on the pharmacophore, due to its conjugation to the bifunctional chelator and coordination to the metal, led to a final complex having a low capability to enter into the cells and/or to interact with EGFR-TK.
In spite of the disappointing in vitro biological results, biodistribution of the 111InL2 complex was studied in healthy mice just to assess its in vivo stability and pharmacokinetics (Fig. S1, ESI†). This study indicated a slow clearance from blood stream (8.4 ± 2.4; 6.7 ± 1.0; 1.0 ± 0.2 of I.D./g at 15 min, 1 h and 4 h respectively) and a relatively low rate of radioactivity excretion from whole body, which occurs predominantly through the hepatobiliar pathway. HPLC analysis of blood and urine samples collected at sacrifice (Fig. 6) demonstrated high in vivo stability (>95%) of 111InL2 making evident that this complex was not significantly metabolized. The biodistribution profile of 111InL2 is significantly different from the one that we have reported for the related 67GaL1 complex, especially concerning the main excretory route and the rate of total excretion.19 At 4 h after administration, the excretion of 111InL2 (34.1 ± 1.0%ID) is much lower than that of 67GaL1 (84.2 ± 1.9% ID), probably due to the change in the predominant way of excretion (hepatobiliar vs. renal). Such a result can certainly be explained by the different hydrophilicity of 111InL2 and 67GaL1, due to the use of linkers with a different number of carbon atoms and also to the presence of a free carboxylate group in 67GaL1.
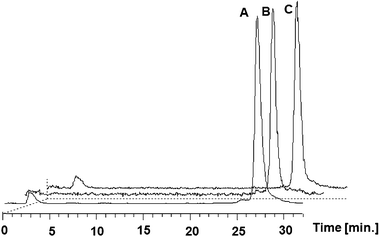 | ||
| Fig. 6 In vitro stability of 111InL2 in serum (A) and in vivo stability in murine serum (B) and in urine (C) at 3 h, 4 h and 1 h, respectively. | ||
5. Conclusion
A DOTA-like bifunctional chelator (H3L2) containing a five-carbon spacer between the macrocycle backbone and a quinazoline moiety has been successfully synthesized and fully characterized. The respective protonation constants were determined by potentiometry and UV-Vis spectrophotometry. H3L2 and the previously reported H3L1, a related ligand containing a smaller ethylene linker for the linkage of the same pharmacophore, were used to prepare the complexes InL (L = L1 and L2). The formation of these complexes was studied in solution by potentiometry and the respective stability constants were determined. InL1 and InL2 were isolated in the solid form and characterized by the common spectroscopic techniques, which included X-ray diffraction analysis in the case of InL1. The characterization of the complexes confirmed the presence of a heptadentate (N4O3) macrocycle, having a dangling pendant arm that contains the quinazoline moiety. The radioactive congener 111InL2 has been prepared with high radiochemical purity. InL2 is a hydrophilic complex that presents a high stability in vitro. Taking into consideration our previous results with the complex 67GaL1, the use of 111In and the lengthening of the alkyl chain between the macrocycle backbone and the quinazoline moiety promoted the increase of the log P values of the complexes. However, the ability of 111InL2 to cross the cell membrane and/or to recognize the EGFR-TK was not improved relatively to 67GaL1, as verified by the negligible in vitro uptake of 111InL2 in the A431 EGFR-expressing cell line, and by the absence of inhibition of cell growth. Biodistribution in healthy mice indicated high in vivo stability and a relatively low clearance and total excretion rate. The low cell uptake and/or non-recognition of EGFR-TK as well as the in vivo behavior did not motivate further studies in tumor bearing mice to evaluate the utility of these complexes for EGFR-TK imaging, as a low retention in target tumor tissues is predicted.Acknowledgements
This work was partially supported by COST Action D38. Raquel Garcia would like to thank the Fundação para a Ciência e Tecnologia for a post-doctoral research grant. We wish to acknowledge Joaquim Marçalo from the Laboratório de Espectrometria de Massa from the ITN, Sacavém, Portugal for the ESI-MS analysis. The quadrupole ion trap mass spectrometer was acquired with the support of the Programa Nacional de Reequipamento Científico (contract REDE/15037/REM/2005-ITN) of Fundação para a Ciência e a Tecnologia and is part of RNEM-Rede Nacional de Espectrometria de Massa.References
- J. J. Laskin and A. B. Sandler, Cancer Treat. Rev., 2004, 30, 1 CrossRef CAS.
- A. Levitzki, Lung Cancer, 2003, 41, 9 CrossRef.
- J.-y. Song, S.-w. Lee, J. P. Hong, S. E. Chang, H. Choe and J. Choi, Cancer Lett., 2009, 283, 135 CrossRef CAS.
- H. M. Shepard, C. M. Brdlik and H. Schreiber, J. Clin. Invest., 2008, 118, 3574 CrossRef CAS.
- W. Cai, G. Niu and X. Chen, Eur. J. Nucl. Med. Mol. Imaging, 2008, 35, 186 CrossRef.
- Z. Yu, T. J. Boggon, S. Kobayashi, C. Jin, P. C. Ma, A. Dowlati, J. A. Kern, D. G. Tenen and B. Halmos, Cancer Res., 2007, 67, 10417 CrossRef CAS.
- T. A. Bonasera, G. Ortu, Y. Rozen, R. Krais, N. M. Freedman, R. Chisin, A. Gazit, A. Levitzki and E. Mishani, Nucl. Med. Biol., 2001, 28, 359 CrossRef CAS.
- S. Dissoki, D. Laky and E. Mishani, J. Labelled Compd. Radiopharm., 2006, 49, 533 CrossRef CAS.
- P. Johnström, A. Fredriksson, J.-O. Thorell and S. Stone-Elander, J. Labelled Compd. Radiopharm., 1998, 41, 623 CrossRef CAS.
- I. Ben-David, Y. Rozen, G. Ortu and E. Mishani, Appl. Radiat. Isot., 2003, 58, 209 CrossRef CAS.
- E. Mishani, G. Abourbeh, Y. Rozen, O. Jacobson, D. Laky, D. I. Ben, A. Levitzki and M. Shaul, Nucl. Med. Biol., 2004, 31, 469 CrossRef CAS.
- E. Mishani, G. Abourbeh, Y. Rozen, O. Jacobson, S. Dissoki, R. B. Daniel, Y. Rozen, M. Shaul and A. Levitzki, J. Med. Chem., 2005, 48, 5337 CrossRef CAS.
- G. Ortu, B. Ben-David, Y. Rozen, M. T. Freedman, R. Chisin, A. Levitski and E. Mishani, Int. J. Cancer, 2002, 101, 360 CrossRef CAS.
- M. Shaul, G. Abourbeh, O. Jacobson, Y. Rozen, D. Laky, A. Levitzki and E. Mishani, Bioorg. Med. Chem., 2004, 12, 3421 CrossRef CAS.
- E. Mishani and G. Abourbeh, Curr. Top. Med. Chem., 2007, 7, 1755 CrossRef CAS.
- N. Breza, J. Pató, L. Orfi, B. Hegymegi- Barakonyi, P. B. Anhegyi, E. Varkondi, G. Borbely, I. Petak and G. Keri, J. Recept. Signal Transduction, 2008, 28, 361 Search PubMed.
- C. Fernandes, C. Oliveira, L. Gano, A. Bourkoula, I. Pirmettis and I. Santos, Bioorg. Med. Chem., 2007, 15, 3974 CrossRef CAS.
- C. Fernandes, I. C. Santos, I. Santos, H.-J. Pietzsch, J.-U. Kunstler, W. Kraus, A. Rey, N. Margaritis, A. Bourkoula, A. Chiotellis, M. Paravatou-Petsotas and I. Pirmettis, Dalton Trans., 2008, 3215 RSC.
- R. Garcia, P. Fousková, L. Gano, A. Paulo, P. Campello, E. Tóth and I. Santos, J. Biol. Inorg. Chem., 2009, 14, 261 CrossRef CAS.
- S. Liu, Adv. Drug Delivery Rev., 2008, 60, 1347 CrossRef CAS.
- A. Beeby, L. M. Bushby, D. Maffeo and J. A. G. Williams, J. Chem. Soc., Dalton Trans., 2002, 48 RSC.
- J. F. Desreux, Inorg. Chem., 1980, 19, 1319–1324 CrossRef CAS.
- M. Kývala and I. Lukeš, International Conference, Chemomotrics '95, Pardubice, Czech Republic, 1995, p. 63; full version of “OPIUM” is available on http: //www.natur.cuni.cz/∼kyvala/opium.html Search PubMed.
- SADABS; Area-Detector Absorption Correction; Brukee AXS Inc., Madison, WI, 2004.
- SAINT: Area-Detector Integration Software (Version7.23); Brukee AXS Inc Madison, WI, 2004.
- A. Altomare, M. C. Burla, M. Camalli, G. Cascarano, G. Giacovazzo, A. Guagliardi, A. G. G. Moliterini, G. Polidoro and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115 CrossRef CAS.
- G. M. Sheldrick, SHELXL-97: Program for the Refinement of Crystal Structure, University of Gottingen, Germany, 1997 Search PubMed.
- L. J. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- L. J. Farrugia, ORTEP-3, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- D. E. Troutner, W. A. Volkert, T. J. Hoffman and R. A. Holmes, Int. J. Appl. Radiat. Isot., 1984, 35, 467 Search PubMed.
- A. E. Martell and R. M. Smith, NIST standard reference database 46 (critically selected stability constants of metal complexes), version 7.0, 2003.
- E. T. Clarke and A. E. Martell, Inorg. Chim. Acta, 1991, 190, 37 CrossRef CAS.
- A. Riesen, T. A. Kaden, W. Ritter and H. R. Mäcke, J. Chem. Soc., Chem. Commun., 1989, 460 RSC.
- C.-T. Yang, Y. Li and S. Liu, Inorg. Chem., 2007, 46, 8988 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Biodistribution data for 111InL2. CCDC reference number 772423. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c004797j |
| This journal is © The Royal Society of Chemistry 2010 |