Comparison of selenohomolanthionine and selenomethionine in terms of selenium distribution and toxicity in rats by bolus administration
Yoshiro
Tsuji
a,
Takahiro
Mikami
a,
Yasumi
Anan
a and
Yasumitsu
Ogra
*bc
aGraduate School of Pharmaceutical Sciences, Chiba University, Chuo, Chiba 260-8675, Japan
bLaboratory of Chemical Toxicology and Environmental Health, Showa Pharmaceutical University, 3-3165 Higashi-Tamagawagakuen, Machida, Tokyo 194-8543, Japan
cHigh Technology Research Center, Showa Pharmaceutical University, 3-3165 Higashi-Tamagawagakuen, Machida, Tokyo 194-8543, Japan. E-mail: ogra@ac.shoyaku.ac.jp; Fax: +81 42 721 1563; Tel: +81 42 721 1563
First published on 13th May 2010
Abstract
The distribution and metabolism of selenohomolanthionine (4,4′-selenobis[2-aminobutanoic acid], SeHLan), a newly identified selenoamino acid in selenized Japanese pungent radish, were compared with those of selenomethionine (SeMet) in rats. Either selenoamino acid was injected intravenously at a bolus dose of 1.0 mg Se/kg body weight. SeMet was preferably accumulated in the pancreas, increasing the serum amylase level, an index of pancreatic damage. SeHLan was preferably accumulated in the kidneys, raising the serum creatinine level, an index of kidney damage. On the other hand, the levels of two major urinary selenometabolites, i.e., Se-methylseleno-N-acetyl-galactosamine and trimethylselenonium, were comparable between SeHLan- and SeMet-administered rats, suggesting that there may be no differences in the efficiency of metabolism of these two selenoamino acids to the urinary selenometabolites despite the difference in distribution. SeHLan is expected to be a potential supplemental source of Se without inducing the onset of pancreatic damage. The specific toxicity of SeHLan to the kidneys may be avoided if its dose is lower than the one used in the present study.
Introduction
Inorganic selenium (Se) salts and selenoamino acid derivatives can be utilized as a nutritional and supplemental source.1 Inorganic Se salts, selenite and selenate, are metabolized to selenoproteins, suggesting that animals can de novo synthesize selenocysteine (SeCys), which is required as an active center of selenoproteins, from the inorganic Se salts.2,3 Selenite is also used for therapeutic purposes. The effects of selenite on patients with severe systemic inflammatory response syndrome, sepsis, and septic shock have been reported.4,5 The bolus administration of selenite has been associated with mortality decrease in septic shock animals and patients.6,7 In that study, selenite was administered at a maximum dose of 4.0 mg Se/day.7 On the other hand, selenoamino acid derivatives, such as SeCys, selenomethionine (SeMet), Se-methylselenocysteine (MeSeCys), γ-glutamylmethylselenocysteine (GluMeSeCys)8–10 and selenohomolanthionine (SeHLan),11 are found in nature12,13 and are, in general, less harmful than inorganic Se salts. Thus, selenoamino acid derivatives are expected to be a better nutritional source of Se than the inorganic ones. In addition to their being a nutritional source, their cancer preventive effects are also expected.14,15 Indeed, SeMet was subjected to a clinical trial for the prevention of prostate cancer, called SELECT (the Se and vitamin E cancer prevention trial). However, no significant effects on the prevention of prostate cancer were observed and instead, a non-significant increase in the risk of diabetes mellitus compared with placebo was noted in the Se alone group at a dose of 0.2 mg Se/day.16 Thus, SELECT has definitively demonstrated that Se, vitamin E, or Se plus vitamin E did not prevent prostate cancer. It is known that SeMet accumulates in pancreas17 and 75Se-labeled SeMet is used as a radiocontrast medium for pancreas.18 This may indicate that the chemical form of SeMet is not suitable for use as a supplement. Consequently, the development of a third-generation Se source that is safer than currently available species, such as selenite and SeMet, is highly awaited.SeHLan (4,4′-selenobis[2-aminobutanoic acid]) is a newly identified selenoamino acid in selenized Japanese pungent radish and shows less toxicity to human cultured cells than SeMet.11 The proposed metabolic pathway of both SeMet and SeHLan is summarized in Scheme 1.19 It is suggested that SeMet participates in three metabolic pathways. SeMet mimics methionine (Met) and shares metabolic pathways with Met. First, SeMet is used for peptide synthesis in place of Met because the translation machinery of animals cannot distinguish SeMet from Met. The pathways mentioned above may not contribute to Se utilization (selenoprotein synthesis). Second, SeMet is utilized for the biosynthesis of Se-adenosylselenomethionine (SeAM), an Se analogue of S-adenosylmethionine.20,21 Then, SeAM is converted into SeCys via Se-adenosylselenohomocysteine (SeAH), selenohomocysteine (SeHCy) and selenocystathionine (Se-cystathionine) in the trans-selenation pathway. The third route is a more specific pathway to SeMet than Met, i.e., it involves the cleavage of methylselenol from SeMet by α,γ-lyase.22,23 Cleaved methylselenol is demethylated to form selenide or further methylated to form dimethylselenide and trimethylselenonium.24 In contrast to SeMet, the metabolic pathway of SeHLan is thought to be simpler, i.e., SeHLan is utilized only in the trans-selenation pathway for selenoprotein synthesis. Hence, SeHLan is expected to less affect the metabolic pathway of Met than SeMet, when it is administered at a large dose.
 | ||
| Scheme 1 Proposed metabolic pathway of SeHLan and SeMet in animal cells. | ||
Although the intracellular metabolism of these selenoamino acids was investigated as mentioned above, the pharmacokinetics of the selenoamino acids, in particular, SeHLan, have not been elucidated. In this study, we evaluated the distribution of Se in rats after the administration of SeHLan at a bolus dose of 1.0 mg Se/kg body weight. This dose was comparable to that of selenite to be used as a therapeutic agent and might be suitable to highlight the difference in distribution between SeHLan and SeMet. The metabolism of SeHLan in rats was also evaluated by speciation. In addition, the toxicity of SeHLan was evaluated by measuring serum biochemical parameters such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), creatinine (CRE), albumin (ALB), lactate dehydrogenase (LDH), total bilirubin (T-BIL), amylase (AMY), and choline esterase (ChE). Then, these data were compared with those of SeMet and finally, the pharmacological availability of SeHLan was discussed.
Experimental
Chemicals
Powdered elemental Se, nitric acid, ammonium acetate, N,N-dimethylformamide dehydrate (DMF), sodium tetrahydroborate, hydrochloric acid, nitric acid, and Wakogel® 100C18 were purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). L-Selenomethionine (SeMet) and (S)-(+)-2-amino-4-bromobutyric acid hydrobromide were purchased from Tokyo Chemical Industry (Tokyo, Japan). Amberlite® 120-R was purchased from Sigma-Aldrich (St. Louis, NJ, USA). All reagents were of the highest or analytical grade. Deionized water (18.3 MΩcm−1) was used throughout.Apparatus
An Agilent7500ce ICP-MS (Agilent Technologies, Hachiouji, Japan) equipped with an octopole reaction system was used to detect Se. Important parameters for operation were as follows: plasma RF power, 1500 W; plasma gas flow, 15.0 L min−1; auxiliary gas flow, 1.15 L min−1; nebulizer gas flow, 1.05 L min−1; m/z monitored, 77 and 82; dwell time, 100 ms; and point per peak, 1.Synthesis of selenohomolanthionine
SeHLan was synthesized in our laboratory by the method previously reported.11 Briefly, elemental Se (powder, 80 mg) was suspended in 2 mL of DMF under nitrogen atmosphere. Then, sodium tetrahydroborate (600 mg) in DMF (12 mL) was added and the mixture was stirred at room temperature for 60 min. (S)-(+)-2-amino-4-bromobutyric acid hydrobromide (574 mg) dissolved in DMF (2 mL) was gradually added and the reaction mixture was left to stand at room temperature for 48 h. The reaction was stopped by the addition of 1 mol L−1 HCl and the reaction mixture was evaporated in vacuo. The residue was dissolved in deionized water and purified on a cation-exchange column (Amberlite® 120-R) and an ODS column (Wakogel® 100C18) to afford SeHLan. Obtained SeHLan was further purified by HPLC (Shodex Asahipak GS-520P; 20 i.d. × 500 mm; Showa Denko, Tokyo). Chemical purity was confirmed by HPLC-ICP-MS as described below.Animal experiments
All animal experiments were carried out according to the “Principles of Laboratory Animal Care” (NIH version, revised 1996) and the Guidelines of the Animal Investigation Committee, Graduate School of Pharmaceutical Sciences, Chiba University, Japan.Six-week-old male Wistar rats were purchased from CLEA Japan (Tokyo). After a one-week acclimation period, the rats were individually housed in a metabolic cage at 22 ± 2 °C with a light/dark cycle of 12/12 h to collect urine and feces, and fed a standard diet (MF; Oriental Yeast, Tokyo) and tap water ad libitum.
Each group of three or four rats received an intravenous injection of SeHLan or SeMet dissolved in saline at a bolus dose of 1.0 mg Se/kg body weight. Blood was collected under light ether anesthesia 1, 6, and 24 h after the injection, and clotted blood was centrifuged at 1600 × g for 10 min to obtain serum. Then, the liver, kidneys, pancreas, lungs, heart, spleen, brain, and testes were excised. The tissues and serum were preserved at −20 °C prior to use. Four rats receiving saline served as control.
Determination of Se concentration in samples
The organs, serum, and urine were wet-ashed with concentrated nitric acid (HNO3) and 30% H2O2, and then the ashed samples were diluted with deionized water. Se concentration in the samples was determined by ICP-MS at m/z 82.HPLC-ICP-MS Analysis
The same volume of urine and serum from three or four rats in each group was combined for HPLC-ICP-MS analysis. The HPLC system consisted of an on-line degasser (DG660B-2, GL Science Inc., Tokyo), an HPLC pump (PU713, GL Science Inc., Tokyo), a six-port injector (model 7125, Rheodyne, CA) with 20 μL and 200 μL sample loops, and a column. A multi-mode gel filtration column, Shodex Asahipak GS-520HQ (7.5 i.d. × 300 mm, with a guard column, 7.5 i.d. × 75 mm, Showa Denko), was injected with a 200 μL aliquot of serum and then eluted with 50 mmol L−1 Tris–HCl, pH 7.4, at a flow rate of 0.6 mL min−1. Another gel filtration column, Shodex Asahipak GS-320HQ (7.5 i.d. × 300 mm, with a guard column, 7.5 i.d. × 75 mm, Showa Denko), was injected with a 20 μL aliquot of urine and then eluted with 50 mmol L−1 ammonium acetate, pH 6.5, at a flow rate of 0.6 mL min−1. The eluate was introduced directly into the Babington nebulizer of the ICP-MS, and Se distribution was monitored at m/z 82. Low molecular weight Se-containing molecules were assigned by retention time matching with authentic standards.Measurement of serum biochemical parameters
Measurement of serum biochemical parameters, i.e., alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood urea nitrogen (BUN), creatinine (CRE), albumin (ALB), lactate dehydrogenase (LDH), total bilirubin (T-BIL), amylase (AMY), and choline esterase (ChE), was performed using a Hitachi7170 model auto-analyzer (Hitachi High-Technologies Corporation, Tokyo). Kits used for ALT, AST, BUN, CRE, ALB, LDH, T-BIL, AMY and ChE were L-type Wako GPT-J2 (Wako), L-type Wako GOT-J2 (Wako), L-type Wako UN (Wako), L-type Wako CRE-M (Wako), albumin II-HA rest Wako (Wako), L-type Wako LDH-J (Wako), T-Bil reagent A (Sysmex Corporation, Kobe, Japan), L-type Wako amylase (Wako) and liquitech choline esterase (Roche Diagnostics, Tokyo), respectively.Statistics
The results are presented as means ± standard deviation (S.D.) of three or four samples. Statistical analysis was performed with the Student’s t-test. The significance level was set at p < 0.05 (*).Results and discussion
Se distribution in rats after administration of selenoamino acids
Se concentration in blood of rats administered SeHLan was significantly higher than that of rats administered SeMet at 1 h after the injection (Fig. 1a). Se concentration in serum showed the same kinetics as that in blood (Fig. 1b). The peak corresponding to the intact form of SeHLan was detected at a retention time of 19.2 min (Fig. 2). A small amount of SeMet was observed in serum 1 h after the injection. These indicate that the increased Se in serum is due to the intact form of SeHLan, and SeHLan less efficiently disappeared from serum than SeMet despite the decrease in intensity of the peak corresponding to SeHLan at 6 h. The peak appearing at a retention time of 13.8 min and corresponding to selenoprotein P (Sel P)25 started to increase from 1 h after the SeMet injection and became apparent at 6 h after the SeHLan injection, suggesting that SeMet was more rapidly utilized for selenoprotein synthesis than SeHLan. The peak appearing at a retention time of 11.1 min and corresponding to extracellular glutathione peroxidase (eGPx) did not show any apparent changes throughout the experiment. In this experiment, the rats were fed an Se-adequate diet; thus, extra amounts of selenoamino acid did not boost the amount of eGPx protein. The peak appearing at a retention time of 12.5 min was found in the chromatogram of serum of rats administered SeMet. Although the protein(s) were not assigned yet, this peak could be assigned to SeMet-containing protein(s) whose sequence(s) show the replacement of Met with SeMet. Peaks appearing at the retention times of 15.0 and 16.0 min were found in the chromatograms of either serum of rats administered SeHLan or SeMet.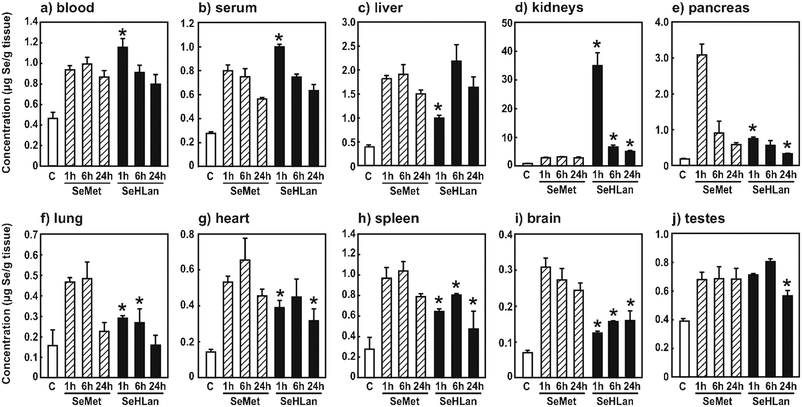 | ||
| Fig. 1 Se concentration in organs, blood, and serum. The rats were intravenously administered SeMet or SeHLan at a bolus dose of 1 mg Se/kg body weight, and blood (a), serum (b), liver (c), kidneys (d), pancreas (e), lungs (f), heart (g), spleen (h), brain (i), and testes (j) were collected 1, 6, and 24 h after administration. “C” means the sample collected from rats without any treatments. Values are expressed as means ± S.D. of three or four rats. Asterisk indicates significant difference between SeHLan-administered group and SeMet-administered group at the same time point at p < 0.05. | ||
 | ||
| Fig. 2 Elution profiles of Se in the serum of rats intravenously administered SeMet or SeHLan at a bolus dose of 1 mg Se/kg body weight. Sera were collected 1, 6, and 24 h after administration, combined in each group of three or four rats, and then subjected to HPLC-ICP-MS analysis on a multi-mode gel filtration column (Shodex Asahipak GS-520HQ) eluted with 50 mmol L−1 Tris-HCl. Se in the eluate was monitored at m/z 82. The concentrations of standard SeHLan and SeMet were 1.0 μg mL−1. | ||
Se concentration in the liver of rats administered SeHLan was significantly lower than that of rats administered SeMet at 1 h after administration, although there were no significant differences between the SeHLan and SeMet groups at the later experimental period (Fig. 1c). This also suggests that SeHLan is retained for a longer time in the bloodstream and more slowly incorporated into the liver than SeMet. It is known that Sel P is biosynthesized in the liver.26,27 Hence, the slower incorporation of SeHLan into the liver is coincident with the slower increase in Sel P in the serum.
Se concentration in the kidneys of rats administered SeHLan was significantly higher than that of rats administered SeMet throughout the experimental period (Fig. 1d). In particular, Se concentration in the kidneys of SeHLan-treated rats was 13 times higher than that of SeMet-treated rats at 1 h after the administration. Considering the results in Fig. 2, the accumulated Se seemed to be an intact form of SeHLan.
As reported previously,17 SeMet was rapidly accumulated in the pancreas (Fig. 1e). Se concentration in the pancreas of SeMet-treated rats was 4.1 times higher than that of SeHLan-treated rats. SeMet was accumulated in the pancreas even under our experimental protocol, i.e., bolus administration.
Se concentration seemed to show the same tendency in lungs, heart, spleen, brain, and testes (Fig. 1f–j): Se concentrations in SeHLan-treated rats were lower than those in SeMet-treated rats. These also indicate that SeHLan is less efficiently incorporated into those organs than SeMet. In other words, SeMet is more efficiently accumulated in those organs because it acts as not only a selenoamino acid but also Met. Thus, SeMet may not be thoroughly utilized as an Se source and may disturb Met metabolism in the organs.
Speciation of urinary selenometabolites
Two major urinary selenometabolites are known: Se-methylseleno-N-acetyl-galactosamine (Se-sugar) and trimethylselenonium (TMSe).28–30 Before the bolus administration of selenoamino acids, one major peak appearing at the retention time of 20.1 min was detected, which corresponded to Se-sugar (Fig. 3). Two major urinary selenometabolites were detected within the initial 6 h after the administration of SeMet, and then, the intensities of the peaks increased in the subsequent 18 h (6–24 h after the administration). No intact form of SeMet at retention time of 21.6 min was detected in the urine. In the urine of SeHLan-treated rats, the largest Se peak appearing at the retention time of 17.6 min was detected, and its retention time matched that of authentic SeHLan. Thus, the intact form of SeHLan was excreted into urine within the initial 6 h after the administration. In addition to the intact SeHLan, comparable amounts of two major selenometabolites, Se-sugar and TMSe, were also detected. Although two additional selenometabolites were detected at the retention times at 15.4 and 16.3 min, they were not assigned yet. Fig. 4 shows the cumulative amounts of Se species in the urine of treated rats. The amounts of the two urinary selenometabolites were comparable between SeHLan- and SeMet- treated rats. This indicates that the efficiency of biotransformation/metabolism from these selenoamino acids to the urinary selenometabolites was comparable. In addition, the results indicate that the increased amount of Se in the urine of SeHLan-treated rats is attributed to the intact SeHLan.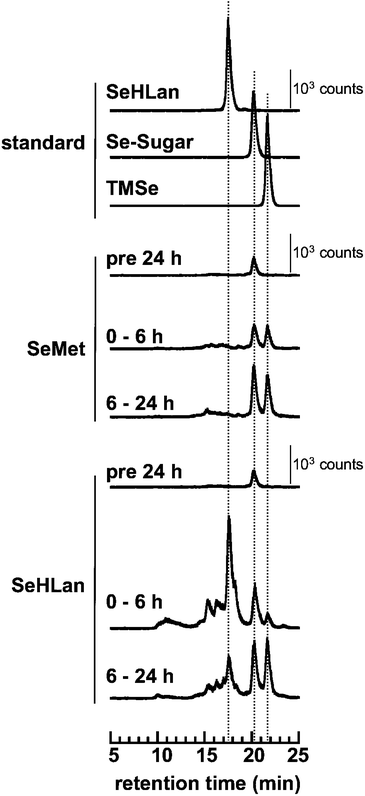 | ||
| Fig. 3 Elution profiles of Se in the urine of rats intravenously administered SeMet or SeHLan at a bolus dose of 1 mg Se/kg body weight. Urine was collected 24 h before and 6 and 24 h after administration, combined in each group of three or four rats, and then subjected to HPLC-ICP-MS analysis on a multi-mode gel filtration column (Shodex Asahipak GS-320HQ) eluted with 50 mmol L−1 ammonium acetate. Se in the eluate was monitored at m/z 82. The concentrations of standard SeHLan, Se-sugar and TMSe were 0.2, 0.1 and 0.2 μg mL−1, respectively. | ||
 | ||
| Fig. 4 Cumulative amounts of urinary selenocompounds (Se-sugar, TMSe, and SeHLan) The amounts of selenocompounds were determined from the results obtained in the speciation study (Fig. 3). | ||
Biochemical parameters of selenoamino-acid-treated rats
LDH level in SeMet-treated rats at 1 h after administration was significantly higher than that in the control (Fig. 5a). The bolus administration of SeMet induced hemolysis in the rats at 1 h after the intravenous injection, whereas SeHLan bolus administration did not do so. As erythrocytes contain LDH, the LDH level was increased by hemolysis. The increases in AST, ALT, and ChE levels indicate liver damage. The bolus administration of these selenoamino acids caused the increases of these parameters (Fig. 5b, c, and i). ALB level was slightly decreased and T-BIL level was significantly but not markedly increased by the bolus administration of the two selenoamino acids (Fig. 5f and g). The results suggest that the bolus dose (1.0 mg Se/kg body weight) of these selenoamino acids induces non-specific and weak damage on the liver and the biliary duct. | ||
| Fig. 5 Serum biochemical parameters of rats intravenously administered SeMet or SeHLan at a bolus dose of 1 mg Se/kg body weight. Sera were collected 1, 6, and 24 h after administration and the serum biochemical parameters were determined using an auto-analyzer and a kit. Values are expressed as means ± S.D. of three or four rats. * indicates significant difference between control group and treated groups at p < 0.05. | ||
CRE level was significantly increased at 6 h after SeHLan administration, but was not increased by SeMet administration. The increase in CRE level indicates kidney damage, hence suggesting that the bolus dose of SeHLan induced nephrotoxicity as a result of the renal accumulation of SeHLan (Fig. 5e). BUN level reflects liver and kidney damages. The significant increase in BUN level was only observed at 1 h after SeMet administration, whereas SeHLan showed the tendency to the increase in BUN level through the experimental period (Fig. 5d). This also suggests that SeMet and SeHLan induce the liver or kidney damages. On the other hand, AMY level was significantly increased at 1 h after SeMet administration (Fig. 5h). As AMY is a pancreatic enzyme, the increase in serum AMY level indicates leakage from the pancreas as a result of pancreatic damage.31 Thus, it can be concluded that SeMet administration at the bolus dose of 1.0 mg kg−1 body weight causes pancreatic damage.
Conclusion
The newly identified selenoamino acid, SeHLan, showed different pharmacokinetics and toxicity from SeMet by bolus administration. Although SeMet was preferably distributed to the pancreas, SeHLan was accumulated in the kidneys. These specific distributions are reflected by their own toxicities, i.e., SeMet and SeHLan caused pancreatic and kidney damage, respectively. SeHLan was excreted into urine in the intact form but SeMet was not. However, the levels of the two major selenometabolites, i.e., Se-sugar and TMSe, in urine were comparable between SeHLan- and SeMet-treated rats at 6 h after the administration. The results demonstrate that there may be no differences in the efficiency of metabolism of SeHLan and SeMet to the urinary selenometabolites despite the minor urinary selenometabolites have not been identified yet. Consequently, SeHLan is expected to be a potential supplemental source of Se without inducing the onset of pancreatic damage. The specific toxicity of SeHLan to the kidneys may be avoided when it is administered at a lower dose than that used in the present study.Acknowledgements
We thank Mr Nobuo Emi (Otsuka Pharmaceutical Factory, Inc.) for the measurement of biochemical parameters. We would like to acknowledge a Grant-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (No. 19390033), and the financial support from Agilent Technologies Foundation, USA.References
- M. P. Rayman, Lancet, 2000, 356, 233–241 CrossRef CAS.
- Y. Shiobara, Y. Ogra and K. T. Suzuki, Life Sci., 2000, 67, 3041–3049 CrossRef CAS.
- Y. Shiobara, Y. Ogra and K. T. Suzuki, Analyst, 1999, 124, 1237–1241 RSC.
- X. Forceville, B. Laviolle, D. Annane, D. Vitoux, G. Bleichner, J. M. Korach, E. Cantais, H. Georges, J. L. Soubirou, A. Combes and E. Bellissant, Crit. Care, 2007, 11, R73 CrossRef.
- M. W. Angstwurm, L. Engelmann, T. Zimmermann, C. Lehmann, C. H. Spes, P. Abel, R. Strauss, A. Meier-Hellmann, R. Insel, J. Radke, J. Schüttler and R. Gärtner, Crit. Care Med., 2007, 35, 118–126 CrossRef CAS.
- Z. Wang, X. Forceville, P. Van Antwerpen, M. Piagnerelli, D. Ahishakiye, P. Macours, D. De Backer, J. Neve and J. L. Vincent, Shock, 2009, 32, 140–146 CrossRef CAS.
- X. Forceville, J. Trace Elem. Med. Biol., 2007, 21(Suppl. 1), 62–65 CrossRef CAS.
- Y. Ogra, K. Ishiwata, Y. Iwashita and K. T. Suzuki, J. Chromatogr., A, 2005, 1093, 118–125 CrossRef CAS.
- M. Montes-Bayón, E. G. Yanes, C. Ponce de León, K. Jayasimhulu, A. Stalcup, J. Shann and J. A. Caruso, Anal. Chem., 2002, 74, 107–113 CrossRef CAS.
- S. McSheehy, J. Szpunar, V. Haldys and J. Tortajada, J. Anal. At. Spectrom., 2002, 17, 507–514 RSC.
- Y. Ogra, T. Kitaguchi, K. Ishiwata, N. Suzuki, Y. Iwashita and K. T. Suzuki, J. Anal. At. Spectrom., 2007, 22, 1390–1396 RSC.
- H. Goenaga Infante, R. Hearn and T. Catterick, Anal. Bioanal. Chem., 2005, 382, 957–967 CrossRef.
- Z. Pedrero and Y. Madrid, Anal. Chim. Acta, 2009, 634, 135–152 CrossRef CAS.
- C. Ip, M. Birringer, E. Block, M. Kotrebai, J. F. Tyson, P. C. Uden and D. J. Lisk, J. Agric. Food Chem., 2000, 48, 2062–2070 CrossRef CAS.
- C. Ip, Y. Dong and H. E. Gänther, Cancer Metastasis Rev., 2002, 21, 281–289 CrossRef CAS.
- S. M. Lippman, E. A. Klein, P. J. Goodman, M. Scott Lucia, I. M. Thompson, L. G. Ford, H. L. Parnes, L. M. Minasian, J. Michael Gaziano, J. A. Hartline, J. Kellogg Parsons, J. D. Bearden III, E. David Crawford, G. E. Goodman, J. Claudio, E. Winquist, E. D. Cook, D. D. Karp, P. Walther, M. M. Lieber, A. R. Kristal, A. K. Darke, K. B. Arnold, P. A. Ganz, R. M. Santella, D. Albanes, P. R. Taylor, J. L. Probstfield, T. J. Jagpal, J. J. Crowley, F. L. Meyskens Jr., L. H. Baker and C. A. Coltman Jr., JAMA, J. Am. Med. Assoc., 2009, 301, 39–51 Search PubMed.
- K. T. Suzuki, C. Doi and N. Suzuki, Toxicol. Appl. Pharmacol., 2006, 217, 185–195 CrossRef CAS.
- R. Bergmann, P. Brust, G. Kampf, H. H. Coenen and G. Stöcklin, Nucl. Med. Biol., 1995, 22, 475–481 CrossRef CAS.
- Y. Ogra and Y. Anan, J. Anal. At. Spectrom., 2009, 24, 1477–1488 RSC.
- Y. Ogra, T. Kitaguchi, K. Ishiwata, N. Suzuki, T. Toida and K. T. Suzuki, Metallomics, 2009, 1, 78–86 RSC.
- K. Wróbel, K. Wróbel and J. A. Caruso, J. Anal. At. Spectrom., 2002, 17, 1048–1054 RSC.
- T. Okuno, S. Motobayashi, H. Ueno and K. Nakamuro, Biol. Trace Elem. Res., 2005, 106, 77–94 CrossRef CAS.
- T. Okuno, S. Motobayashi, H. Ueno and K. Nakamuro, Biol. Trace Elem. Res., 2005, 108, 245–257 CrossRef CAS.
- Y. Ohta and K. T. Suzuki, Toxicol. Appl. Pharmacol., 2008, 226, 169–177 CrossRef CAS.
- S. Yoneda and K. T. Suzuki, Biochem. Biophys. Res. Commun., 1997, 231, 7–11 CrossRef CAS.
- R. F. Burk and K. E. Hill, Biol. Trace Elem. Res., 1992, 33, 151–153 CrossRef CAS.
- K. E. Hill, P. R. Lyons and R. F. Burk, Biochem. Biophys. Res. Commun., 1992, 185, 260–263 CAS.
- Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuki, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2002, 767, 301–312 CrossRef CAS.
- Y. Kobayashi, Y. Ogra, K. Ishiwata, H. Takayama, N. Aimi and K. T. Suzuki, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15932–15936 CrossRef CAS.
- K. A. Francesconi and F. Pannier, Clin. Chem., 2004, 50, 2240–2253 CrossRef CAS.
- T. D. Kinney, N. Kaufman, J. V. Klavins, R. W. Marsters and C. Y. Tseng, Am. J. Pathol., 1960, 37, 137–160 Search PubMed.
| This journal is © The Royal Society of Chemistry 2010 |