Synthesis and biological applications of ionic triphenyltin(IV) chloride carboxylate complexes with exceptionally high cytotoxicity†
Goran N.
Kaluđerović
*ab,
Harish
Kommera
b,
Evamarie
Hey-Hawkins
c,
Reinhard
Paschke
b and
Santiago
Gómez-Ruiz
*cd
aBiozentrum, Martin-Luther-Universität Halle-Wittenberg, Weinbergweg 22, 06120 Halle, Germany. E-mail: goran.kaluderovic@chemie.uni-halle.de; Fax: +49-345 5527028
bInstitut für Chemie, Martin-Luther-Universität Halle-Wittenberg, Kurt-Mothes-Straße 2, D-06120 Halle, Germany
cInstitut für Anorganische Chemie der Universität Leipzig, Johannisallee 29, D-04103 Leipzig, Germany. E-mail: santiago.gomez@urjc.es; Fax: +34-914888143
dDepartamento de Química Inorgánica y Analítica, E.S.C.E.T., Universidad Rey Juan Carlos, 28933 Móstoles, Madrid, Spain
First published on 20th May 2010
Abstract
The reaction of N-phthaloylglycine (P-GlyH), N-phthaloyl-L-alanine (P-AlaH), and 1,2,4-benzenetricarboxylic 1,2-anhydride (BTCH) with triethylamine led to the formation of the corresponding ammonium salts [NHEt3][P-Gly] (1), [NHEt3][P-Ala] (2) and [NHEt3][BTC] (3) in very high yields. The subsequent reaction of 1–3 with triphenyltin(IV) chloride (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) yielded the compounds [NHEt3][SnPh3Cl(P-Gly)] (4), [NHEt3][SnPh3Cl(P-Ala)] (5), and [NHEt3][SnPh3Cl(BTC)] (6), respectively. The molecular structure of 4 was determined by X-ray diffraction studies. The cytotoxic activity of the ammonium salts (1–3) and the triphenyltin(IV) chloride derivatives (4–6) were tested against human tumor cell lines from five different histogenic origins: 8505C (anaplastic thyroid cancer), A253 (head and neck cancer), A549 (lung carcinoma), A2780 (ovarian cancer) and DLD-1 (colon cancer). Triphenyltin(IV) chloride derivatives (4–6) show very high activity against these cell lines while the ammonium salts of the corresponding carboxylic acids (1–3) are totally inactive. The most active compound is 4 which is 50 times more active than cisplatin. Compound 4 is found to induce apoptosis via extrinsic pathways on DLD-1 cell lines, probably by accumulation of caspases 2, 3 and 8. Furthermore, compound 4 seems to cause disturbances in G1 and G2/M phases in cell cycle of DLD-1 cell line.
:1) yielded the compounds [NHEt3][SnPh3Cl(P-Gly)] (4), [NHEt3][SnPh3Cl(P-Ala)] (5), and [NHEt3][SnPh3Cl(BTC)] (6), respectively. The molecular structure of 4 was determined by X-ray diffraction studies. The cytotoxic activity of the ammonium salts (1–3) and the triphenyltin(IV) chloride derivatives (4–6) were tested against human tumor cell lines from five different histogenic origins: 8505C (anaplastic thyroid cancer), A253 (head and neck cancer), A549 (lung carcinoma), A2780 (ovarian cancer) and DLD-1 (colon cancer). Triphenyltin(IV) chloride derivatives (4–6) show very high activity against these cell lines while the ammonium salts of the corresponding carboxylic acids (1–3) are totally inactive. The most active compound is 4 which is 50 times more active than cisplatin. Compound 4 is found to induce apoptosis via extrinsic pathways on DLD-1 cell lines, probably by accumulation of caspases 2, 3 and 8. Furthermore, compound 4 seems to cause disturbances in G1 and G2/M phases in cell cycle of DLD-1 cell line.
1. Introduction
The fight against cancer is one of the primary targets concerning medicinal chemistry, in which bioorganometallic chemistry has become an interesting and fruitful research field.1 The initial efforts in the evaluation of platinum-based anticancer drugs, have been shifted to non-platinum metal-based agents.2–14 Many different metals, for example Ti, Ga, Ge, Pd, Au, Co, Ru and Sn have been already used for these purposes and have helped in the minimization of the side effects associated with the use of platinum compounds as anticancer drugs.2–11 In this context, many organotin(IV) complexes have shown interesting in vivo anticancer activity as new chemotherapy agents.15–20 However, the possible application of the synthesized organotin derivatives was very limited by their poor water solubility.7 Thus, solubility seems to be the one of the main problems for the use of tin(IV) in anticancer drugs, however, with a rational design and functionalization of the corresponding complexes, some new tin(IV) complexes with higher water solubility have been reported.21–23 Additional interest, due to solubility reasons, have been paid to the synthesis and evaluation of the cytotoxicity of organotin(IV) compounds containing carboxylate ligands.24–31 In addition, coordination of carboxylates to organotin moieties offers the possibility of studying the variations of the coordination mode from monodentate or chelate (symmetric or asymmetric) to bridging which may give rise to oligomeric or polymeric structures.32 The moieties RnSn(4–n)+ (n = 2 or 3) may bind to membrane proteins or glycoproteins, or to cellular proteins; e.g. to hexokinase, ATPase, acetylcholinesterase of human erythrocyte membrane or to skeletal muscle membranes26 or may interact directly with DNA.33Following our research on the synthesis, characterization and cytotoxic properties of metal-based anticancer drugs,34–43 and using carboxylato ligands which have shown interesting biological properties when bound to metals,31,38,39 here we present the synthesis, characterization and the study of the cytotoxicity of three different ionic triphenyltin(IV) chloride complexes containing carboxylate ligands which present, according to their ionic nature, higher solubility in polar solvents. These compounds represent the first examples of triethylammonium salts of triphenyltin(IV) carboxylates which have shown an extremely high cytotoxic activity.
The carboxylic acids used in this study (Fig. 1) have been previously used in the synthesis of different neutral triorganotin(IV) carboxylates,44–52 however, the synthesis and characterization of more convenient ionic tin(IV) complexes containing these ligands have not yet been reported and their cytotoxicity has not been studied. Thus, as numerous reports show that there is not a single or a well-defined number of pathways for the interaction of tin compounds with the membrane or constituents within the cell53–56 and many other studies indicate the possible mechanisms of action of tin compounds, the study of the biological properties and anticancer mechanisms of this new group of anionic organotin carboxylates, which may give rise to intermolecular interactions with an effect on the overall and the local tin environment and in the final cytotoxicity, is very useful for gaining novel insights on the cellular action of organotin complexes.
 | ||
| Fig. 1 Carboxylic acids used in the study. | ||
2. Experimental
2.1 General manipulations
All experiments were performed under an atmosphere of dry nitrogen using standard Schlenk techniques. Solvents and triethylamine were distilled from the appropriate drying agents and degassed before use. NMR spectra were recorded on a Bruker AVANCE DRX 400 spectrometer. 1H NMR (400.13 MHz): internal standard solvent, external standard TMS; 13C{1H} NMR (100.6 MHz): internal standard solvent, external standard TMS; 119Sn{1H} NMR (149.2 MHz): internal standard solvent, external standard tetramethyltin. The splitting of proton resonances in the reported 1H NMR spectra are defined as s = singlet, d = doublet, t = triplet, q = quadruplet, br = broad and m = multiplet. IR spectra: KBr pellets were prepared in a nitrogen-filled glove box and the spectra were recorded on a Perkin-Elmer System 2000 FTIR spectrometer in the range 350–4000 cm−1. ESI MS were recorded with a FT-ICR MS Bruker-Daltonics (APEX II, 7 Tesla, MASPEC II), and solutions of ca. 1 mg mL−1 of the compounds in a mixture of dry CHCl3–CH3CN (1:1) were injected. All solvents were purified by distillation, dried, saturated with nitrogen and stored over potassium mirror. N-phthaloylglycine (P-GlyH), N-phthaloyl-L-alanine (P-AlaH), and 1,2,4-benzenetricarboxylic 1,2-anhydride (BTCH) and triphenyltin(IV) chloride were purchased from Alfa Aesar or Acros Organics. All the commercial reagents were used directly, without further purification.
2.2 Synthesis of [NHEt3][P-Gly] (1)
A solution of NEt3 (1.76 mL, 12.19 mmol) in THF (20 mL) was added dropwise over 15 min to a solution of P-GlyH (2.50 g, 12.19 mmol) in THF (130 mL) at room temperature. The reaction mixture was stirred under reflux overnight. The solvent was then evaporated under reduced pressure and the resulting white solid was washed twice with hexane (40 mL) to give a white solid characterized as 1. Yield: 3.32 g, 89%. FT-IR (KBr): 3491 (s) (υ NH), 1720 (s) (υ CO) 1616 (s) (υa COO−), 1383 (s) (υs COO−); 1H NMR (400 MHz, CDCl3, 25 °C): δ 1.25 (t, 9H, 3J(1H-1H) = 7.6 Hz, CH3 of Et), 3.01 (q, 6H, 3J(1H-1H) = 7.6 Hz, CH2 of Et), 4.28 (s, 2H, CH2 of P-Gly), 7.67 (m, 2H, aromatic H in P-Gly), 7.83 (m, 2H, aromatic H in P-Gly), 14.31 (br, 1H, NHEt3); 13C{1H} NMR (100.6 MHz, CDCl3, 25 °C): δ 8.5 (CH3 of Et), 41.3 (CH2 of P-Gly), 44.9 (CH2 of Et), 123.1, 133.5 (aromatic CH of P-Gly) 132.7 (aromatic ipso-C of P-Gly), 168.3 (CO of P-Gly), 172.3 (COO); ESI-MS (negative)(m/z): 203.8 [M+-NHEt3], 159.9 [M+-NHEt3-COO]; ESI-MS (positive)(m/z): 102.1 [M+-(P-Gly) + H]; elemental analysis for C16H22N2O4 (306.4): calc. C, 62.73; H, 7.24; N, 9.14, found C, 62.40; H, 7.19; N 9.09%.2.3 Synthesis of [NHEt3][P-Ala] (2)
The synthesis of 2 was carried out in an identical manner to 1. NEt3 (1.65 mL, 11.46 mmol) and P-AlaH (2.50 g, 11.46 mmol). Yield: 3.41 g, 93%. FT-IR (KBr): 3460 (s) (υ NH), 1710 (s) (υ CO) 1650 (s) (υa COO−), 1390 (s) (υs COO−); 1H NMR (400 MHz, CDCl3, 25 °C): δ 1.25 (t, 9H, 3J(1H-1H) = 7.2 Hz, CH3 of Et), 1.56 (d, 3H, 3J(1H-1H) = 7.6 Hz, CH3 of P-Ala), 3.08 (q, 6H, 3J(1H-1H) = 7.6 Hz, CH2 of Et), 4.73 (q, 1H, 3J(1H-1H) = 7.2 Hz, CH of P-Ala), 7.67 (m, 2H, aromatic H in P-Ala), 7.79 (m, 2H, aromatic H in P-Ala), 11.88 (br, 1H, NHEt3); 13C{1H} NMR (100.6 MHz, CDCl3, 25 °C): δ 8.9 (CH3 of Et), 16.3 (CH3 of P-Ala) 41.9 (CH of P-Ala), 44.8 (CH2 of Et), 123.7, 133.2 (aromatic CH of P-Ala) 132.9 (aromatic ipso-C of P-Ala), 168.9 (CO of P-Ala), 172.5 (COO); ESI-MS (negative)(m/z): 217.9 [M+-NHEt3], 174.2 [M+-NHEt3-COO]; ESI-MS (positive)(m/z): 102.1 [M+-(P-Ala) + H]; elemental analysis for C17H24N2O4 (320.4): calc. C, 63.73; H, 7.55; N, 8.74, found C, 63.55; H, 7.39; N 8.79%.2.4 Synthesis of [NHEt3][BTC] (3)
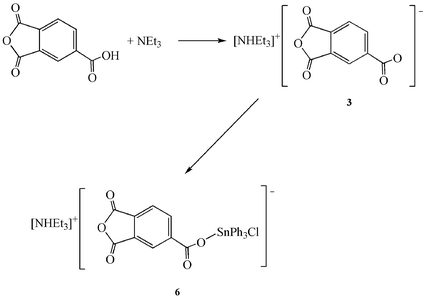
The synthesis of 3 was carried out in an identical manner to 1. NEt3 (1.88 mL, 13.01 mmol) and BTCH (2.50 g, 13.01 mmol). Yield: 3.09 g, 81%. FT-IR (KBr): 3435 (s) (υ NH), 1778, 1715 (s) (υ CO) 1615 (s) (υa COO−), 1351 (s) (υs COO−); 1H NMR (400 MHz, CDCl3, 25 °C): δ 1.39 (t, 9H, 3J(1H-1H) = 7.2 Hz, CH3 of Et), 3.23 (q, 6H, 3J(1H-1H) = 7.2 Hz, CH2 of Et), 7.99 (d, 1H, 3J(1H-1H) = 8.0 Hz, aromatic H in BTC), 8.58 (d, 1H, 3J(1H-1H) = 8.0 Hz, aromatic H in BTC), 8.64 (s, 1H, aromatic H in BTC), 13.33 (br, 1H, NHEt3); 13C{1H} NMR (100.6 MHz, CDCl3, 25 °C): δ 8.6 (CH3 of Et), 45.6 (CH2 of Et), 125.1, 126.8, 137.2 (aromatic CH of BTC), 145.5 (aromatic C–COO of BTC), 162.8, 162.9 (C–CO of BTC), 169.3 (COO of BTC), 170.7, 170.8 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of anhydride of BTC); ESI-MS (negative)(m/z): 190.8 [M+-NHEt3], 164.9 [M+-NHEt3-O], 146.9 [M+-NHEt3-COO]; ESI-MS (positive)(m/z): 102.1 [M+-BTC + H]; elemental analysis for C15H19NO5 (293.3): calc. C, 61.42; H, 6.53; N, 4.78, found C, 61.21; H, 6.44; N 4.79%.
O of anhydride of BTC); ESI-MS (negative)(m/z): 190.8 [M+-NHEt3], 164.9 [M+-NHEt3-O], 146.9 [M+-NHEt3-COO]; ESI-MS (positive)(m/z): 102.1 [M+-BTC + H]; elemental analysis for C15H19NO5 (293.3): calc. C, 61.42; H, 6.53; N, 4.78, found C, 61.21; H, 6.44; N 4.79%.
2.5 Synthesis of [NHEt3][SnPh3Cl(P-Gly)] (4)
A solution of [NHEt3][P-Gly] (1) (0.79 g, 2.59 mmol) in THF (20 mL) was added dropwise over 15 min to a solution of SnPh3Cl (1.00 g, 2.59 mmol) in THF (80 mL) at room temperature. The reaction mixture was stirred under reflux overnight. The solvent was then evaporated under reduced pressure and the resulting white solid was washed twice with hexane (40 mL) to give a white solid characterized as 4. The product is totally soluble in EtOH, DMSO, CHCl3, CH2Cl2,·CH3CN and in mixtures EtOH–H2O and is partially soluble in toluene. Crystallization in toluene afforded crystals suitable for X-ray diffraction analysis. Yield: 1.32 g, 74%. FT-IR (KBr): 3436 (s) (υ NH), 1718 (s) (υ CO) 1623 (s) (υa COO−), 1370 (s) (υs COO−), 457 (m) (υ Sn–O); 1H NMR (400 MHz, CDCl3, 25 °C): δ 1.05 (t, 9H, 3J(1H-1H) = 7.2 Hz, CH3 of Et), 2.70 (q, 6H, 3J(1H-1H) = 7.6 Hz, CH2 of Et), 4.15 (s, 2H, CH2 of P-Gly), 7.35 (br m, 9H, m- and p-protons in SnPh3), 7.66 (m, 2H, aromatic H in P-Gly), 7.73 (br m, 6H, o-protons in SnPh3, 3J(1H–Sn) = ca. 60 Hz); 7.80 (m, 2H, aromatic H in P-Gly), 11.62 (br, 1H, NHEt3), 13C{1H} NMR (100.6 MHz, CDCl3, 25 °C): δ 8.4 (CH3 of Et), 40.9 (CH2 of P-Gly), 45.4 (CH2 of Et), 123.2, 133.8 (aromatic CH of P-Gly), 128.6 (C-3 and C-5 of SnPh3, 3J(13C–Sn) = 67.2 Hz), 129.4 C-4 of SnPh3, 4J(13C–Sn) = 46.5 Hz), 132.4 (aromatic ipso-C of P-Gly), 136.4 (C-2 and C-6 of SnPh3, 2J(13C–Sn) = 48.7 Hz), 140.4 (C-1 of SnPh3, 1J(13C–Sn) not observed), 167.9 (CO of P-Gly), 171.9 (COO); 119Sn{1H} NMR (149.2 MHz, CDCl3, 25 °C): δ −144.5; ESI-MS (negative)(m/z): 590.02 [M+-NHEt3]; elemental analysis for C34H37ClN2O4Sn (691.8): calc. C, 59.03; H, 5.39; N, 4.05, found C, 58.59; H, 5.29; N 4.09%.2.6 Synthesis of [NHEt3][SnPh3Cl(P-Ala)] (5)
The synthesis of 5 was carried out in an identical manner to 4. [NHEt3][P-Ala] (2) (0.83 g, 2.59 mmol) and SnPh3Cl (1.00 g, 2.59 mmol). Yield: 1.42 g, 78%. FT-IR (KBr): 3447 (s) (υ NH), 1713 (s) (υ CO) 1618 (s) (υa COO−), 1389 (s) (υs COO−), 455 (m) (υ Sn–O); 1H NMR (400 MHz, CDCl3, 25 °C): δ 1.22 (t, 9H, 3J(1H-1H) = 7.2 Hz, CH3 of Et), 1.71 (d, 3H, 3J(1H-1H) = 7.2 Hz, CH3 of P-Ala), 2.93 (q, 6H, 3J(1H-1H) = 7.6 Hz, CH2 of Et), 4.99 (q, 1H, 3J(1H-1H) = 7.6 Hz, CH of P-Ala), 7.44 (br m, 9H, m- and p- protons in SnPh3), 7.68 (m, 2H, aromatic H in P-Ala), 7.70 (br m, 6H, o- protons in SnPh3, 3J(1H–Sn) = ca. 52 Hz); 7.83 (m, 2H, aromatic H in P-Ala), 11.70 (br, 1H, NHEt3), 13C{1H} NMR (100.6 MHz, CDCl3, 25 °C): δ 8.5 (CH3 of Et), 16.0 (CH3 of P-Ala) 45.6 (CH of P-Ala), 48.2 (CH2 of Et), 123.3, 133.9 (aromatic CH of P-Ala), 129.0 (C-3 and C-5 of SnPh3, 3J(13C–Sn) = 65.1 Hz), 130.2 (C-4 of SnPh3, 4J(13C–Sn) = 13.6 Hz), 132.1 (aromatic ipso-C of P-Gly), 136.3 (C-2 and C-6 of SnPh3, 2J(13C–Sn) = 48.7 Hz), 139.8 (C-1 of SnPh3, 1J(13C–Sn) not observed), 167.5 (CO of P-Ala), 178.1 (COO); 119Sn{1H} NMR (149.2 MHz, CDCl3, 25 °C): δ −94.0; ESI-MS (negative)(m/z): 604.03 [M+-NHEt3]; elemental analysis for C35H39ClN2O4Sn (705.9): calc. C, 59.56; H, 5.57; N, 3.97, found C, 59.49; H, 5.48; N 3.98%.2.7 Synthesis of [NHEt3][SnPh3Cl(BTC)] (6)
The synthesis of 6 was carried out in an identical manner to 4. [NHEt3][BTC] (3) (0.76 g, 2.59 mmol) and SnPh3Cl (1.00 g, 2.59 mmol). Yield: 1.16 g, 66%. FT-IR (KBr): 3433 (s) (υ NH), 1780, 1719 (s) (υ CO) 1616 (s) (υa COO−), 1362 (s) (υs COO−), 454 (m) (υ Sn–O); 1H NMR (400 MHz, CDCl3, 25 ºC): δ 1.33 (t, 9H, 3J(1H-1H) = 7.2 Hz, CH3 of Et), 3.07 (q, 6H, 3J(1H-1H) = 7.2 Hz, CH2 of Et), 7.46 (br m, 9H, m- and p- protons in SnPh3), 7.75 (br m, 6H, o- protons in SnPh3, 3J(1H–Sn) = 61.1 Hz), 7.98 (d, 1H, 3J(1H-1H) = 7.9 Hz, aromatic H in BTC), 8.56 (d, 1H, 3J(1H-1H) = 7.9 Hz, aromatic H in BTC), 8.65 (s, 1H, aromatic H in BTC), 12.67 (br, 1H, NHEt3); 13C{1H} NMR (100.6 MHz, CDCl3, 25 °C): δ 8.6 (CH3 of Et), 45.9 (CH2 of Et), 125.3, 127.1, 137.5 (aromatic CH of BTC), 128.9 (C-3 and C-5 of SnPh3, 3J(13C–Sn) = 63.8 Hz), 130.1 (C-4 of SnPh3, 4J(13C–Sn) = 13.6 Hz), 136.4 (C-2 and C-6 of SnPh3, 2J(13C–Sn) = 48.8 Hz), 138.9 (C-1 of SnPh3, 1J(13C–Sn) not observed), 142.6 (aromatic C-COO of BTC), 162.5, 162.6 (C-CO of BTC), 168.7 (COO of BTC), 170.6, 170.8 (CO of anhydride of BTC); 119Sn{1H} NMR (149.2 MHz, CDCl3, 25 °C): δ −95.0; ESI-MS (negative)(m/z): 576.99 [M+-NHEt3]; elemental analysis for C33H34ClNO5Sn (705.9): calc. C, 58.39; H, 5.05; N, 2.06, found C, 58.10; H, 4.97; N 2.19%.
2.8 Data Collection and Structural Refinement of 4
The data of 4 were collected with a CCD Oxford Xcalibur S (λ(Mo-Kα) = 0.71073 Å) using ω and φ scans mode. Semi-empirical from equivalents absorption corrections were carried out with SCALE3 ABSPACK.57 The structure was solved by Patterson methods.58 Structure refinement was carried out with SHELXL-97.59 All non-hydrogen atoms were refined anisotropically, H atom bonded to nitrogen was localized and refined freely and the other hydrogen atoms were placed in calculated positions and refined with calculated isotropic displacement parameters. Table 1 lists crystallographic details. Crystallographic data for the structural analysis 4 have been deposited with the Cambridge Crystallographic Data Centre, CCDC-759465 (4).†| 4 | |
| Formula | C34H37ClN2O4Sn |
| Fw | 691.80 |
| T/K | 130(2) |
| Cryst syst | Monoclinic |
| Space group | P21/n |
| a/pm | 990.82(5) |
| b/pm | 3066.76(9) |
| c/pm | 1121.89(4) |
| α (°) | 90 |
| β (°) | 107.661(5) |
| γ (°) | 90 |
| V (nm3) | 3.2483(2) |
| Z | 4 |
| Dc (Mg m−3) | 1.415 |
| μ/mm−1 | 0.908 |
| F(000) | 1416 |
| Cryst dimens/mm | 0.3 × 0.2 × 0.2 |
| θ range (°) | 2.66 to 28.28 |
| hkl ranges | −13 ≤ h ≤ 13, |
| −37 ≤ k ≤ 40, | |
| −14 ≤ l ≤ 14 | |
| Data/parameters | 8049/350 |
| goodness-of-fit on F2 | 1.055 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0558, |
| wR2 = 0.1264 | |
| R indices (all data) | R 1 = 0.0837, |
| wR2 = 0.1352 | |
| largest diff. peak and hole/e Å−3 | 1.769 and −1.353 |
2.9. In vitro studies
2.9.4.1 Trypan blue exclusion test. Apoptotic cell death was analyzed by trypan blue dye (Sigma Aldrich, Germany) on DLD-1 cell line. The cell culture flasks with 70% to 80% confluence were treated with IC90 dose of 4 for 24 h. The supernatant medium with floating cells was collected after treatment and centrifuged to collect the dead and apoptotic cells. The cell pellet was resuspended in serum free media. Equal amounts of cell suspension and trypan blue were mixed and this was analyzed under a microscope. The cells which were viable excluded the dye and were colorless while those whose cell membrane was destroyed were blue. If the proportion of colorless cells is higher than the colored cells, then the death can be characterized as apoptotic.
2.9.4.2 DNA fragmentation assay. Determination of apoptotic cell death was performed by DNA gel electrophoresis. Briefly, DLD-1 cells were treated with the respective IC90 dose of 4 for 24 h. Floating cells induced by drug exposure were collected, washed with PBS and lysed with lysis buffer (100 mM Tris-HCl pH 8.0; 20 mM EDTA; 0.8% SDS; all from Sigma Aldrich). Then they were treated with RNAse A at 37 °C for 2 h and proteinase K at 50 °C (both from Roche Diagnostics chemical company, Mannheim, Germany). DNA laddering was observed by running the samples on 2% agarose gel followed by ethidium bromide (Sigma Aldrich) staining.
3. Results and discussion
3.1 Synthesis and spectroscopic studies
The ammonium salts [NHEt3][P-Gly] (1), [NHEt3][P-Ala] (2) (Scheme 1) and [NHEt3][BTC] (3) (Scheme 2) were prepared, in very high yields, by the reaction of N-phthaloylglycine (P-GlyH), N-phthaloyl-L-alanine (P-AlaH), or 1,2,4-benzenetricarboxylic 1,2-anhydride (BTCH) with triethylamine respectively. The NMR, mass and IR spectra showed that all the complexes, isolated as crystalline solids, were of high purity. In the 1H NMR spectra of 1–3, a set of three signals consisting in a triplet at ca. 1.3 ppm, a quadruplet at ca. 3.2 and a broad signal between 11 and 13 ppm corresponding to protons of the triethylammonium cation were observed. For each compound, signals corresponding to the different carboxylate anions were recorded. For 1 one singlet at 4.28 ppm corresponding to the –CH2– protons and two multiplets at 7.67 and 7.83 ppm assigned to the aromatic protons of the N-phthaloyl group were observed. In the case of 2, two multiplets (at 7.67 and 7.79 ppm) were also recorded for the phthaloyl group and a doublet at 1.56 and a quadruplet at 3.08 ppm were assigned for the CH–CH3 moiety of the N-phthaloyl-L-alanine. In the 1H NMR spectrum of compound 3, two doublets and one singlet between 7.9 and 8.7 ppm were assigned to the aromatic protons of the carboxylate, in addition to the signals corresponding to the cation. The 13C{1H} NMR spectra of 1–3 showed the expected signals. The IR spectra of the ionic ligands present strong bands in two different regions between 1650–1610 and 1390–1350 cm−1, which correspond to the asymmetric and symmetric vibrations, respectively, of the COO moiety. The difference between the asymmetric and symmetric vibrations of more than 200 cm−1 in all cases, indicates monodentate coordination of the carboxylate ligand,62 in addition to these signals, a typical band at ca. 3450 cm−1 assigned to the N–H vibration was observed in all the spectra.63 The ESI-MS spectra of 1–3 recorded in negative mode showed the peaks of the anionic part of these molecules, while recorded in positive mode, presented a peak corresponding to the triethylammonium cation. | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
The subsequent reaction of 1–3 with triphenyltin chloride (1:1) yielded the compounds [NHEt3][SnPh3Cl(P-Gly)] (4), [NHEt3][SnPh3Cl(P-Ala)] (5) (Scheme 1) and [NHEt3][SnPh3Cl(BTC)] (6) (Scheme 2), respectively.
The organotin(IV) complexes 4–6 were characterized by multinuclear NMR spectroscopy, mass spectrometry, IR spectroscopy and elemental analysis. In the 1H NMR spectra of all the compounds a set of two signals at ca. 7.3 and 7.7 ppm corresponding to the protons of the phenyl groups of the SnPh3Cl moiety, was observed in addition to the signals corresponding to the carboxylate ligands, which presented similar spectral patterns to those observed in the 1H NMR spectra of 1–3. 13C{1H} NMR spectra for 4–6 showed the expected signals for the phenyl groups, as well as the signals corresponding to the different carboxylate ligands. A typical signal at low field (ca. 170 ppm) was observed in all the spectra and assigned to the carbon atom of the carboxylate group. Analogously to the IR spectra of the starting carboxylate 1–3, the spectra of 4–6 present strong bands between 1630–1610 and 1390–1360 cm−1, which correspond to the asymmetric and symmetric vibrations, respectively, of the COO moiety. Again, the difference between the asymmetric and symmetric vibrations of more than 200 cm−1 in all cases, indicates monodentate coordination of the carboxylate ligand,62 as was also confirmed, in the case of complex 4, by single crystal X-ray diffraction studies (see section 3.2). In addition, medium absorptions corresponding to the Sn–O stretching mode of vibration appeared at ca. 455 cm−1. 4–6 were also characterized by ESI-MS observing, in all cases in the negative mode; the peaks corresponding to the anionic parts of 4–6 (see sections 2.2–2.7).
Stability of the synthesized complexes in d6-DMSO and d6-DMSO/D2O solutions has been followed by NMR spectroscopy. After several hours in dry d6-DMSO, the NMR spectra of the studied compounds suffered apparent no changes, indicating their relatively high stability. The analysis of the spectra of 4–6 in d6-DMSO/D2O show a very slow decomposition process which over 72 h leads to the formation of a mixture of tin-containing products which included Ph3SnOSnPh3, SnPh3OH, SnPh3L (L = P-Gly, P-Ala, BTC) and the free carboxylic acids, between other unidentified compounds.
3.2 Structural studies
Complex 4 (Fig. 2) crystallizes in the centrosymmetric monoclinic space group P21/n with four molecules in the unit cell. | ||
| Fig. 2 Molecular structure and atom-labeling scheme for 4 with thermal ellipsoids at 50% probability (hydrogen atoms, except at nitrogen, are omitted for clarity). | ||
The molecular structure of 4 reveals that the central tin atom is pentacoordinated; presenting a almost ideal trigonal bipyramidal geometry with the Cl(1)–Sn(1)–O(1) angle of 177.23(8)° and O(1)–Sn(1)–Cphenyl and Cl(1)–Sn(1)–Cphenyl close to 90°. The Sn–O bond lengths Sn(1)–O(1) 223.1(3) and Sn(1)–O(2) 333.8(3) pm are quite different indicating monodentate coordination to tin of the carboxylate ligand. Weak interionic interactions (O(2)⋯H(2N) ca. 168 pm) between the hydrogen atom of the ammonium cation and the non-coordinated oxygen of the anion were observed. The bond length N(2)–H(1N) yielded reasonable distances of ca. 98 pm. The most interesting structural parameter of this complex is the bond length Sn(1)–Cl(1) of 256.5(2) pm which is in agreement with other Sn–Cl bonds in zwitterionic complexes and about ca. 10 pm longer than the expected for a tryaryltin(IV) chloride derivative,64–68 indicating some repulsive effect of the carboxylate ligand on the Sn–Cl bond which elongates the distance between the two atoms. Selected bond lengths and angles for 4 are summarized in Table 2.
| 4 | |
| Sn(1)–O(1) | 223.1(3) |
| Sn(1)–O(2) | 333.8(3) |
| Sn(1)–Cl(1) | 256.5(2) |
| C(19)–O(1) | 125.8(5) |
| C(19)–O(2) | 123.4(6) |
| C(22)–O(3) | 119.5(5) |
| C(23)–O(4) | 120.4(6) |
| N(2)–H(1N) | 98(2) |
| O(2)⋯H(2N) | 168(2) |
| Cl(1)–Sn(1)–O(1) | 177.23(8) |
| Cl(1)–Sn(1)–O(2) | 141.45(9) |
| Cl(1)–Sn(1)–C(1) | 91.0(2) |
| Cl(1)–Sn(1)–C(7) | 92.7(1) |
| Cl(1)–Sn(1)–C(13) | 91.2(2) |
| O(1)–C(19)–O(2) | 125.3(5) |
3.3 In vitro studies
Triethylammonium salts of carboxylic acids were inactive against the investigated panel of tumor cell lines (IC50 >100 μM). In this way is also proved that neither hypothetic activity of the free thriethylammonium cation nor the uncoordinated carboxylato ligand have influence on the activity of the studied triphenyltin(IV) complexes. The IC50 values of the studied compounds and cisplatin are summarized in Table 3. The triphenyltin(IV) compounds showed a dose-dependent antiproliferative effect toward all the studied cancer cell lines (Fig. 3). Triphenyltin(IV) compounds present lower IC50 values than those of cisplatin. The difference in activity of compounds 4–6 and cisplatin presents a maximum of up to 50 times, in cisplatin resistant cell line DLD-1. A similar efficiency was found against anaplastic thyroid cancer cisplatin resistant cell line 8505C, where triphenyltin(IV) compounds are almost up to 40 times more active than cisplatin. These values become lower in cisplatin sensitive A2780, A253 and A549 cell lines where triphenyltin(IV) compounds are only between 3.2 and 14.8 times more cytotoxic than cisplatin.
 | ||
| Fig. 3 Representative graphs show survival of 8505C, A253, A549, A2780 and DLD-1 cells grown for 96 h in the presence of increasing concentrations of triphenyltin(IV) complexes 4–6. | ||
| Compound | IC50/μM | ||||
|---|---|---|---|---|---|
| 8505C | A253 | A549 | A2780 | DLD-1 | |
| 1–3 | >100 | >100 | >100 | >100 | >100 |
| 4 | 0.129 ± 0.004 | 0.093 ± 0.003 | 0.102 ± 0.004 | 0.121 ± 0.002 | 0.103 ± 0.004 |
| 5 | 0.179 ± 0.003 | 0.139 ± 0.005 | 0.152 ± 0.003 | 0.170 ± 0.002 | 0.165 ± 0.003 |
| 6 | 0.241 ± 0.058 | 0.238 ± 0.002 | 0.236 ± 0.011 | 0.130 ± 0.003 | 0.210 ± 0.006 |
| cisplatin | 5.02 ± 0.23 | 0.81 ± 0.02 | 1.51 ± 0.02 | 0.55 ± 0.03 | 5.14 ± 0.12 |
Triphenyltin(IV) complexes 4 and 5 bearing very similar moieties showed notable differences in the cytotoxic activity. From those results one can envisage that small structural changes, (replacement of H with Me group (4 to give 5)) led to a significant decrease of the final cytotoxic activity. On the other hand, triphenyltin(IV) compound substitution of the phthaloyl aminoacids by 1,2,4-benzenetricarboxylic-1,2-anhydride (6) led to further decreases in the cytotoxicity, except in A2780 cell line, when compared with 4 and 5. Thus, one can think that the incorporation of biocompatible phthaloyl aminoacids as carboxylato ligands may have a positive influence in the final cytotoxicity of the tin(IV) complexes when compared with other different carboxylato ligands.
3.3.2.1 Dye exclusion test and DNA laddering. Apoptosis and cell mediated cytotoxicity are characterized by a fragmentation of the genomic DNA. These DNA fragments have a length of about 180 base pairs or multiples of this number (360, 540, 720, …), this is, the characteristic DNA-length of a nucleosome (DNA-histone-complex). Endonucleases cleave selectively DNA at sites located between nucleosomal units (linker DNA). In agarose gel electrophoresis of these, DNA fragments are resolved to a distinctive ladder pattern. To test whether complex 4 induced cell death mediated by apoptosis, floating cells from DLD-1 colon carcinoma cell line after 24 h treatment with the IC90 concentration were collected and analyzed by DNA laddering technique. In DLD-1 cell line with triphenyltin(IV) compound 4, typical DNA laddering was observed (Fig. 4). Furthermore, DLD-1 cells were exposed to IC90 dose of complex 4, the ability to exclude trypan blue and detached cells were investigated. The treatment with IC90 dose of 4 resulted in apoptotic cell death, in which floating cells showed the ability to exclude the blue dye (Fig. 4).

3.3.2.2 Effect of 4 on the caspase 2, 3, 8 and 9 activities. In order to gain insights into the induced apoptosis mechanism by the most active triphenyltin(IV) complex 4, we analyzed whether caspases were involved as downstream effectors in the induced cell death. The upstream caspases 2, 8 and 9, and the downstream caspase 3 were used for the present study. DLD-1 colon carcinoma cell line was chosen for investigation of the activity of caspases 2, 3, 8 and 9. When treated with compound 4 for 2 h, only caspase 8 was found to be upregulated. Similarly to that, when cisplatin was used for the same time period also activation of only caspase 8 was observed (Fig. 5). The cells treated for 6 h with 4 show activation of caspase 2 and 8 (apoptosis activator) as well as caspase 3 (apoptosis executioner). On the other hand, treatment for 6 h with cisplatin showed activation of caspase 8 and 9 (apoptosis activators). As shown here, complex 4 is activating apoptosis faster than cisplatin within 6 h of action on DLD-1 cell line. Furthermore, complex 4 and cisplatin seem to express a different apoptosis pathway on DLD-1 cell line. In the case of cisplatin, apoptosis is triggered by both internal (intrinsic or mitochondrial pathway, caspase 9 dependent pathway) and external signals (extrinsic or death receptor pathway), while for triphenyltin(IV) complex 4 apoptosis is induced via extrinsic receptor pathway on DLD-1 cell line.
 | ||
| Fig. 5 Activity of caspases 2 (C2), 3 (C3), 8 (C8) and 9 (C9) on DLD-1 colon cancer cell line treated for 2 and 6 h with 4 and cisplatin. | ||
3.3.2.3 Cell cycle perturbations. Cell cycle perturbations were analyzed on colon carcinoma DLD-1 cell line. The cells were treated with IC90 concentration of 4 for 24 h (Fig. 6). When compared to control, compound 4 caused almost no changes S phases, however, a decrease at around 46 and 40% in the number of cells in G1 and G2/M phases, respectively, with an increase in the number of apoptotic cells (SubG1-peak), was observed. Those results indicate that apoptosis caused by 4 on colon carcinoma DLD-1 cell line may be due to disturbances caused in both G1 and G2/M phases in the cell cycle.
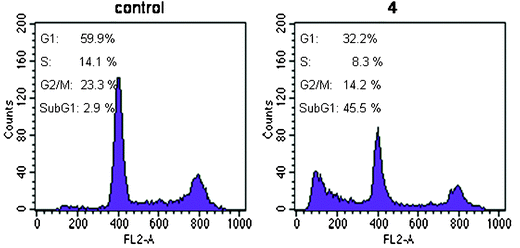 | ||
| Fig. 6 Cell cycle analysis of DLD-1 cells untreated (control) and treated with the IC90 concentration of 4 for 24 h. | ||
4. Conclusions and outlook
Novel triethylammonium salts of triphenyltin(IV) chloride carboxylate complexes have been synthesized and structurally characterized. The complexes were tested for in vitro antiproliferative activity against five cell lines of different histogenic origin. The triphenyltin(IV) complexes exhibited much higher cytotoxicity than cisplatin, up to 50 times (4). Although it seems that the active species of these mixtures beside complexes 4–6 may be SnPh3+ cations, based on the stability studies carried out for these compounds in aqueous solutions. In addition, a positive influence in the final cytotoxicity of the tin(IV) complexes has been observed with the incorporation of biocompatible phthaloyl aminoacids as ligands when compared with other different carboxylato ligands.Complex 4 seems to induce apoptosis via extrinsic or death receptor pathway. The presented data clearly demonstrate a cell-cycle dependence of triphenyltin(IV) compound 4 which induced cell death in DLD-1 cell line. Following compound 4 exposure, cells undergo G1 and G2/M arrest and apoptosis is finally induced in these phases of the cell cycle. Thus, while the phases affected by the tin compounds are G1 (when organelles are being synthesized resulting in a great amount of protein synthesis and a high metabolic rate in the cell) and G2/M (when the protein kinase plays a quite important role), the action mechanism of these compounds seem to be related either to the interaction of SnPh3+ moieties with protein kinases and DNA, as previously reported for other neutral triphenyltin(IV) carboxylates,26,33 or to the possible binding to the phosphate groups in DNA,69–71 changing the intracellular metabolism of the phospholipids of the endoplasmic reticulum.72,73 However, further studies, already in progress in our laboratories, will try to clarify this fact.
Therefore, complex 4 seems to be a very promising candidate for further in vivo tests which will be carried in the near future.
Acknowledgements
We gratefully acknowledge financial support from the Ministerio de Educación y Ciencia, Spain (Grant no. CTQ2008-05892/BQU) the Universidad Rey Juan Carlos and Comunidad de Madrid (postdoctoral fellowship for S.G-R). We would also like to thank Ministerium für Wirtschaft und Arbeit des Landes Sachsen-Anhalt, Deutschland (Grant No. 6003368706) and BioSolutions Halle for cell culture facilities.References
- R. H. Fish and G. Jaouen, Organometallics, 2003, 22, 2166–2177 CrossRef CAS.
- M. Nikolini, Platinum and Other Coordination Compounds in Cancer Chemotherapy, ed. Martinus-Nijhoff Publishing, Boston, MA, 1988 Search PubMed.
- I. Ott and R. Gust, Arch. Pharm., 2007, 340, 117–126 CrossRef CAS.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- M. Gielen, Coord. Chem. Rev., 1996, 151, 41–51 CrossRef CAS.
- P. Yang and M. Guo, Coord. Chem. Rev., 1999, 185–186, 189–211 CrossRef CAS.
- S. K. Hadjikakou and N. Hadjiliadis, Coord. Chem. Rev., 2009, 253, 235–249 CrossRef CAS.
- K. Strohfeldt and M. Tacke, Chem. Soc. Rev., 2008, 37, 1174–1187 RSC.
- P. M. Abeysinghe and M. M. Harding, Dalton Trans., 2007, 3474–3482 RSC.
- R. Gust, D. Posselt and K. Sommer, J. Med. Chem., 2004, 47, 5837–5846 CrossRef CAS.
- C. G. Hartinger and P. J. Dyson, Chem. Soc. Rev., 2009, 38, 391–401 RSC.
- P. C. A. Bruijnincx and P. J. Sadler, Adv. Inorg. Chem., 2009, 61, 1–62 CAS.
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS.
- Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764–4776 RSC.
- L. Nagy, A. Szorcsik and K. Kovacs, Pharm. Hungarica, 2000, 70, 53–71 Search PubMed.
- M. Nath, S. Pokharia and R. Yadav, Coord. Chem. Rev., 2001, 215, 99–149 CrossRef CAS.
- M. Gielen, Tin-Based Antitumor Drugs; NATO ASI Ser., Ser. 2, 1997, 26, 445–455 Search PubMed.
- D. De Vos, R. Willem, M. Gielen, K. E. Van Wingerden and K. Nooter, Met.-Based Drugs, 1998, 5, 179–188 Search PubMed.
- C. Pettinari, Main Group Met. Chem., 1999, 22, 661–692 CAS.
- S. P. Fricker, in Metal Compounds in Cancer Therapy, ed. Chapman & Hall, London, UK, 1994pp. 147–179 Search PubMed.
- M. Gielen, Main Group Met. Chem., 1994, 17, 1–8 CAS.
- M. G. Mirisola, A. Pellerito, T. Fiore, G. C. Stocco, A. Cestelli and I. Di Liegro, Appl. Organomet. Chem., 1997, 11, 499–511 CrossRef CAS.
- M. Kemmer, M. Gielen, M. Biesemans, D. de Vos and R. Willem, Met.-Based Drugs, 1998, 5, 189–196 Search PubMed.
- A. K. Saxena and F. Huber, Coord. Chem. Rev., 1989, 95, 109–123 CrossRef CAS.
- J. Susperregui, M. Bayle, G. Lain, C. Giroud, T. Baltz and G. Deleris, Eur. J. Med. Chem., 1999, 34, 617–623 CAS.
- L. Pellerito and L. Nagy, Coord. Chem. Rev., 2002, 224, 111–150 CrossRef CAS and references therein.
- M. Gielen and E. R. T. Tiekink, Metallotherapeutic Drugs and Metal-Based Diagnostic Agents: The Use of Metals in Medicine, ed. John Wiley & Sons, Ltd., 2005, p. 421 Search PubMed.
- T. S. Basu Baul, W. Rynjah, E. Rivarola, A. Lycka, M. Holcapek, R. Jirasko, D. de Vos, R. J. Butcher and A. Linden, J. Organomet. Chem., 2006, 691, 4850–4862 CrossRef.
- L. Tian, Y. Sun, H. Li, X. Zheng, Y. Cheng, X. Liu and B. Qian, J. Inorg. Biochem., 2005, 99, 1646–1652 CrossRef CAS.
- G. Han and P. Yang, J. Inorg. Biochem., 2002, 91, 230–236 CrossRef CAS.
- S. Gómez-Ruiz, G. N. Kaluđerović, S. Prashar, E. Hey-Hawkins, A. Erić, Ž. Žižak and Z. D. Juranić, J. Inorg. Biochem., 2008, 102, 2087–2096 CrossRef CAS.
- E. R. T. Tiekink, Trends Organomet. Chem., 1994, 1, 71–116 Search PubMed.
- M. L. Falcioni, M. Pellei and R. Gabbianelli, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 2008, 653, 57–62 CrossRef CAS and references therein.
- S. Gómez-Ruiz, G. N. Kaluđerović, D. Polo-Cerón, S. Prashar, M. Fajardo, Ž. Žižak, Z. D. Juranić and T. J. Sabo, Inorg. Chem. Commun., 2007, 10, 748–752 CrossRef.
- S. Gómez-Ruiz, G. N. Kaluđerović, S. Prashar, D. Polo-Cerón, M. Fajardo, Ž. Žižak, T. J. Sabo and Z. D. Juranić, J. Inorg. Biochem., 2008, 102, 1558–1570 CrossRef CAS.
- M. E. Kelly, A. Dietrich, S. Gómez-Ruiz, B. Kalinowski, G. N. Kaluđerović, T. Müller, R. Paschke, J. Schmidt, D. Steinborn, C. Wagner and H. Schmidt, Organometallics, 2008, 27, 4917–4927 CrossRef CAS.
- D. Pérez-Quintanilla, S. Gómez-Ruiz, Ž. Žižak, I. Sierra, S. Prashar, I. del Hierro, M. Fajardo, Z. D. Juranić and G. N. Kaluđerović, Chem.–Eur. J., 2009, 15, 5588–5597 CrossRef CAS.
- S. Gómez-Ruiz, B. Gallego, M. R. Kaluđerović, H. Kommera, E. Hey-Hawkins, R. Paschke and G. N. Kaluđerović, J. Organomet. Chem., 2009, 694, 2191–2197 CrossRef CAS.
- S. Gómez-Ruiz, B. Gallego, Ž. Žižak, E. Hey-Hawkins, Z. D. Juranić and G. N. Kaluđerović, Polyhedron, 2010, 29, 354–360 CrossRef CAS.
- S. Gómez-Ruiz, S. Prashar, T. Walther, M. Fajardo, D. Steinborn, R. Paschke and G. N. Kaluđerović, Polyhedron, 2010, 29, 16–23 CrossRef CAS.
- M. R. Kaluđerović, S. Gómez-Ruiz, B. Gallego, E. Hey-Hawkins, R. Paschke and G. N. Kaluđerović, Eur. J. Med. Chem., 2010, 45, 519–525 CrossRef CAS.
- G. N. Kaluđerović, D. Pérez-Quintanilla, I. Sierra, S. Prashar, I. del Hierro, Ž. Žižak, Z. D. Juranić, M. Fajardo and S. Gómez-Ruiz, J. Mater. Chem., 2010, 20, 806–814 RSC.
- G. N. Kaluđerović, D. Pérez-Quintanilla, Ž. Žižak, Z. D. Juranić and S. Gómez-Ruiz, Dalton Trans., 2010, 39, 2597 RSC.
- M. Ashfaq, J. Organomet. Chem., 2006, 691, 1803–1808 CrossRef CAS.
- S. Verma, A. Joshi, R. B. Gaur and R. R. Sharma, Main Group Met. Chem., 2005, 28, 185–192 CAS.
- M. K. Lo and S. W. Ng, Main Group Met. Chem., 2000, 23, 733–734.
- S. W. Ng and V. G. Kumar Das, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 546–548 CrossRef.
- S. W. Ng, A. J. Kuthubutheen, V. G. Kumar Das, A. Linden and E. R. T. Tiekink, Appl. Organomet. Chem., 1994, 8, 37–42 CrossRef CAS.
- N. W. Ahmad, S. A. Mohd, S. Balabaskaran and V. G. K. Das, Appl. Organomet. Chem., 1993, 7, 583–591 CrossRef CAS.
- V. I. Shcherbakov, E. F. Ivanov, S. Y. Khorshev, M. S. Feldman, E. B. Kopytov and T. N. Konkina, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 1991, 21, 1549–1567 Search PubMed.
- S. W. Ng, C. Wei, V. G. K. Das, R. J. Wang and T. C. W. Mak, J. Crystallogr. Spectrosc. Res., 1991, 21, 375–377 CrossRef CAS.
- V. I. Shcherbakov, I. P. Malysheva, O. N. Druzhkova and T. N. Chulkova, J. Organomet. Chem., 1991, 407, 181–189 CrossRef CAS.
- C. Syng-Ai, T. S. Basu Baul and A. Chatterijee, J. Environ. Pathol. Toxicol. Oncol., 2001, 20, 333–342 Search PubMed.
- F. Barbieri, M. Viale, F. Sparatore, G. Schettini, A. Favre, C. Bruzzo, F. Novelli and A. Alema, Anti-Cancer Drugs, 2002, 13, 599–604 CrossRef CAS.
- H. Seibert, S. Moerchel and M. Guelden, Cell Biol. Toxicol., 2004, 20, 273–283 CrossRef CAS.
- N. Hoti, J. Ma, S. Tabassum, Y. Wang and M. Wu, J. Biochem., 2003, 134, 521–528 CrossRef.
- SCALE3 ABSPACK: Empirical absorption correction, CrysAlis—Software package, Oxford Diffraction Ltd., 2006 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, Göttingen, 1997 Search PubMed.
- P. Skehan, R. Storeng, D. Scudiero, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS.
- A. Dietrich, T. Mueller, R. Paschke, B. Kalinowski, T. Behlendorf, F. Reipsch, A. Fruehauf, H. J. Schmoll, C. Kloft and W. Voigt, J. Med. Chem., 2008, 51, 5413–5422 CrossRef CAS.
- G. B. Deacon and R. J. Philips, Coord. Chem. Rev., 1980, 33, 227–250 CrossRef CAS.
- O. Knop, T. S. Cameron, M. A. James and M. Falk, Can. J. Chem., 1981, 59, 2550–2555 CrossRef CAS.
- M. C. Carcelli, C. Ferrari, C. Pelizzi, G. Pelizzi and G. Predieri, J. Chem. Soc., Dalton Trans., 1992, 2127–2128 RSC.
- M. Suzuki, I. H. Son and R. Noyori, Organometallics, 1990, 9, 3043–3053 CrossRef CAS.
- D. Dakternieks, T. S. Basu Baul, S. Duta and E. R. T. Tiekink, Organometallics, 1998, 17, 3058–3062 CrossRef.
- J. P. Charland, F. L. Lee, E. J. Gabe, L. E. Khoo and F. E. Smith, Inorg. Chim. Acta, 1987, 130, 55–60 CrossRef CAS.
- E. J. Gabe, F. L. Lee, L. E. Khoo and F. E. Smith, Inorg. Chim. Acta, 1985, 105, 103–106 CrossRef CAS.
- Q. Li, N. Jin, P. Yang, J. Wan, W. Wu and J. Wan, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 1997, 27, 811–823 Search PubMed.
- Q. Li, P. Yang, H. Wang and M. Guo, J. Inorg. Biochem., 1996, 64, 181–195 CrossRef CAS.
- J. S. Casas, E. E. Castellano, M. D. Couce, J. Ellena, A. Sanchez, J. L. Sanchez, J. Sordo and C. Taboada, Inorg. Chem., 2004, 43, 1957–1963 CrossRef CAS.
- Y. Arakawa, Biomed. Res. Trace Elem., 1993, 4, 129–130 CAS.
- Y. Arakawa, Biomed. Res. Trace Elem., 2000, 11, 259–286 CAS.
Footnote |
| † CCDC reference number 759465. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c0mt00007h |
| This journal is © The Royal Society of Chemistry 2010 |