Bioinformatics in bioinorganic chemistry†
Ivano
Bertini
*ab and
Gabriele
Cavallaro
a
aMagnetic Resonance Center (CERM) – University of Florence, Via L. Sacconi 6, 50019 Sesto Fiorentino, Italy. E-mail: bertini@cerm.unifi.it; Fax: +39 055 4574271; Tel: +39 055 4574272
bDepartment of Chemistry – University of Florence, Via della Lastruccia 3, 50019 Sesto Fiorentino, Italy
First published on 29th September 2009
Abstract
Bioinformatics is a central discipline in modern life sciences aimed at describing the complex properties of living organisms starting from large-scale data sets of cellular constituents such as genes and proteins. In order for this wealth of information to provide useful biological knowledge, databases and software tools for data collection, analysis and interpretation need to be developed. In this paper, we review recent advances in the design and implementation of bioinformatics resources devoted to the study of metals in biological systems, a research field traditionally at the heart of bioinorganic chemistry. We show how metalloproteomes can be extracted from genome sequences, how structural properties can be related to function, how databases can be implemented, and how hints on interactions can be obtained from bioinformatics.
 Ivano Bertini | Ivano Bertini is Professor of General and Inorganic Chemistry at the University of Florence and is Director of the Magnetic Resonance Center (CERM). He has received several honors, among which are three Laurea Honoris Causa (from the universities of Stockholm, Ioannina and Siena). He is a member of the Academia Europaea and the Italian Accademia dei Lincei, and is, or has been, on the editorial staff or advisory board of over 20 of the most authoritative chemistry and biochemistry journals. Since 1975 he has studied the structure–function relationships of metalloproteins through biophysical methods. In 1990, he created an NMR lab for structural biology of metalloproteins, and eventually pioneered the exploitation of genome data banks. He has pursued advancements in technology for solution structure determination and developed specific software applications. He has also established a molecular biology department for high-throughput protein expression in structural genomics projects on metalloproteins. He has published over 600 research articles and has solved more than 100 protein structures. In 1999 he founded the CERM in an independent and prestigious building hosting an impressive battery of NMR spectrometers. The Center constitutes a major NMR infrastructure in the Life Sciences. |
 Gabriele Cavallaro | Gabriele Cavallaro was born in Florence in 1973. He graduated in chemistry from the University of Florence (Italy), where he received his PhD in structural biology in 2004. He is now a postdoctoral fellow at the Magnetic Resonance Center (CERM) in Florence. His research interests include several areas of computational biology ranging from molecular dynamics simulations to protein structure calculation. At present, he is mainly involved in the development of bioinformatics tools for sequence and structure-based analysis of metalloproteins, and is collaborating with the European Bioinformatics Institute (EBI) to annotate and classify metal-binding sites in proteins. |
Introduction
Bioinorganic chemistry has the aim of investigating inorganic elements in living organisms.1,2 In recent years, the development of high-throughput technologies capable of providing large-scale data sets for several cellular biomolecules (e.g., genomics , proteomics) has renewed the idea that biological systems should be studied as a whole to understand their complex properties.3–5 The possibility of having virtually complete lists of the key molecular components of cells ignited an explosion of efforts to understand how these components interact in space and time, and how these complex interactions result in the concerted set of biological processes which sustain life.6 In this new scenario, therefore, bioinorganic chemistry must also be placed in a system-wide perspective.7 In the first place, this requires that comprehensive data sets similar to those produced, e.g., by genomics and proteomics are constructed for the cellular components relevant to bioinorganic chemistry: the emerging areas of metallomics and metalloproteomics do address this issue by attempting to identify all metal species and all metalloproteins, respectively, in a cell.8,9 These data sets must then be annotated with functional information in order for metal-binding molecules to be integrated into the framework of biological networks. The physiological functions of metalloproteins, in particular, crucially depend on binding to specific metals, which can play different roles including structural (metals required for the protein to correctly fold), catalytic (metals required for the catalytic activity of an enzyme), and regulatory (metals whose binding to the protein modulates its functionality). Furthermore, appropriately structured databases must be developed to allow efficient data storage, access and analysis. It is apparent that the accomplishment of all these tasks will strongly rely on bioinformatics, which is essential to manage, interpret and make sense of large amounts of experimental data, and can be very helpful in complementing such data with predictions.10,11In this review, we describe some bioinformatics approaches tailored for application in the various stages of the study of metals in biological systems, as outlined above. We focus on metalloproteins rather than on metal species in general, because data sets of metalloproteins are most suitable to be integrated with other types of biological information, which are mostly centred on proteins. First, we illustrate how bioinformatics can be used to identify, on the basis of sequence alone, which proteins of a given organism are metalloproteins, thus providing a prediction of its metalloproteome. Second, we show how bioinformatics can exploit the available protein structure information for more reliable identification and classification, including functional inference, of metalloproteins. Third, we describe actual and potential bioinformatics resources designed to contain and organize chemical and biological information on metalloproteins. Finally, we discuss how metalloproteomics data can be combined with other genome -scale data sets into models of various complexity, with the ultimate perspective of obtaining an overall picture of metals in living organisms.
Tools for predicting metalloproteomes
Whole-genome sequencing projects are revealing the full set of genes of an ever-increasing number of organisms, at a rate that far exceeds the capability to characterize experimentally the products of these genes. Therefore, a broad variety of computational approaches have been developed to infer functional information from the amino acid sequences of proteins derived from the translation of nucleotide sequences.12–15 In particular, predicting whether a protein binds a metal, and which metal it binds, can provide important insights into its biological role. Furthermore, performing this kind of prediction for all the proteins encoded by the genome of an organism allows one to estimate the metalloproteome of that organism in a metal-specific manner. The computational identification of metalloproteomes is of special value in that the major experimental techniques used in metalloproteomics, including mass spectrometry approaches such as ESI-MS and ICP-MS and synchrotron-based approaches such as XAS and XRF, are not yet routinely available.9In the last few years, our group has developed a bioinformatics method, applicable on a whole-genome scale, to predict the metal-binding properties of a protein from its amino acid sequence.16–20 The method, which is schematically depicted in Fig. 1, is based on the recognition of two different signatures diagnostic for metal binding, which are used in combination. Specifically, the protein sequence under consideration is analysed for the presence of (i) metal-binding domains, and (ii) metal-binding patterns, as detailed below.
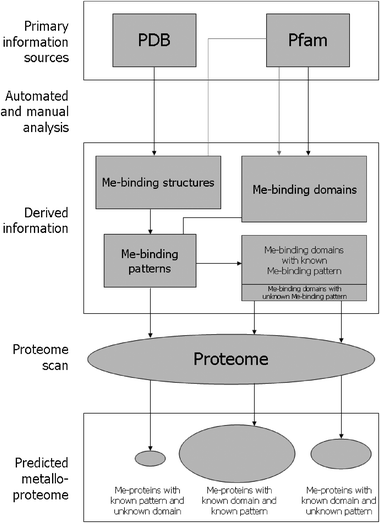 | ||
| Fig. 1 Scheme depicting our method for the identification of metalloproteins from amino acid sequences (Me = any metal). Primary information sources for protein structures (PDB) and domains (Pfam) are examined by a mixture of manual and automated analysis to build libraries of metal-binding patterns (in the form of regular expressions) and metal-binding domains (in the form of profile HMMs). On average, about 80% of the latter have a pattern associated with them. The complete predicted proteome of an organism is then automatically scanned for protein sequences containing metal-binding domains (using HMMER) or metal-binding patterns (using flexible pattern matching). Protein sequences containing a metal-binding domain but lacking the associated pattern (if available) are filtered out. The relative proportion of metalloproteins detected on average by the identification of both domains and patterns (about 75%), of domains only (about 20%), and of patterns only (about 5%) is shown by the size of each circle. | ||
The identification of conserved domains is a well-established approach for the classification of protein sequences, which are scanned for matches against libraries of known domains.21 A widely used resource for this purpose is the Pfam database,22 a comprehensive collection of protein domains represented as profile hidden Markov models (HMMs)23 which can be analyzed for similarity to a query sequence using the HMMER software package.24 The assignment of the query sequence to a specific domain depends on whether the similarity score calculated by HMMER, usually expressed as an E-value, exceeds an appropriate threshold (e.g., E < 0.001). In order for this approach to be used for predicting whether a protein binds a given metal, therefore, it is necessary to construct a library of all Pfam domains that bind that metal. In our method, such metal-specific libraries are constructed by querying Pfam for those domains whose annotation contains the name and/or the symbol of the metal, and then checking the literature to filter out incorrectly extracted domains. Additionally, domains that have been structurally characterized as metal-binding but are not annotated as such in Pfam (and thus cannot be detected as above) are also included. These domains are identified by retrieving from the Protein Data Bank (PDB) all protein structures that bind the metal of interest, and then using HMMER to compare their sequences against the Pfam database. Pfam domains recovered by this procedure typically represent a small, but not negligible fraction of the metal-specific library: in the case of zinc-binding domains, for example, they were found to be 25 out of a total of 314 (i.e., approximately 8%).18 It is important to note that the collection of metal-binding PDB structures also requires analysis of the literature, so as to discard the proteins bound to non-physiological metals (e.g., due to adventitious binding during crystallization or purification procedures). Therefore, when the approach is applied for the first time to a given metal, the construction of the domain library turns out to be the most time-consuming stage of the entire protocol, requiring manual curation by experts in bioinorganic chemistry. In subsequent implementations, on the other hand, libraries just need to be updated to reflect changes in Pfam and in the PDB, and much less human intervention is needed. It is worth noting that in some cases literature analysis may not lead to definite conclusions about the metal-binding properties of a given domain, particularly in the absence of relevant structural information. For example, biochemical studies25 indicated that CHCH (standing for coiled coil-helix-coiled coil-helix) domains can bind copper by two conserved Cys residues which, however, appear to serve a purely structural role in a recently determined CHCH structure,26 suggesting that CHCH domains may not generally bind copper. Therefore, updates of domain libraries may involve not only the addition of previously uncharacterized metal-binding domains, but also the removal of domains whose metal-binding properties are proven incorrect by new evidence.
With a library of metal-binding Pfam domains available, HMMER can be used in a systematic manner on large protein sequence data sets, such as the complete predicted proteome of an organism, to select those containing at least one such domain. The selected proteins would thus represent the estimated metalloproteome of the organism. However, there is a major problem in predicting the metal-binding capability of a protein based solely on the fact that it contains a domain which is commonly associated with a certain metal. In fact, since this capability depends on the presence in the domain of a metal-binding site formed by a (small) number of residues in the proper spatial arrangement, evolutionary changes affecting these residues (e.g., non-conservative mutations) may cause the domain to lose its metal-binding properties while retaining its overall structure and sequence features. This is the case, for instance, of certain members of the ADAMs family of proteins27 which, despite containing a conserved protease domain implicated in zinc-dependent catalysis, lack the His residues responsible for zinc binding and are thus unlikely to be proteolytically active.28 Therefore, using the occurrence of metal-binding domains as the only criterion to identify metalloproteins may lead to a substantial rate of false positives, i.e., of proteins incorrectly predicted to be metalloproteins. For this reason, it is necessary to supplement this criterion with additional requirements, which can reduce the number of false positives. In our method, such additional restraints are provided by metal-binding patterns.
The identification of conserved patterns is another common, and possibly the simplest, approach for the classification of protein sequences. It makes use of regular expressions to represent motifs of amino acid residues which correspond to functionally important, and thus highly conserved, regions of proteins, such as catalytic or binding sites. Regular expressions specify which residues may or may not occur at each position of the pattern: the motif characterizing the zinc-binding members of the abovementioned ADAMs family of proteins, for instance, is represented as HEXXHXXGXXH, where amino acids are given as one-letter codes, and X indicates any amino acid.29 Simple string matching algorithms can then be used to determine whether a protein sequence contains a given pattern or not. Similarly to what is described above for metal-binding domains, constructing a library of metal-binding patterns is the first step to take to exploit this approach in the prediction of metalloproteins. In our method, this library is built starting from the PDB, taking advantage of the curated selection of metal-binding structures performed to enrich the library of metal-binding domains (see above). Metal-binding patterns are automatically derived from PDB structures by mapping the metal ligands onto the protein sequence, and are expressed in the general form AXnBXmC…, where A, B, C… are the metal-binding amino acids and n, m… are the number of amino acids in between two subsequent ligands. Importantly, the extraction of metal-binding patterns from the same PDB structures that were assigned to Pfam domains allows us to associate each pattern with the domain in which it is found. As a result, all the metal-binding domains for which at least one representative with known structure is available have one metal-binding pattern (or more, if different patterns are found in different structures of the same domain) associated with them. With this information, it is then possible to filter the metalloproteins predicted on the basis of metal-binding domains by requiring that the domains identified by HMMER contain at least one of their associated metal-binding patterns. When performing pattern matching, we allow the distance in sequence between two subsequent metal ligands to vary within ±20% (or ±1 amino acid for distances less than five residues) to take into account the observation that there is little evolutionary pressure to maintain a specific sequence spacing between two metal ligands when the spacing is large (i.e., of the order of tens of residues).16 The addition of the pattern-based selection criterion dramatically improves the quality of the predicted metalloproteomes, by eliminating false positives such as the abovementioned ADAM proteins lacking the zinc-binding site. In the case of non-heme iron, for instance, the precision of the results was improved by 30% with respect to the sole domain-based prediction.19
In our method, metal-binding patterns are used not only to refine the results obtained from the recognition of metal-binding domains, but also to identify possible further metalloproteins whose sequences match one of the patterns but which do not contain any known Pfam domain, and thus cannot be detected by domain searches. The rationale behind this supplementary search is that the same metal-binding site can occur as a common element in a broad variety of proteins (e.g., due to convergent evolution),30 including proteins whose structure (and thus sequence) is unrelated to any known domain. On the other hand, however, the occurrence of a metal-binding pattern in the sequence of a protein whose structure is different from that from which the pattern was extracted may not correspond to an actual metal-binding site in three-dimensional space (see Fig. 2 for example). Therefore, simply looking for metal-binding patterns in protein sequences with no similarity to known domains can generate a high proportion of false positives. To discern such false positives, we use a parameter which accounts for the degree of local sequence similarity around the metal-binding pattern between the known metalloprotein (from which the pattern was extracted) and that predicted (where the pattern was retrieved), which are aligned using the PHI-BLAST program.31 The presence of local sequence similarity is in fact indicative of local structural similarity, implying similar conformation for the metal ligands and thus formation of the metal-binding site.32 This parameter is called IdGlobal, and is defined as the ratio between the number of amino acids aligned by PHI-BLAST and the entire sequence length of the known metalloprotein. When IdGlobal is higher than 0.2, metalloproteins are predicted with a level of confidence of over 99%, dropping to about 50% when IdGlobal is between 0.2 and 0.1, and to about 25% when it is below 0.1.16 In genome -wide predictions of metalloproteins, we select sequences with IdGlobal > 0.2, typically contributing an additional 5% to the size of the predicted metalloproteomes.19
 | ||
| Fig. 2 Example of a metal-binding pattern which does not correspond to an actual metal-binding site in a protein whose structure is different from that from which the pattern was extracted. The CX2CX11CX13H pattern is associated with a zinc-binding site in the RING finger protein Rbx1 (PDB code 1ldj,129 left), but is not so in the Herpes virus entry mediator HveA (PDB code 1jma,130 right). | ||
To summarize, at the conclusion of the above procedure a prediction of the metalloproteome of an organism is obtained which consists of three subsets: one includes metalloproteins predicted by the identification of both a metal-binding domain and a metal-binding pattern, one includes metalloproteins predicted by the identification of only a metal-binding domain, and one includes metalloproteins predicted by the identification of only a metal-binding pattern. The first subset has the highest degree of confidence, fulfilling both criteria used to recognize metal-binding capabilities. The second subset follows from the fact that not all Pfam domains have a representative structure in the PDB, precluding the possibility of defining metal-binding patterns to be used as a selection criterion. From what is described above, this subset (whose size will decrease with time as more structures become available) is expected to overestimate the number of genuine metalloproteins. The size of this overestimation can be evaluated by assuming that the fraction of false positives in this subset is the same as that observed for the pattern-filtered subset. On this assumption, it can be approximated as the product of the percentage of metalloproteins filtered out by the pattern filter times the percentage contribution of the non-filtered subset to the total metalloproteome. As shown in Table 1, such percentages are different for different metals, but the overestimation is expected to be less than 5% in all cases. Table 1 also reports the estimated loss of genuine metalloproteins which would result from entirely discarding the non-filtered subset. Although this loss can be as high as 28%, such a conservative choice may be more appropriate for applications in which only high-confidence predictions are needed. Finally, the third subset follows from the fact that the space of protein sequences is not yet fully covered by Pfam domains,33 but this limitation can be overcome, at least in part, by attempting to recognize sequence regions (i.e., those corresponding to the metal-binding site) which are smaller than whole domains. The adoption of the IdGlobal parameter ensures a confidence level close to 100% for this subset, although the choice of a discrimination threshold necessarily involves discarding a number of genuine metalloproteins. The impact of this underestimation on the total metalloproteome is expected to be less than 2%, given that the fraction of true metalloproteins when IdGlobal is lower than the threshold is about 37%,16 and this subset, as mentioned above, represents around 5% of the predicted metalloproteome.
| Metal | Percentage of proteins containing a metal-binding domain that were filtered out because they lack a metal-binding pattern | Percentage of metalloproteins predicted by the identification of metal-binding domains only | Metalloproteome overestimation due to the inclusion of metalloproteins predicted by the identification of metal-binding domains only | Metalloproteome underestimation due to the exclusion of metalloproteins predicted by the identification of metal-binding domains only |
|---|---|---|---|---|
| Zinc | 40% | 10% | 4% | 6% |
| Iron | 30% | 15% | 5% | 11% |
| Copper | 10% | 30% | 3% | 27% |
| Resource | Type of resource | Web address |
|---|---|---|
| CATH | Database of hierarchically classified protein structures | http://www.cathdb.info/ |
| DALI | Web server for protein structure comparison | http://ekhidna.biocenter.helsinki.fi/dali_server/ |
| DIP | Database of experimentally determined protein–protein interactions | http://dip.doe-mbi.ucla.edu/dip/Main.cgi |
| HMMER | Tool for identifying protein sequence similarity based on profile HMMs | http://hmmer.janelia.org/ |
| IntAct | Database of known and predicted protein–protein interactions | http://www.ebi.ac.uk/intact/main.xhtml |
| Integr8 | Database of integrated information about deciphered genomes and their corresponding proteomes | http://www.ebi.ac.uk/integr8/EBI-Integr8-HomePage.do |
| InterPro | Integrated documentation resource for protein families, domains, regions and sites | http://www.ebi.ac.uk/interpro/ |
| JESS | Tool for searching structural motifs in protein structures | Available from the authors64 |
| MDB | Database of metal-binding sites in protein structures | http://metallo.scripps.edu/ |
| Metal-MACiE | Database of information on the catalytic mechanisms of metal-dependent enzymes | http://www.ebi.ac.uk/thornton-srv/databases/Metal_MACiE/home.html |
| MINT | Database of experimentally determined protein–protein interactions | http://mint.bio.uniroma2.it/mint/Welcome.do |
| PDB | Database of experimentally determined structures of proteins, nucleic acids and complex assemblies | http://www.rcsb.org/pdb/home/home.do |
| PDBeMotif | Integrated database and analysis tool of structural motifs in protein structures | http://www.ebi.ac.uk/pdbe-site/pdbemotif/ |
| PDBSiteScan | Web server for searching structural motifs in protein structures | http://wwwmgs.bionet.nsc.ru/mgs/gnw/pdbsitescan/ |
| Pfam | Database of protein domains, represented by multiple sequence alignments and HMMs | http://pfam.sanger.ac.uk/ |
| PHI-BLAST | Web server for searching protein databases for proteins that contain a specific pattern and are similar to the query sequence in the vicinity of the pattern | http://blast.ncbi.nlm.nih.gov/Blast.cgi |
| PINTS | Web server for searching structural motifs in protein structures | http://www.russell.embl.de/pints/ |
| ProFunc | Web server for structure-based prediction of protein function | http://www.ebi.ac.uk/thornton-srv/databases/ProFunc/ |
| PROMISE | Database of literature-based annotation of metalloproteins | http://metallo.scripps.edu/PROMISE/ |
| SCOP | Database of hierarchically classified protein structures | http://scop.mrc-lmb.cam.ac.uk/scop/ |
| SSM | Web server for protein structure comparison | http://www.ebi.ac.uk/msd-srv/ssm/ |
| STRING | Database of known and predicted protein–protein interactions | http://string-db.org/ |
The most obvious limitation of our method is that it cannot detect metalloproteins which do not contain either a known metal-binding domain or a known metal-binding pattern. In a way, this can be seen as a general limitation of predictive computational approaches, all of which ultimately depend on the available knowledge. However, the number of structurally novel metalloproteins being discovered each year, which may extend the knowledge on which our method is based, is progressively reducing, suggesting that future predictions will bring about relatively small corrections to those made today.32 An alternative means to tackle the challenge of identifying metalloproteins, which in principle can detect also those with unknown domains and patterns, is provided by machine learning techniques such as neural networks and support vector machines (SVMs).34–36 These methods do not, in fact, escape the rule that what can be predicted depends on what is known, but are often referred to as ‘de novo’ methods because they are able to find non-trivial correlations between sequence features and protein properties, extrapolating regularities which cannot be encoded in profiles or regular expressions. A number of machine learning approaches to predict metalloproteins have been developed in the last few years,37–40 sometimes coupled to standard similarity-based predictions,41 and have been shown to be indeed able to identify unprecedented metal-binding sites.39,40 Other attempts have also been reported based on the identification of metal-binding domains alone,42 as well as of metal-binding patterns alone.43 It is likely, however, that such predictions based on the recognition of a single signature may suffer from the limitations described here, which need to be overcome by the integration of independent, complementary tools.
Tools to exploit PDB to characterize metalloproteins
In the previous section, we illustrated how methods that predict the metal-binding properties of a protein from its amino acid sequence can be used to obtain genome -scale, metal-specific data sets of metalloproteins. As mentioned in the Introduction, for these data to be exploited in the context of biological studies, it is necessary to enrich them with functional information. Given the size of the data sets involved, this means in practice that descriptive annotation must be inferred through automated computational approaches. Computational tools can provide insights into the function of proteins based on various sources of information besides the mere amino acid sequence, including for instance protein structure,44,45 genomic context,46,47 and protein–protein interactions.48,49 In the past few years, for example, a combination of sequence and genomic context analyses was used to investigate the biological roles of metalloproteins such as cytochrome c50 and Sco.51 Here, we focus on methods to characterize metalloproteinsin silico that rely on three-dimensional structural information.It is well known that the function of proteins is closely associated with their specific tertiary structures and, as a consequence, structural knowledge is of the utmost importance to understand protein function in detail. This statement is particularly relevant in the case of metalloproteins, where the structural and chemical features of the protein environment around the metal-binding site can finely modulate the functionality of the bound metal, which in turn is often responsible for protein function (e.g., in metalloenzyme catalysis).52 Furthermore, functional similarity can be inferred from structural similarity even when sequence similarity is undetectable, because proteins with a common evolutionary origin can maintain similar folds and functions over time even though their sequences diverge to a point where their relatedness cannot be recognized.53 For these reasons, the methods for protein function prediction based on structural information are generally more powerful than those based on sequence alone.14
Not unlike their sequence-based counterparts, structure-based approaches for predicting protein function typically rely on detecting structural similarity, global or local, between the protein under consideration and a protein of known function.54 Global similarity searches involve the use of structure alignment tools such as SSM55 and DALI56 to compare the query structure against the PDB, or against databases like CATH57 and SCOP58 where PDB structures are classified into groups depending on structural and functional similarity. The functional clues provided by these searches, however, may be of limited significance in some cases, because certain folds occur very commonly in nature and are shared by proteins carrying out a wide variety of different functions.59,60 Therefore, even proteins with very similar global structures can have very different functions as the result of local differences in functionally important regions such as catalytic or binding sites. On the other hand, convergent evolution can produce similar functional sites in proteins with unrelated folds, thus causing proteins with very different global structures to perform similar functions.61 For these reasons, it is convenient to combine global similarity searches with local ones, in which the query structure is compared against data sets of local structural motifs associated with known functional sites. In these comparisons, structural motifs are typically represented as three-dimensional templates containing the spatial coordinates of the residues that form the site (e.g., the Ser-His-Asp catalytic triad of serine proteases),62 which are matched against the query structure using tools such as PINTS63 or JESS.64 In the case of metalloproteins, in particular, local similarity searches can provide key functional hints by detecting known metal-binding sites with defined functions (e.g., electron transfer, oxygen binding). Therefore, in silico characterization of metalloproteins can especially benefit from the availability of metal-specific libraries of metal-binding structural motifs annotated with functional information.
We have recently developed a method, which is schematically depicted in Fig. 3, to build libraries of metal-binding motifs in a largely automated fashion.65 The key feature of these libraries is that metal-binding motifs are represented as three-dimensional templates that contain not only the metal ligands (i.e., the first coordination sphere of the metal) but also the residues interacting with the metal ligands (i.e., the second coordination sphere of the metal), as shown in Fig. 4. This choice reflects the notion that the second coordination sphere can contribute to modulate metal function,66,67 therefore it must be included in templates to properly describe the structural and chemical features of metal-binding sites.
 | ||
| Fig. 3 Scheme depicting our method for the construction of libraries of metal-binding structural motifs (Me = any metal). Primary information sources for protein structures (PDB), protein structure classification (CATH, SCOP) and function (literature) are examined by a mixture of manual and automated analysis to build libraries of metal-binding structural motifs in the form of three-dimensional templates containing the coordinates of the first and the second coordination sphere of the metal. The motifs are grouped according to CATH and SCOP classification, and annotated with respect to function. Protein structures can then be scanned for similarity to known metal-binding motifs using structural alignment tools (e.g., FAST)131 to obtain functional information. Also, the data set can be automatically analysed to obtain information on various characteristics (e.g., amino acid composition) of metalloproteins binding a given metal. | ||
 | ||
| Fig. 4 Example of a three-dimensional template that includes the first and the second coordination sphere of the metal to represent a metal-binding site. The iron site in phenylalanine hydroxylase (PDB code 1ltz)132 is described by the spatial coordinates of the iron ligands (shown as red sticks) plus those of the residues interacting with such ligands (magenta sticks). Iron is shown as a yellow sphere. | ||
In our method, protein structures that physiologically bind the metal of interest are selected from the PDB in the same way described in the previous section (in fact, the exact same collection can be used), and the structural domains present in these proteins are identified and classified according to the CATH and SCOP databases. Three-dimensional templates representing the metal-binding sites of these proteins are automatically generated from the PDB structures by extracting the spatial coordinates of the residues forming the first and the second coordination sphere of the metal, as defined above. The metal-binding sites (and thus the corresponding templates) are then associated with the structural domain in which they are found, by mapping the metal ligands onto the domains defined by CATH and SCOP. For example, the iron-binding protein desulfoferrodoxin68 contains an iron-binding site in the N-terminal domain, which is classified in SCOP as a rubredoxin-like domain, and an iron-binding site in the C-terminal domain, which is classified in SCOP as an immunoglobulin -like beta-sandwich domain. The N-terminal site is then associated with the rubredoxin-like domain, and the C-terminal site with the immunoglobulin -like beta-sandwich domain. Metal-binding sites found in the same structural domain are grouped together, allowing one to evaluate the redundancy and eventually adjust the size of the data set by selecting a number of representative sites from each group. The PDB is in fact highly redundant in that it may contain many structures of the same protein (e.g., determined under various conditions), as well as of highly similar proteins (e.g., mutants, homologues from closely related species). In the context of function prediction, the representative sites of a group must be selected so as to cover the range of different functions performed by the members of the group, i.e., by the metal-binding sites found in a given structural domain. Importantly, this grouping procedure also allows a substantial reduction in the time required to annotate the functions of metal-binding sites, because the annotation process can be performed on a per-group basis rather than for individual proteins. Still, literature analysis represents the most costly step in terms of manual effort, although (as discussed in the previous section for sequence-based libraries), it is much more time-consuming in the initial construction of the data set than in subsequent updates.
The result of the above procedure is a library, organized on a structural and functional basis, of metal-binding motifs represented by three-dimensional templates and annotated with functional information, against which protein structures can be compared to gain detailed insight into their metal-dependent functional properties. On a large scale, such comparisons can involve experimental protein structures produced by structural genomics projects,69 which are typically devoid of functional information, as well as protein structures predicted from amino acid sequences using homology modelling or fold recognition techniques.70 The extent to which complete proteomes can be analysed by this approach, therefore, is determined by the fraction of protein sequences in the proteome for which a reliable structural model can be obtained. Homology modelling can currently achieve about 65% structural coverage of whole proteomes, although this fraction can vary widely from organism to organism.71,72
In addition to serving as a reference for structural similarity searches, a library of metal-binding motifs designed as above lends itself to a number of analyses which can themselves provide useful information. For example, the templates included in the library can be compared against each other, so as to reveal common metal-binding motifs in structurally unrelated proteins, and highlight fine differences in the metal-binding sites of proteins with similar structures but different functions. This analysis can thus give interesting hints on the mechanism of action and the evolution of metalloproteins. Also, the library can be examined for the amino acid composition of metal-binding sites, yielding indications on which residues are important to modulate the properties and determine the specific functions of a metal. In a case study conducted on non-heme iron sites,65 we illustrated all the above applications of a library of metal-binding motifs constructed by our method and, in particular, we obtained functional hints for 14 of 15 iron proteins with unknown function, showing that local similarity searches based on these libraries can be advantageously used for function prediction of unannotated metalloproteins.
The fundamental concepts underlying our approach are common to the large majority of tools that have been developed to recognize metal-binding motifs in protein structures. Generally speaking, all such methods extract information on known metal-binding motifs from the PDB, encode this information in some form, and then scan protein structures for matches to the motifs. What differs among them is the type of information used to describe the metal-binding motifs and, consequently, the way protein structures are analysed for matches. The three-dimensional templates used in our approach, which are generated directly from PDB coordinates and can be compared to query structures by structural alignment, represent a straightforward, intuitive way to describe structural motifs. Analogous templates, which however contain only the residues that coordinate the metal, are employed to detect metal-binding sites in structure-based function prediction servers such as PDBSiteScan73 and ProFunc,74 and have also been used to investigate the evolution of zinc-binding and calcium-binding sites involved in protein structure stabilization.75
In other approaches, the structures of metal-binding sites are not specified by spatial coordinates, but are described abstractly in terms of various selected parameters, which collectively make up the “fingerprint” of a site. Relatively simple descriptions consider only geometrical parameters such as combinations of distances between atoms of metal-coordinating residues,76 whereas more complex descriptions also take into account features like secondary structure and solvent accessibility, as well as information not obtainable from a single structure such as the degree of conservation of metal-binding residues across homologues.77,78 A singular description, which has been used to identify magnesium-binding sites, involves the conversion of structures into sequences of letters drawn from a so-called structural alphabet based on the conformation of five-residue protein fragments.79 In a study aimed at detecting zinc-binding sites, no less than 43 parameters describing the physicochemical properties of the protein in six concentric shells around the metal were considered.80 These approaches typically exploit machine learning techniques because, as mentioned in the previous section, such techniques are able to infer complex rules from the analysis of many different parameters: in a nutshell, the combinations of parameters that characterize metal-binding sites are derived from the analysis of known metalloproteins, and query structures are then examined for the occurrence of such combinations. A conspicuous exception in this scenario is represented by the Fold-X method, in which the information derived from known metalloproteins is translated into empirical force field parameters, and metal-binding sites are detected by calculating energetically favourable positions for metals in protein structures.81 Some of the above approaches have also been shown to be effective when applied to structures obtained from homology modelling, suggesting that they may be used on a genome -wide scale.80,82
The need for specific databases
Comprehensive analyses of metalloproteins focusing on various aspects of metal-binding sites such as geometry and amino acid composition have been undertaken several times, even recently.67,83–87 The guiding premise of these analyses, which are primarily motivated by the rapidly growing number of structures available in the PDB, is that the systematic overview of all known metalloproteins can reveal the basic principles governing their structural and functional properties. General rules derived from these principles could then be used, e.g. to improve the prediction and characterization of metal-binding sites in proteins, to generate restraints for metal-binding sites in structure calculation programs, and to engineer metalloproteins with programmed features.67,84,87 Such broad surveys are typically carried out starting from primary sources of information such as the PDB that are not specifically devoted to metalloproteins, and thus require substantial efforts to process and organize the relevant knowledge contained in these sources. This holds also for the methods described in the previous two sections, whose application leads to accumulation of a wealth of information consisting not only of their end results (e.g., data sets of predicted metalloproteins), but also of the curated data sets constructed during their implementation (e.g., metal-specific libraries of Pfam domains). For this information to be fully exploited, it is crucial that it can be adequately accessed, shared and kept up-to-date: in a word, it must be stored in databases.Databases lie at the heart of modern genome -based biology, forming the infrastructure necessary for the collection, maintenance and provision of biological information. They are indispensable resources in an increasingly information-rich science, in which formidable amounts of data coming from different research fields need to be catalogued to be analysed and interpreted. Also, databases represent a long-term protection of the research efforts and investments made to generate the data, which due to their size can no longer be published in a conventional sense. Despite the large number of web-accessible data resources created over the last years, public databases devoted to metalloproteins have been surprisingly scarce.88 We have recently developed one such database, called Metal-MACiE, which aims to organize the available knowledge on the catalytic mechanisms of metalloenzymes .89 It presents a detailed description of the properties and the roles of metals involved in enzyme reactions, and has provided a basis for analysing the specific functions of different metals in relation to their individual chemical properties and availability in the environment.90 In the past, the MDB91 and the PROMISE92 databases were created with the aim of providing a comprehensive resource for metalloproteins. They were intended to serve as complementary databases, in that MDB provided quantitative information on the structural and chemical features of metal-binding sites retrieved from PDB structures, and PROMISE provided qualitative information on metalloproteins in the form of descriptive annotations derived from the literature. Unfortunately, both of them were discontinued some years ago. At present, a functionality similar to that of MDB is available through the PDBeMotif (formerly called MSDmotif)93 resource at the PDBe database (formerly called MSD),94 which can be used to interactively analyse several types of protein sites and motifs with known structure. However, PDBeMotif, as well as its predecessor MSDsite,95 is not a tool designed to investigate metalloproteins, and its use for this purpose is limited by its complexity and the lack of specific information provided on metal-binding sites.
In light of the above, it appears that a comprehensive, up-to-date database which collects, organizes and makes easily available the current knowledge on metalloproteins does not presently exist. In our opinion, the key to achieving the ambitious goal of gathering, on a single platform, the exceptional variety of metalloproteins is to design a database architecture based on a rigorous classification of metal-binding sites. A similar concept was put forward by the developers of the COMe ontology (i.e., a formal definition of concepts of a given area of knowledge such as bioinorganic chemistry, described in a standardized form),96 who proposed to catalogue metalloproteins by describing their metal-binding motifs according to a standard formalism.97 We have suggested that the representation of metal-binding sites by way of the three-dimensional templates discussed in the previous section can provide a useful basis for their classification, because it allows them to be compared in a systematic and largely automated fashion.65 The structure-based categorization of metal-binding sites could provide the fundamental framework for the database, in which other kinds of information such as functional and sequence information could be integrated as sketched in Fig. 5. This information should also be endowed with a defined ontology (see above), so as to design an efficient and flexible query system, thus optimizing database access, and to automate as much as possible the generation and processing of data, thus facilitating database maintenance and updates. Nevertheless, this task can be far from trivial for certain kinds of data: in the case of functional information, for example, there is a lot of active research going on to define controlled vocabularies such as the Gene Ontology (GO) system.98
 | ||
| Fig. 5 Scheme depicting a possible architecture for a comprehensive database of metalloproteins. At the core of the database are three-dimensional templates representing all known metal-binding sites, which are classified based on their structural features (see also Fig. 3). Metal-binding sites and the metalloproteins containing them are annotated with functional descriptions derived from literature analysis and following the GO terminology as much as possible. Sequence information may include the amino acid sequences of the metalloproteins (taken from UniProt), the Pfam domains identified within these sequences, and the sequences of related metalloproteins predicted using the methods described in the first section (e.g., containing the same Pfam domain, or the same metal-binding pattern). In principle, any other kind of information may also be incorporated, such as information regarding the sub-cellular localization of metalloproteins (e.g. in mitochondria, derived from the MitoP2 database).133 | ||
A comprehensive resource on metalloproteins like that envisaged above would provide an unprecedented, unified vision of these systems, and would thus constitute an important reference for many research areas also outside bioinorganic chemistry. In the context of today’s systems biology approaches, however, the knowledge contained in such a database would be fully exploited only by connecting it to that contained in other, diverse resources. At a minimum, connections may be made by suitable cross-references to other databases such as the PDB and Pfam, which in turn could be linked to the metalloprotein database. Ideally, the information on metalloproteins would be incorporated into integrated resources such as InterPro and Integr8, where several heterogeneous data are combined together and can be accessed through a single interface.13
The interactome as a perspective
Biological processes in living organisms are the result of the complex interactions between the various constituents of cells, such as proteins, nucleic acids and small molecules. For this reason, the determination and analysis of molecular interaction networks has become a central element of studies aimed at understanding the properties and behaviour of biological systems.99–101 There are several types of interaction networks underlying cellular processes, such as protein–protein interaction, metabolic, and signalling networks, which overlap and complement each other forming what has been called a ‘network of networks’.102 Out of these types of network, those most widely used in the investigation of biological systems are protein–protein interaction (or ‘interactome’) networks, for which many large-scale data sets are now available. These data sets are generated by high-throughput experimental approaches such as yeast two-hybrid (Y2H) systems103–105 and affinity purification followed by mass spectrometry (AP/MS),106–108 as well as by computational approaches.109Protein–protein interaction data are contained in several databases, including databases focusing on experimentally determined interactions such as DIP110 and MINT,111 and databases comprising also predicted interactions such as IntAct112 and STRING.113Interactome data are typically represented in the form of network diagrams, where any two interacting proteins are represented as two nodes connected by an edge. This highly abstract representation allows one to examine networks by using concepts from graph theory, thus obtaining important information on their topological properties.102,114 For example, graph analysis can be used to identify densely connected sub-networks which are likely to represent proteins involved in the same biological process.115–117 However, such a simplified formal description of interactions has only a limited relationship with physical reality, and appears to be inadequate for obtaining meaningful insight into cellular functioning. Furthermore, currently available interactome data are still largely incomplete and affected by high error rates, thus causing a need for effective means to discriminate between true positive and false positive interactions.118 These issues can be addressed by integrating various types of biological information within the framework provided by the interaction network. For example, protein structure is often regarded as crucial information to validate interactions, determining which interactions are compatible with each other and which ones are mutually exclusive, as well as to estimate quantitative interaction parameters such as affinity constants.119–122 Also, information on protein sub-cellular localization and specificity of expression can be used to single out interactions between proteins which, despite having the potential to interact, are never found in the same cellular compartment, or even (in multi-cellular organisms) in the same cell, or tissue. Incorporating the information associated with the metal-binding properties of proteins, therefore, can be very useful to characterize individual protein–protein interactions in networks (see Fig. 6 for example). In particular, this knowledge may be essential to assess metal-mediated interactions, which are transient protein–protein interactions occurring only in the presence of the metal (e.g., between the copper-binding Atx1 and Ccc2 proteins)123 and which may be overlooked in large-scale approaches for interactome mapping. On the other hand, positioning metal-binding proteins in an interaction network can help to assess the metal-binding properties that were predicted for those proteins. Computational predictions indicate in fact the potential capability of a protein to bind a given metal, whereas the binding of specific metals by proteinsin vivo may involve elaborate mechanisms relying on controlled metal delivery mediated by metal-specific carriers, and protein compartmentalization.124,125 Therefore, a valuable indication as to the metal actually bound in vivo by a predicted metalloprotein can be obtained from the knowledge of the cellular context in which it acts.
 | ||
| Fig. 6 Example of protein–protein interaction network analysis supported by integrating the information on the metal-binding properties of proteins. In this hypothetical case, proteins binding a given metal (shown as grey boxes) form a highly connected sub-network (sub-network A) which is linked by protein P9 to another sub-network (sub-network B) comprising proteins that do not bind that metal (shown as white boxes). This suggests that P9 is a multifunctional protein playing a role in both the cellular management of the metal (the function associated with sub-network A) and a different cellular process (associated with sub-network B). Also, sub-network A contains a protein (protein P3) that was not predicted to bind the metal. Protein P3 is thus an interesting target for characterization, which may reveal a novel metalloprotein. | ||
A major goal of systems biology approaches is to translate biological networks into mathematical models, linking the behaviour of a system to the whole of the interactions between its molecular components.10,126 Mathematical modelling provides an essential framework to simulate the complex, space- and time-dependent processes taking place in biological systems, and requires a comprehensive, quantitative description of the networks underlying the modelled processes. It is becoming increasingly clear118 that such a comprehensive description, which also involves the experimental determination of several parameters such as kinetic constants and diffusion coefficients, can currently be achieved only for small-scale biological systems such as certain signalling pathways127 and cell motility machineries.128 In this framework, the integration of metalloprotein information into interactome networks can be used to isolate sub-networks relevant to specific metals, highlighting the sets of interacting proteins responsible for the management and the utilization of those metals. Modelling and experimental efforts could be then focused on these sets, setting up an iterative cycle in which predictions formulated by the model are tested by experiments, whose results in turn allow the refinement of the model. Bioinorganic chemistry and bioinformatics would thus support each other in a synergistic fashion, towards achieving the ultimate goal of describing the mechanisms by which metals are framed in living organisms.
Concluding remarks
Bioinformatics is central to modern biology, which aims to describe living organisms at the system level and therefore must take into account a huge amount of data and information, put them together and extract rules and generalities that may account for the complex, macroscopic properties of cells and also of multi-cellular organisms. Here we focused on bioinformatics approaches and resources (Table 2) dedicated to obtain, store and interpret data on metals in proteins, which are key components of living organisms and must thus be taken into account to achieve the picture of cell functioning which is the goal of systems biology. Proper integration of metals into this picture requires that bioinformatics is supported by the knowledge of bioinorganic chemistry.Acknowledgements
This work was supported by the Ministero Italiano dell’Universita’ e della Ricerca (MIUR) through the FIRB Project RBRN07BMCT and the PRIN Project 2007M5MWM9.References
- J. J. R. Frausto da Silva and R. J. P. Williams, The Biological Chemistry of the Elements: The Inorganic Chemistry of Life, Oxford University Press, New York, 2001 Search PubMed.
- I. Bertini, H. B. Gray, E. I. Stiefel and J. S. Valentine, Biological Inorganic Chemistry, University Science Books, Sausalito, California, 2006 Search PubMed.
- A. R. Joyce and B. O. Palsson, Nat. Rev. Mol. Cell Biol., 2006, 7, 198–210 CrossRef CAS.
- F. J. Bruggeman and H. V. Westerhoff, Trends Microbiol., 2007, 15, 45–50 CrossRef CAS.
- M. L. Mo and B. O. Palsson, Trends Biotechnol., 2009, 27, 37–44 CrossRef CAS.
- M. E. Cusick, N. Klitgord, M. Vidal and D. E. Hill, Hum. Mol. Genet., 2005, 14, R171–R181 CrossRef CAS.
- I. Bertini and G. Cavallaro, JBIC, J. Biol. Inorg. Chem., 2008, 13, 3–14 CAS.
- J. Szpunar, Analyst, 2005, 130, 442–465 RSC.
- W. Shi and M. R. Chance, Cell. Mol. Life Sci., 2008, 65, 3040–3048 CrossRef CAS.
- H. Kitano, Nature, 2002, 420, 206–210 CrossRef CAS.
- P. Kersey and R. Apweiler, Nat. Cell Biol., 2006, 8, 1183–1189 CrossRef CAS.
- A. Valencia, Curr. Opin. Struct. Biol., 2005, 15, 267–274 CrossRef CAS.
- N. J. Mulder, P. Kersey, M. Pruess and R. Apweiler, Mol. Biotechnol., 2008, 38, 165–177 Search PubMed.
- M. Punta and Y. Ofran, PLoS Comput. Biol., 2008, 4, e1000160 Search PubMed.
- Y. Loewenstein, D. Raimondo, O. C. Redfern, J. Watson, D. Frishman, M. Linial, C. Orengo, J. Thornton and A. Tramontano, GenomeBiology, 2009, 10, 207 CrossRef.
- C. Andreini, I. Bertini and A. Rosato, Bioinformatics, 2004, 20, 1373–1380 CrossRef CAS.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2006, 5, 196–201 CrossRef CAS.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2006, 5, 3173–3178 CrossRef CAS.
- C. Andreini, L. Banci, I. Bertini, S. Elmi and A. Rosato, Proteins: Struct., Funct., Bioinf., 2007, 67, 317–324 Search PubMed.
- C. Andreini, L. Banci, I. Bertini and A. Rosato, J. Proteome Res., 2008, 7, 209–216 CrossRef CAS.
- P. Coggill, R. D. Finn and A. Bateman, Curr. Protoc. Bioinformatics, 2008 Search PubMed , ch. 2, Unit 5.
- R. D. Finn, J. Tate, J. Mistry, P. C. Coggill, S. J. Sammut, H. R. Hotz, G. Ceric, K. Forslund, S. R. Eddy, E. L. Sonnhammer and A. Bateman, Nucleic Acids Res., 2008, 36, D281–D288 CAS.
- S. R. Eddy, Nat. Biotechnol., 2004, 22, 1315–1316 CrossRef CAS.
- S. R. Eddy, Bioinformatics, 1998, 14, 755–763 CrossRef CAS.
- K. Rigby, L. Zhang, P. A. Cobine, G. N. George and D. R. Winge, J. Biol. Chem., 2007, 282, 10233–10242 CrossRef CAS.
- L. Banci, I. Bertini, C. Cefaro, S. Ciofi-Baffoni, A. Gallo, M. Martinelli, D. P. Sideris, N. Katrakili and K. Tokatlidis, Nat. Struct. Mol. Biol., 2009, 16, 198–206 CrossRef CAS.
- D. F. Seals and S. A. Courtneidge, Genes Dev., 2003, 17, 7–30 CrossRef CAS.
- C. Andreini, L. Banci, I. Bertini, S. Elmi and A. Rosato, J. Proteome Res., 2005, 4, 881–888 CrossRef CAS.
- W. Bode, F. X. Gomis-Ruth and W. Stockler, FEBS Lett., 1993, 331, 134–140 CrossRef CAS.
- D. J. Rigden and M. Y. Galperin, J. Mol. Biol., 2004, 343, 971–984 CrossRef CAS.
- Z. Zhang, A. A. Schaffer, W. Miller, T. L. Madden, D. J. Lipman, E. V. Koonin and S. F. Altschul, Nucleic Acids Res., 1998, 26, 3986–3990 CrossRef CAS.
- I. Bertini and A. Rosato, Eur. J. Inorg. Chem., 2007, 2546–2555 CrossRef CAS.
- S. J. Sammut, R. D. Finn and A. Bateman, Briefings Bioinf., 2008, 9, 210–219 Search PubMed.
- W. S. Noble, Nat. Biotechnol., 2006, 24, 1565–1567 CrossRef CAS.
- G. B. Fogel, Briefings Bioinf., 2008, 9, 307–316 Search PubMed.
- A. Ben Hur, C. S. Ong, S. Sonnenburg, B. Scholkopf and G. Ratsch, PLoS Comput. Biol., 2008, 4, e1000173 Search PubMed.
- C. T. Lin, K. L. Lin, C. H. Yang, I. F. Chung, C. D. Huang and Y. S. Yang, Int. J. Neural Syst., 2005, 15, 71–84 CrossRef.
- A. Passerini, M. Punta, A. Ceroni, B. Rost and P. Frasconi, Proteins: Struct., Funct., Bioinf., 2006, 65, 305–316 Search PubMed.
- H. H. Lin, L. Y. Han, H. L. Zhang, C. J. Zheng, B. Xie, Z. W. Cao and Y. Z. Chen, BMC Bioinformatics, 2006, 7, S13 CrossRef.
- A. Passerini, C. Andreini, S. Menchetti, A. Rosato and P. Frasconi, BMC bioinformatics, 2007, 8, 39 CrossRef.
- N. Shu, T. Zhou and S. Hovmoller, Bioinformatics, 2008, 24, 775–782 CrossRef CAS.
- C. L. Dupont, S. Yang, B. Palenik and P. E. Bourne, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17822–17827 CrossRef CAS.
- R. Thilakaraj, K. Raghunathan, S. Anishetty and G. Pennathur, Bioinformatics, 2007, 23, 267–271 CAS.
- J. D. Watson, R. A. Laskowski and J. M. Thornton, Curr. Opin. Struct. Biol., 2005, 15, 275–284 CrossRef CAS.
- D. Lee, O. Redfern and C. Orengo, Nat. Rev. Mol. Cell Biol., 2007, 8, 995–1005 CrossRef CAS.
- M. Huynen, B. Snel, W. 3. Lathe and P. Bork, Genome Res., 2000, 10, 1204–1210 CrossRef CAS.
- T. Gabaldon and M. A. Huynen, Cell. Mol. Life Sci., 2004, 61, 930–944 CrossRef CAS.
- M. Huynen, B. Snel, C. von Mering and P. Bork, Curr. Opin. Cell Biol., 2003, 15, 191–198 CrossRef CAS.
- B. A. Shoemaker and A. R. Panchenko, PLoS Comput. Biol., 2007, 3, e43 Search PubMed.
- I. Bertini, G. Cavallaro and A. Rosato, Chem. Rev., 2006, 106, 90–115 CrossRef CAS.
- L. Banci, I. Bertini, G. Cavallaro and A. Rosato, J. Proteome Res., 2007, 6, 1568–1579 CrossRef CAS.
- R. H. Holm, P. Kennepohl and E. I. Solomon, Chem. Rev., 1996, 96, 2239–2314 CrossRef CAS.
- C. Chothia and A. M. Lesk, EMBO J., 1986, 5, 823–826 CAS.
- J. C. Whisstock and A. M. Lesk, Q. Rev. Biophys., 2003, 36, 307–340 CrossRef CAS.
- E. Krissinel and K. Henrick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2256–2268 CrossRef CAS.
- L. Holm and C. Sander, Trends Biochem. Sci., 1995, 20, 478–480 CrossRef CAS.
- C. A. Orengo, A. D. Michie, S. Jones, D. T. Jones, M. B. Swindells and J. M. Thornton, Structure, 1997, 5, 1093–1108 CrossRef CAS.
- A. G. Murzin, S. E. Brenner, T. Hubbard and C. Chothia, J. Mol. Biol., 1995, 247, 536–540 CrossRef CAS.
- J. M. Thornton, A. E. Todd, D. Milburn, N. Borkakoti and C. A. Orengo, Nat. Struct. Biol., 2000, 7, 991–994 CrossRef CAS.
- C. A. Orengo and J. M. Thornton, Annu. Rev. Biochem., 2005, 74, 867–900 CrossRef CAS.
- R. B. Russell, J. Mol. Biol., 1998, 279, 1211–1227 CrossRef CAS.
- A. C. Wallace, R. A. Laskowski and J. M. Thornton, Protein Sci., 1996, 5, 1001–1013 CAS.
- A. Stark and R. B. Russell, Nucleic Acids Res., 2003, 31, 3341–3344 CrossRef CAS.
- J. A. Barker and J. M. Thornton, Bioinformatics, 2003, 19, 1644–1649 CrossRef CAS.
- C. Andreini, I. Bertini, G. Cavallaro, R. J. Najmanovich and J. M. Thornton, J. Mol. Biol., 2009, 388, 356–380 CrossRef CAS.
- T. Dudev, Y. L. Lin, M. Dudev and C. Lim, J. Am. Chem. Soc., 2003, 125, 3168–3180 CrossRef CAS.
- T. Dudev and C. Lim, Annu. Rev. Biophys., 2008, 37, 97–116 Search PubMed.
- C. Ascenso, F. Rusnak, I. Cabrito, M. J. Lima, S. Naylor, I. Moura and J. J. Moura, JBIC, J. Biol. Inorg. Chem., 2000, 5, 720–729 CrossRef CAS.
- J. M. Chandonia and S. E. Brenner, Science, 2006, 311, 347–351 CrossRef CAS.
- Y. Zhang, Curr. Opin. Struct. Biol., 2008, 18, 342–348 CrossRef CAS.
- F. Kiefer, K. Arnold, M. Kunzli, L. Bordoli and T. Schwede, Nucleic Acids Res., 2009, 37, D387–D392 CrossRef CAS.
- U. Pieper, N. Eswar, B. M. Webb, D. Eramian, L. Kelly, D. T. Barkan, H. Carter, P. Mankoo, R. Karchin, M. A. Marti-Renom, F. P. Davis and A. Sali, Nucleic Acids Res., 2009, 37, D347–D354 CrossRef CAS.
- V. A. Ivanisenko, S. S. Pintus, D. A. Grigorovich and N. A. Kolchanov, Nucleic Acids Res., 2004, 32, W549–W554 CrossRef CAS.
- R. A. Laskowski, J. D. Watson and J. M. Thornton, Nucleic Acids Res., 2005, 33, W89–W93 CrossRef CAS.
- J. W. Torrance, M. W. MacArthur and J. M. Thornton, Proteins: Struct., Funct., Bioinf., 2008, 71, 813–830 Search PubMed.
- K. Goyal and S. C. Mande, Proteins: Struct., Funct., Bioinf., 2008, 70, 1206–1218 Search PubMed.
- J. S. Sodhi, K. Bryson, L. J. McGuffin, J. J. Ward, L. Wernisch and D. T. Jones, J. Mol. Biol., 2004, 342, 307–320 CrossRef CAS.
- M. Babor, S. Gerzon, B. Raveh, V. Sobolev and M. Edelman, Proteins: Struct., Funct., Bioinf., 2008, 70, 208–217 Search PubMed.
- M. Dudev and C. Lim, BMC Bioinformatics, 2007, 8, 106 CrossRef.
- J. C. Ebert and R. B. Altman, Protein Sci., 2008, 17, 54–65 CAS.
- J. W. Schymkowitz, F. Rousseau, I. C. Martins, J. Ferkinghoff-Borg, F. Stricher and L. Serrano, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10147–10152 CrossRef CAS.
- R. Levy, M. Edelman and V. Sobolev, Proteins: Struct., Funct., Bioinf., 2008, 76, 365–374 Search PubMed.
- M. M. Harding, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 401–411 CrossRef CAS.
- I. N. Kasampalidis, I. Pitas and K. Lyroudia, Proteins: Struct., Funct., Bioinf., 2007, 68, 123–130 Search PubMed.
- K. Patel, A. Kumar and S. Durani, Biochim. Biophys. Acta, Proteins Proteomics, 2007, 1774, 1247–1253 CrossRef CAS.
- I. Dokmanic, M. Sikic and S. Tomic, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 257–263 CrossRef.
- H. Zheng, M. Chruszcz, P. Lasota, L. Lebioda and W. Minor, J. Inorg. Biochem., 2008, 102, 1765–1776 CrossRef CAS.
- I. Bertini and A. Rosato, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3601–3604 CrossRef CAS.
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, Bioinformatics, 2009, 25, 2088–2089 CrossRef CAS.
- C. Andreini, I. Bertini, G. Cavallaro, G. L. Holliday and J. M. Thornton, JBIC, J. Biol. Inorg. Chem., 2008, 13, 1205–1218 CrossRef CAS.
- J. M. Castagnetto, S. W. Hennessy, V. A. Roberts, E. D. Getzoff, J. A. Tainer and M. E. Piquet, Nucleic Acids Res., 2002, 30, 379–382 CrossRef CAS.
- K. N. Degtyarenko, A. C. North, D. N. Perkins and J. B. Findlay, Nucleic Acids Res., 1998, 26, 376–381 CrossRef CAS.
- A. Golovin and K. Henrick, BMC Bioinformatics, 2008, 9, 312 CrossRef.
- M. Tagari, J. Tate, G. J. Swaminathan, R. Newman, A. Naim, W. Vranken, A. Kapopoulou, A. Hussain, J. Fillon, K. Henrick and S. Velankar, Nucleic Acids Res., 2006, 34, D287–D290 CrossRef CAS.
- A. Golovin, D. Dimitropoulos, T. Oldfield, A. Rachedi and K. Henrick, Proteins: Struct., Funct., Bioinf., 2005, 58, 190–199 Search PubMed.
- O. Carugo and S. Pongor, Trends Biotechnol., 2002, 20, 498–501 CrossRef CAS.
- K. Degtyarenko and S. Contrino, BMC Struct. Biol., 2004, 4, 3 CrossRef.
- D. P. Hill, B. Smith, M. S. McAndrews-Hill and J. A. Blake, BMC Bioinformatics, 2008, 9, S2 CrossRef.
- S. Bader, S. Kuhner and A. C. Gavin, FEBS Lett., 2008, 582, 1220–1224 CrossRef CAS.
- R. B. Russell and P. Aloy, Nat. Chem. Biol., 2008, 4, 666–673 CrossRef CAS.
- A. M. Feist, M. J. Herrgard, I. Thiele, J. L. Reed and B. O. Palsson, Nat. Rev. Microbiol., 2009, 7, 129–143 Search PubMed.
- A. L. Barabasi and Z. N. Oltvai, Nat. Rev. Genet., 2004, 5, 101–113 CrossRef CAS.
- P. Uetz, L. Giot, G. Cagney, T. A. Mansfield, R. S. Judson, J. R. Knight, D. Lockshon, V. Narayan, M. Srinivasan, P. Pochart, A. Qureshi-Emili, Y. Li, B. Godwin, D. Conover, T. Kalbfleisch, G. Vijayadamodar, M. Yang, M. Johnston, S. Fields and J. M. Rothberg, Nature, 2000, 403, 623–627 CrossRef CAS.
- J. F. Rual, K. Venkatesan, T. Hao, T. Hirozane-Kishikawa, A. Dricot, N. Li, G. F. Berriz, F. D. Gibbons, M. Dreze, N. Ayivi-Guedehoussou, N. Klitgord, C. Simon, M. Boxem, S. Milstein, J. Rosenberg, D. S. Goldberg, L. V. Zhang, S. L. Wong, G. Franklin, S. Li, J. S. Albala, J. Lim, C. Fraughton, E. Llamosas, S. Cevik, C. Bex, P. Lamesch, R. S. Sikorski, J. Vandenhaute, H. Y. Zoghbi, A. Smolyar, S. Bosak, R. Sequerra, L. Doucette-Stamm, M. E. Cusick, D. E. Hill, F. P. Roth and M. Vidal, Nature, 2005, 437, 1173–1178 CrossRef CAS.
- H. Yu, P. Braun, M. A. Yildirim, I. Lemmens, K. Venkatesan, J. Sahalie, T. Hirozane-Kishikawa, F. Gebreab, N. Li, N. Simonis, T. Hao, J. F. Rual, A. Dricot, A. Vazquez, R. R. Murray, C. Simon, L. Tardivo, S. Tam, N. Svrzikapa, C. Fan, A. S. de Smet, A. Motyl, M. E. Hudson, J. Park, X. Xin, M. E. Cusick, T. Moore, C. Boone, M. Snyder, F. P. Roth, A. L. Barabasi, J. Tavernier, D. E. Hill and M. Vidal, Science, 2008, 322, 104–110 CrossRef CAS.
- A. C. Gavin, M. Bosche, R. Krause, P. Grandi, M. Marzioch, A. Bauer, J. Schultz, J. M. Rick, A. M. Michon, C. M. Cruciat, M. Remor, C. Hofert, M. Schelder, M. Brajenovic, H. Ruffner, A. Merino, K. Klein, M. Hudak, D. Dickson, T. Rudi, V. Gnau, A. Bauch, S. Bastuck, B. Huhse, C. Leutwein, M. A. Heurtier, R. R. Copley, A. Edelmann, E. Querfurth, V. Rybin, G. Drewes, M. Raida, T. Bouwmeester, P. Bork, B. Seraphin, B. Kuster, G. Neubauer and G. Superti-Furga, Nature, 2002, 415, 141–147 CrossRef CAS.
- Y. Ho, A. Gruhler, A. Heilbut, G. D. Bader, L. Moore, S. L. Adams, A. Millar, P. Taylor, K. Bennett, K. Boutilier, L. Yang, C. Wolting, I. Donaldson, S. Schandorff, J. Shewnarane, M. Vo, J. Taggart, M. Goudreault, B. Muskat, C. Alfarano, D. Dewar, Z. Lin, K. Michalickova, A. R. Willems, H. Sassi, P. A. Nielsen, K. J. Rasmussen, J. R. Andersen, L. E. Johansen, L. H. Hansen, H. Jespersen, A. Podtelejnikov, E. Nielsen, J. Crawford, V. Poulsen, B. D. Sorensen, J. Matthiesen, R. C. Hendrickson, F. Gleeson, T. Pawson, M. F. Moran, D. Durocher, M. Mann, C. W. Hogue, D. Figeys and M. Tyers, Nature, 2002, 415, 180–183 CrossRef CAS.
- A. C. Gingras, M. Gstaiger, B. Raught and R. Aebersold, Nat. Rev. Mol. Cell Biol., 2007, 8, 645–654 CrossRef CAS.
- L. Skrabanek, H. K. Saini, G. D. Bader and A. J. Enright, Mol. Biotechnol., 2008, 38, 1–17 Search PubMed.
- I. Xenarios, D. W. Rice, L. Salwinski, M. K. Baron, E. M. Marcotte and D. Eisenberg, Nucleic Acids Res., 2000, 28, 289–291 CrossRef CAS.
- A. Chatr-aryamontri, A. Ceol, L. M. Palazzi, G. Nardelli, M. V. Schneider, L. Castagnoli and G. Cesareni, Nucleic Acids Res., 2007, 35, D572–D574 CrossRef CAS.
- S. Kerrien, Y. Alam-Faruque, B. Aranda, I. Bancarz, A. Bridge, C. Derow, E. Dimmer, M. Feuermann, A. Friedrichsen, R. Huntley, C. Kohler, J. Khadake, C. Leroy, A. Liban, C. Lieftink, L. Montecchi-Palazzi, S. Orchard, J. Risse, K. Robbe, B. Roechert, D. Thorneycroft, Y. Zhang, R. Apweiler and H. Hermjakob, Nucleic Acids Res., 2007, 35, D561–D565 CrossRef CAS.
- L. J. Jensen, M. Kuhn, M. Stark, S. Chaffron, C. Creevey, J. Muller, T. Doerks, P. Julien, A. Roth, M. Simonovic, P. Bork and C. von Mering, Nucleic Acids Res., 2009, 37, D412–D416 CrossRef CAS.
- S. Brohee, K. Faust, G. Lima-Mendez, G. Vanderstocken and J. van Helden, Nat. Protoc., 2008, 3, 1616–1629 Search PubMed.
- L. H. Hartwell, J. J. Hopfield, S. Leibler and A. W. Murray, Nature, 1999, 402, C47–C52 CrossRef CAS.
- B. Snel, P. Bork and M. A. Huynen, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5890–5895 CrossRef CAS.
- V. Spirin and L. A. Mirny, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 12123–12128 CrossRef CAS.
- W. Kelly and M. Stumpf, Curr. Opin. Biotechnol., 2008, 19, 396–403 CrossRef CAS.
- P. Aloy and R. B. Russell, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5896–5901 CrossRef CAS.
- P. Aloy and R. B. Russell, Nat. Rev. Mol. Cell Biol., 2006, 7, 188–197 CrossRef CAS.
- A. Campagna, L. Serrano and C. Kiel, FEBS Lett., 2008, 582, 1231–1236 CrossRef CAS.
- C. Kiel, P. Beltrao and L. Serrano, Annu. Rev. Biochem., 2008, 77, 415–441 CrossRef CAS.
- L. Banci, I. Bertini, F. Cantini, I. C. Felli, L. Gonnelli, N. Hadjiliadis, R. Pierattelli, A. Rosato and P. Voulgaris, Nat. Chem. Biol., 2006, 2, 367–368 CrossRef CAS.
- S. Tottey, D. R. Harvie and N. J. Robinson, Acc. Chem. Res., 2005, 38, 775–783 CrossRef CAS.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 7, 25–35 Search PubMed.
- B. Di Ventura, C. Lemerle, K. Michalodimitrakis and L. Serrano, Nature, 2006, 443, 527–533 CrossRef CAS.
- P. Uetz and I. Stagljar, Mol. Syst. Biol., 2006, 2, 0006.
- S. V. Rajagopala, B. Titz, J. Goll, J. R. Parrish, K. Wohlbold, M. T. McKevitt, T. Palzkill, H. Mori, R. L. Finley, Jr and P. Uetz, Mol. Syst. Biol., 2007, 3, 128.
- N. Zheng, B. A. Schulman, L. Song, J. J. Miller, P. D. Jeffrey, P. Wang, C. Chu, D. M. Koepp, S. J. Elledge, M. Pagano, R. C. Conaway, J. W. Conaway, J. W. Harper and N. P. Pavletich, Nature, 2002, 416, 703–709 CrossRef CAS.
- A. Carfi, S. H. Willis, J. C. Whitbeck, C. Krummenacher, G. H. Cohen, R. J. Eisenberg and D. C. Wiley, Mol. Cell, 2001, 8, 169–179 CrossRef CAS.
- J. Zhu and Z. Weng, Proteins: Struct., Funct., Bioinf., 2005, 58, 618–627 Search PubMed.
- H. Erlandsen, J. Y. Kim, M. G. Patch, A. Han, A. Volner, M. M. Abu-Omar and R. C. Stevens, J. Mol. Biol., 2002, 320, 645–661 CrossRef CAS.
- M. Elstner, C. Andreoli, T. Klopstock, T. Meitinger and H. Prokisch, Methods Enzymol., 2009, 457, 3–20 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Example protocol for the identification of zinc proteins based on our method. See DOI: 10.1039/b912156k |
| This journal is © The Royal Society of Chemistry 2010 |