Analytical methodologies for metallomics studies of antitumor Pt-containing drugs
Diego
Esteban-Fernández
b,
Estefanía
Moreno-Gordaliza
a,
Benito
Cañas
a,
María
Antonia Palacios
a and
María
Milagros Gómez-Gómez
*a
aDepartment of Analytical Chemistry, Universidad Complutense de Madrid, Av. Complutense s/n, 28040, Madrid, Spain. E-mail: mmgomez@quim.ucm.es; Fax: +34 91-394-43-29; Tel: +34 91-394-51-46
bDepartment of Chemistry, Humboldt-Universitaet zu Berlin, Brook-Taylor Str. 2, 12489 Berlin, Germany
First published on 9th November 2009
Abstract
Pt-containing drugs are nowadays essential components in cancer chemotherapy. However, drug resistance and side effects limit the efficiency of the treatments. In order to improve the response to Pt-based drugs, different administration strategies or new Pt-compounds have been developed with little success. The reason for this failure could be that the mechanism of action of these drugs is not completely understood. In this way, metallomics studies may contribute to clarify the interactions of Pt-containing drugs within the organism. This review is mainly focused on the role of Analytical Chemistry on the study of the interactions between Pt-based drugs and biomolecules. A summary of the analytical techniques and the most common sample treatment procedures currently used in metallomics studies of these drugs is presented. Both are of paramount importance to study these complex samples preserving the drug–biomolecule interaction. Separation and detection techniques must be carefully selected in order to achieve the intended goals. The use of multidimensional hyphenated techniques is usually necessary for a better understanding of the Pt-based drugs interactions in the organism. An overview of Pt-drugs biological interactions is presented, considering the different sample matrices and the drugs course through the organism. Samples analysed in the included studies are blood, urine, cell cytosol, DNA as well as the drugs themselves and their derivatives. However, most of these works are based on in vitro experiments or incubations of standards, leading in some cases to contradictory results depending on the experimental conditions used. Though in vivo experiments represent a great challenge due to the high complexity and the low concentrations of the Pt-adducts in real samples, these studies must be undertaken to get a deeper understanding of the real interactions concerning Pt-containing drugs.
1. Introduction
Most of the trace elements in biological systems are bound to biomolecules. Metal-binding compounds play essential roles acting as biological catalysts that regulate reactions and physiological functions in cells and organs. For instance, the metalloproteins which catalyse biological reactions, called metalloenzymes , participate in important biological processes.1 Metal-binding biomolecules are also present in degeneration processes: traces of Fe, Cu and Zn are involved in the growth of neurotoxic amyloid fibrils which allow the progression of Alzheimer’s disease.2 Furthermore, drugs containing metal atoms are frequently employed in medicine.3 Bi complexes have been successfully used therapeutically in antiulcer treatments;4 Au compounds are used on the treatment of rheumatoid arthritis,5 also exhibiting in vivo antitumor activity when gold is in oxidation state +1 or +3;6 and, finally, compounds with Ga,7 Ru,8 Rh,9 Sn10 and As11 present antitumor properties. Nowadays, the most powerful metallodrugs used are the Pt-containing compounds cisplatin, carboplatin or oxaliplatin, which are applied worldwide in the clinical practice.The antitumor properties of cisplatin (cis-diaminedichloroplatinum(II)) were discovered in the 60’s by Rosenberg et al.12 and its clinical use was approved by the FDA in 1978. When DNA reacts with cisplatin, cross-linked adducts are produced, resulting in the distortion of its double-helix structure, with a global bending in the duplex of 35–40° and a local unwinding of 25° in the double helix.13 As a consequence of the DNA damage several cellular processes are disrupted, including transcription and replication, being cell death, either by apoptosis or necrosis, finally induced.14 As it has been recently summarised, there are four consecutive stages involved in the inhibition of the transcription by Pt-containing drugs: (1) cellular accumulation by both passive and active uptake; (2) activation of the Pt(II) complex; (3) binding to nucleic acids to form a variety of Pt-DNA adducts , and (4) the cellular response to DNA damage.14
Clinical treatments are limited by the toxic side effects such as nephrotoxicity, emetogenesis and neurotoxicity. Nephrotoxicity may be partially inhibited using administration protocols including a previous hydration of the patients with saline solutions15 as well as varying the extension of the infusion times, volume and number of the dosages.16 Efforts have been made towards the improvement of cisplatin therapeutic properties, looking for minimising its side effects, and as a result, many new Pt-based drugs have been developed, some of them already approved for clinical use or under consideration for approval by regulatory authorities. Carboplatin (cis-diamine(1,1-cyclobutanedicarboxylato)platinum(II)), which was authorised by the FDA in 1989, is a second generation drug widely used to treat ovarian and lung cancers. Compared to cisplatin, carboplatin presents lower activity and toxicity, can be used more easily in combined therapies and is active against the same type of tumours. Oxaliplatin ((trans-RR-cyclohexane-1,2-diamine)oxalatoplatinum(II)), a third generation Pt-drug approved by the FDA in 2004, improves the toxic behaviour of cisplatin, does not present cross-resistance with cisplatin and is very effective against colon cancer.17 The main side effects of carboplatin and oxaliplatin are myelosupression and neuropathy, respectively. Fig. 1 shows some other important Pt-based drugs. Tetraplatin, iproplatin and satraplatin (JM216) are Pt(IV) water soluble compounds, and therefore, suitable for oral administration. Specifically, satraplatin has been extensively studied18 and now is under consideration for approval by the FDA for hormone-refractory prostate cancer.19 Other promising Pt-drugs are nedaplatin, lobaplatin and picoplatin, the latter in Phase III of clinical trials for small-cell lung cancer.20 New generation Pt-based drugs that do not follow the traditional structure–activity rules of platinum cytoxicity established by Cleare and Hoeschele in 197321 are trans-compounds (JM335) or even polynuclear complexes (BBR3464).
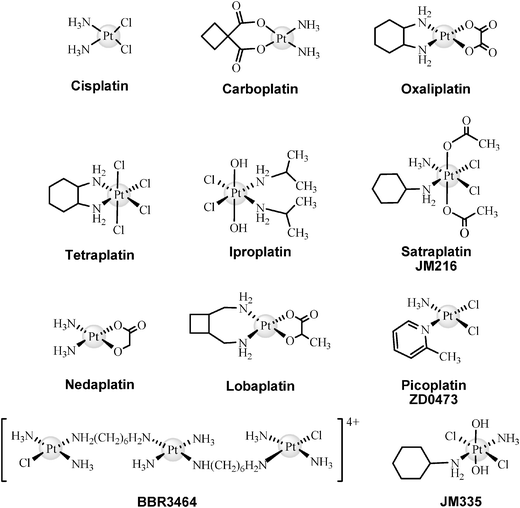 | ||
| Fig. 1 Structures of the most important Pt-based drugs. | ||
The evolution of the number of publications involving cisplatin, oxaliplatin and carboplatin analysis in the last decades shows a great increase. This tendency is due not only to the current interest of the scientific community in Pt-based drugs, but also to the improvement of the analytical tools and methodologies used in such studies. A new discipline, Metallomics,22 is being developed to study the evolution of metal or metalloid species and their interaction in time and space with other type of molecules, including biomolecules, focusing both in qualitative and quantitative aspects. Consequently, the goals of metallomics are the determination of the metallome, which is defined as the entirety of metal and metalloid species within a cell or tissue type, and the elucidation of the physiological roles and functions in which any metallo-species may be implicated in a biological system.23 Analytical Chemistry methodologies, techniques and instrumentation are essential for the study of metallomes.24 The chemical forms and amounts of trace elements present in biological systems are responsible for their bioavailability, toxicity or functionality. For that reason, elemental biospeciation and structural determination studies are essential for the comprehension of the effects produced by metal-containing compounds in living organisms. The high complexity of Pt-based drugs metallomics studies arise from factors like the poor stability of the drugs and drug–biomolecule adducts , their low concentrations in real samples, the different complex sample matrices to be analysed and the high amount of potential biomolecule targets.25,26
Pt(II), as a soft Lewis acid, presents a high affinity for soft bases with donor atoms. It shows a preferential binding to S-donor groups, but also other groups such as N-donors represent significant coordination points, being the affinity to the latter higher than for O-donors. Thus, aside from DNA, peptides and proteins with S- and N-containing aminoacidic residues are potentially reactive molecules towards Pt-drugs.26 It has been suggested that on reaction with proteins, cisplatin may bind as a bidentate ligand, for instance, with one of the leaving groups replaced by a S-donor and the other by a neighbouring N- or O- donor. Since the SH → SS bonding is important for the protein conformation, the binding of Pt-drugs to cystines may produce an alteration on the structure and the biological functions of proteins. This fact may be related to the side effects observed during the antitumor treatment.
When Pt-based drugs enter the blood stream, a series of interactions with blood components and later on with other molecules on cell membranes and inside the cells take place. The low chloride concentration in cytosols triggers the hydrolysis of cisplatin, producing a series of adducts as a result of the interaction of the evolved drug with cytoplasmic biomolecules. It is believed that some of these adducts are the key to understand the antitumor and toxic effects of Pt-drugs. The extent of the DNA-adducts formation may be limited by the interaction of the drug with cytosolic biomolecules like MT (metallothioneins) or GSH (glutathione), resulting in detoxification or resistance effects. Moreover, the binding of the drugs to blood proteins or the efficacy of the cell uptake could also alter the supply of the drug to the DNA. Other proposed resistance mechanism is related with the capacity of the cell to recognise and repair the damage produced by the drug–DNAadducts avoiding cell death.
The low concentration and the high diversity of drug–biomolecule adducts require the use of mass spectrometry analytical techniques, both elemental and molecular, coupled to different separation techniques, for the quantification and structural determination of Pt-adducts.27 Depending on the objective, blood, urine, tissues or cell fractions must be analysed, using at least one high resolution separation technique.28 Both analysis conditions and sample treatment, such as pre-concentration or clean-up steps, must ensure the preservation of the Pt-species identities. Nevertheless, the difficulty to analyse Pt-adducts at the trace levels present in real samples, preserving their identity along the several steps needed to separate and detect the adducts , has precluded, in general, in vivo experiments in bio-speciation. Alternatively, many of the reported articles consist in in vitro experiments that simplify the sample matrix and allow working with more concentrated species. The concordance between in vitro and in vivo experiments is usually a problem because slight differences in the reaction media and analyte concentration results in great differences in the identity and stability of the Pt-species. Results obtained from in vitro experiments must be carefully reviewed evaluating their reproducibility in biological living systems, being in any case a previous step to tackle the challenge of analysing real samples.
Not only speciation studies are important to evaluate the Pt-based drugs mechanism of action. Accumulation and distribution studies of the drug among different organs, tissues and cell compartments impose the use of cell fractionation techniques and cut-off filters to determine the affinity of the drug and to allow the identification of their main biological targets.29 The use of samples from patients with different sort of tumours, ages, administration dosages, etc., for in vivo experiments, have allowed to draw a distribution map of Pt-based drugs in organisms and are reported in a great number of works.29,30
The information collected in this review will help the reader to clarify and sum up the reported data related with the speciation of Pt-containing drugs and the Pt adducts they form with biomolecules and also concerning the determination of total Pt content after the administration of the aforementioned drugs. The most commonly employed analytical tools are presented firstly, including sample preparation, separation and detection techniques. According to the sample matrix, the last section contains a complete summary of the most interesting reported analytical articles.
2. Analytical methodologies
In the last years there has been an extensive scientific activity in order to improve the identification and quantification of metal–biomolecule adducts using novel analytical methodologies. To fulfil this goal a combination of powerful separation techniques with detection techniques based mainly on mass spectrometry, the so called hyphenated techniques, have been employed.28 Furthermore, important efforts have been directed towards the development of soft sample preparation methods in order to extract the target biomolecules without changing their original structures.For several decades the determination of total Pt content in biological samples has been performed using analytical techniques like adsorptive stripping voltammetry (ASV), neutron activation analysis (NAA), or absorption and emission atomic spectroscopy, among others.30–34 Nowadays, inductively coupled plasma mass spectrometry (ICP-MS) seems to be the most powerful elemental technique because of its high sensitivity (reported limit of detection for Pt of 6 μg Kg−1 and 63 ng L−1 for tissues and cytosolic samples, respectively),35 selectivity, lack of significant interferences for Pt determination in clinical samples and multielemental detection. Usually a simple acidic digestion in microwave oven (MO) reactors, followed by a direct flow injection analysis of the digested sample by ICP-MS, is enough to analyse the total content of Pt after fractionation of cellular extracts by ultracentrifugation or ultrafiltration through different cut-off filters.35
Bio-speciation analysis involves highly complex analytical methodologies able to overcome important drawbacks like low concentrations, complex matrices, species transformations or cleavage of the metal–biomolecule bonds. Usually the complexity of biological samples makes necessary to perform several fractionation/separation steps for the isolation of the drug–biomolecule adducts by liquid chromatography36 or electrophoretic techniques37 prior to their determination. Apart from the previously referred procedures using ultracentrifugation and/or ultrafiltration, precipitation of the thermally labile proteins by heating at 55 °C is commonly used.38 Even Size Exclusion Chromatography (SEC) may be considered as a desalting and pre-fractionation technique due to its high protein recovery. SEC, Ion Exchange (IXC) or Reverse Phase (RPC), are the chromatographic separation methods most frequently used. Electrophoretic techniques have also been used for the separation of drug–biomolecule adducts .37 2D-SDS-PAGE (Two Dimensional Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) allows high resolution separations for proteins, although it can not be coupled on-line to detectors. Moreover, denaturing conditions, used in 2D-SDS-PAGE experiments, may affect certain metal–biomolecule bonds. Capillary Electrophoresis (CE) has also been employed in metallomics studies.39 The advantages of this high resolution separation technique are the compatibility with ICP-MS and ESI-MS detection, the simplicity and the short analysis times needed.
Structural information is necessary for the final identification of metal–biomolecule adducts . Traditionally, molecular mass spectrometry has been used, not only for proteomics but also for metallomics studies, where the identification of the biomolecule and its structure is also necessary. The development of “soft” ionisation sources such as matrix-assisted laser desorption/ionisation (MALDI) and electrospray ionisation (ESI) represents a great advance in mass spectrometry for proteins, peptides and other biomolecule studies. These ionisation sources are able to ionise and turn large molecules into gas phase with a minimal change in their integrity. Their coupling to time of flight (TOF), ion trap (IT), triple quadrupole (TQ) and more recently, Fourier transform ion cyclotron resonance (FT-ICR) or Orbitrapmass spectrometry analysers, have allowed to obtain fast and sensitive mass spectra for identification purposes. This great variety of mass spectrometry techniques together with the possibility to combine analysers in tandem instruments, offer different and even complementary ways to identify unknown biomolecules. In metallomics studies, ESI seems to be more suitable than MALDI because the softer ionisation process it involves helps to preserve the integrity of the metal–biomolecule bond. Moreover, ESI-MS can be coupled to different separation techniques more easily than MALDI-MS.
Since the mid 90’s, peptide mass fingerprinting (PMF) or de novo sequencing from peptide fragmentation spectra, have been the most applied procedures for protein identification, constituting the so-called “classical proteomics”.40 However, current demands for high throughput protein identification, quantitative determinations in differential protein expression or characterising post-translational modifications, have promoted a “Second Generation Proteomics”. Some examples of these new methodologies are the MudPIT analysis (Multidimensional ChromatographyProtein Identification Technology) or the quantification approaches by isotope labelling.
The use of elemental mass spectrometry techniques like ICP-MS in biomolecule analysis, can also contribute to the advances in that new generation proteomics. The need of more sensitive determinations and quantitative results makes ICP-MS highly suitable for metallomics studies. ICP-MS provides multielemental information on metals, metalloids and some important non-metals such as halogens, sulfur or phosphorus. The high energy ionisation source produces species- or compound-independent signals, making possible the absolute quantification of biomolecules containing detectable elements. The high sensitivity and the wide concentration linear range of the ICP-MS allow tackling the simultaneous quantification of both low and high abundance biomolecules, as well as discriminating between very close expression levels of proteins. Therefore, ICP-MS is excellent for quantification tasks41 as well as for following the target species during an interaction process or a separation experiment.42 Besides, the multi-isotopic capability of the technique allows performing isotopic dilution analysis (IDA). In the last years it has been pointed out that the combined use of elemental and molecular hyphenated techniques27 is the key to a real improvement in metallomics analysis.
The low concentration level of the metal–biomolecule adducts in most samples, and the lower sensitivity of molecular mass spectrometrydetectors compared with ICP-MS, make necessary the use of preconcentration procedures such as solvent evaporation, freeze-drying, ultrafiltration or column-head focusing prior to high resolution chromatographic separations.43 On-line focusing can be carried out by IEC and RPC, achieving high preconcentration levels with low sample manipulation. Sometimes concentrated chromatographic fractions may present a high saline content that is incompatible with structural mass spectrometry techniques such as ESI-MS or MALDI-MS. Therefore, a previous desalting step is usually performed by SEC, dialysis, cut-off filters, or micro-SPE cartridges.44
Each of the described procedures should keep the integrity of the original metal–biomolecule adducts . For this reason, the use of protease inhibitors or the careful control of experimental conditions like pH, temperature, salinity or storage time, are mandatory to avoid bond cleavage, species transformation and protein denaturing or aggregation.28
The characterisation of drug–proteinadducts may be undertaken by either bottom-up or top-down mass spectrometry based approaches. Both methodologies have already been performed45,46 resulting in the elucidation of the Pt binding sites within several proteins. In fact, there is a significant research activity in an attempt to establish the suitability of each methodology for the full characterisation of Pt adducts in real samples, keeping in mind the need to preserve the drug–protein binding along the whole process. Whenever results are attainable using a bottom-up strategy, it is the method of choice, avoiding the interpretation of the highly complex mass spectra generated by top-down methodologies. Moreover, in a bottom-up approach the characteristic isotopic distribution of platinum can be useful for finding peptides bound to Pt because it modifies the typical isotopic pattern of a peptide, this being clearly detectable by mass spectrometry. Therefore, the recognition of Pt-containing peptides resulting from protein digests facilitates the identification of the platinated proteins from which they derive and their Pt binding sites. This is not the case of whole large proteins involved in top-down approaches because the contribution of this element to their isotopic pattern is less significant.
On the other hand, the accurate mass measurement of FT-ICR analysers and the fragmentation of isolated intact proteins are outstandingly contributing to improve top-down applications. However, the lack of high resolution separation techniques for proteins that can be easily coupled to mass spectrometrydetectors, the signal suppression effects in ESI sources and the not well-controlled fragmentation mechanisms inside the ICR cell, are fundamental drawbacks of the top-down approaches. Still, top-down experiments allow studying post-translational modifications, site-specific mutations and minimise the alteration of the binding of Pt-containing drugs to biomolecules.
3. Pt-based drug interactions
3.1 Stability in aqueous media
In their way to the therapeutic targets, changes in the original chemical form of the administered drugs are often produced. Physiological aqueous medium leads to hydrolysed forms of the Pt-drugs, which may react to a different extent with blood components or cell biomolecules. These transformations increase the reactivity of the drug and have an influence on their accumulation, excretion and on their toxic or antitumor effects. However, the formation of the active hydrolysis products is time-dependent and thus the reactivity of the drug follows the hydrolysis kinetics.The hydrolysed forms of cisplatin, as shown in Fig. 2, are well known since several decades.25,47–51 However, the challenge is their detection in physiological media at therapeutic levels. Andersson et al.52,53 detected and separated cisplatin and the monoaqua derivate in plasma and blood samples53,54 by HPLC (combining Strong Cation Exchange (SCX) and Strong Anion Exchange (SAX) mechanisms in a coupled column system) with post-column derivatisation with DDTC and UV detection. The detection of these two cisplatin chemical forms in the same type of samples was also reported using a more sensitive method based on anion-exchange chromatography and off-line detection by GFAAS.33 Other cisplatin derivates like the dimer and the diaqua species were studied by RP-HPLC coupled to UV or ICP-MSdetectors in aqueous solutions of the drug.55 As in other reported articles,49 there is no evidence on the presence of the latter species in aqueous solution at room temperature, and it could only be obtained by removing the chlorides from the solution (with AgNO3 for instance). All the studies agree on the fast evolution of cisplatin to the monoaqua form in water, being inhibited by the addition of NaCl.56 Using RP-HPLC-ESI-MS, Cui and Mester57 demonstrated that at basic pH the excess of OH− displaces the chloride ions, promoting the formation of the dihydroxo derivative, while at acid pH only the monoaqua is produced. However, when performing in vitro or in vivo experiments, only physiological pH must be considered. The formation of the dimers is discarded in clinical samples.
 | ||
| Fig. 2 Hydrolysis of cisplatin. | ||
Looking for more sensitive methodologies, HPLC-ICP-MS experiments have achieved excellent detection limits (between 0.09 and 0.15 μg L−1) from biological and environmental samples58 for cisplatin and the two main hydrolysed derivatives, as well as for carboplatin and oxaliplatin. Other authors have reported detection limits of 0.7 μg L−1 for cisplatin by high-field asymmetric waveform ion mobility spectrometry (ESI-FAIMS-IT-MS).59 This mass spectrometry technique allows detecting and quantifying the intact drug and its most important hydrolysed forms without any previous separation technique.
The interaction between cisplatin and the chromatographic mobile phasesolvents and modifiers has been reported. Some interesting works by El-Khateeb et al.60 using RP-HPLC-UV and NMR conclude that MeOH is a better choice as mobile phase for HPLC than ACN, due to its lower interaction with the drug. The use of trifluoromethanosulfonic acid to adjust the mobile phase pH is recommended instead of carboxylic acids, and it is also reported that SDS does not interact with any of the drug forms. Other authors have evaluated the possibility to use formic or phosphoric acid as reagents for the mobile phase,55 being the later highly reactive with cisplatin and its derivatives.
Carboplatin was designed with the aim of keeping the antitumoral properties of cisplatin and reducing its toxicity with the incorporation of more stable leaving groups. The result was a less toxic drug for the gastrointestinal tract, less neurotoxic and lacking of nephrotoxicity, being myelosuppression the dosage limiting factor. The same amount of carboplatin produces less DNA–Pt adducts than cisplatin with a slower formation rate.61 Nevertheless, when used at their appropriate dosage, which is higher for carboplatin than for cisplatin, the clinical efficacy is similar for both drugs.62 A low percentage of the intact carboplatin evolves to the hydrolysed forms in biological media because of the high stability of the drug in aqueous or saline solution.58 Up to eight minor hydrolysis products of carboplatin derivatives have been detected by RP-HPLC-UV and three of those species have been identified by ESI-MS.63 Other authors have also detected the dimer by RP-HPLC-UV-ESI-MS.64
Unlike cisplatin, oxaliplatin is less stable on growing the chloride concentration. Oxaliplatin undergoes a spontaneous transformation in aqueous biological media, being Pt (DACH)Cl2 its main product.65 The oxalate group can be shifted by nucleophile groups as Cl− or HCO3− and either by H2O. Some of the hydrolysed or chloride structures of oxaliplatin, which may be found in urine and blood, are described in the literature66,67 and shown in Fig. 3.
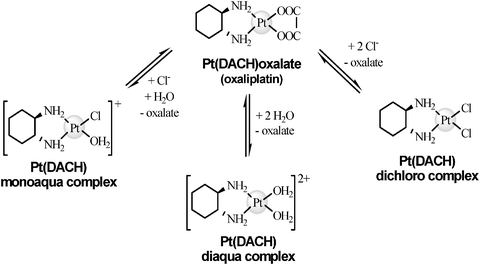 | ||
| Fig. 3 Hydrolisis of oxaliplatin. | ||
3.2 Interaction of Pt-based drugs with blood biomolecules
Since intravenous injection is the usual way to administer Pt-containing drugs, their interaction with blood biomolecules will determine aspects like toxicity, antitumor efficiency, transport, accumulation and excretion.68Cisplatin interacts to a high extent with blood biomolecules69 remaining free only about 20% of the drug 24 h after its administration.70 This fact and the high saline content of blood prevent cisplatin from significantly evolving to the monoaqua form.53,54 Furthermore, the low amount of monoaqua derivative found in blood comes from the originally administered drug itself. The analysis of blood from patients, treated with cisplatin between 5 and 17 years before, showed levels of Pt which were between 2 and 3 orders of magnitude higher than basal.71 Such persistence is in agreement with the statement by some authors that the drug–proteinadducts formed in blood act as an storage for the drug.72
The low reactivity of carboplatin with blood biomolecules explains the lack of works describing this interaction. A great amount of the drug is excreted intact and some authors have reported that about 60% of the drug incubated with rat ultrafiltrated plasma is recovered intact.73 Even so, carboplatinadducts with proteins having molecular weights similar to HSA and γ-globulin were recently detected by SEC-ICP-MS.74
When oxaliplatin enters the blood stream, its oxalate group is lost, resulting in new and highly reactive species of the drug. The intensive interaction of oxaliplatin with blood biomolecules66 explains that after 5 h of incubation, between 85 and 88% of the drug is bound to blood plasma biomolecules.75In vivo experiments confirm this trend, showing that up to 95%76 and 98%77 of the detected drug are bound to blood biomolecules after 5 and 21 days from the administration, respectively. The irreversibility of the oxaliplatin bond with red cells75 and the lack of Pt flow from erytrocithes76 seem to indicate that blood does not work as an active drug-delivering system.
SEC-ICP-MS analysis of ultrafiltrated plasma from patients treated with oxaliplatin shows that about 40% of the Pt elutes in the range of 160–200 KDa, 40% around 60 KDa and 15% over 2 KDa, which are equal to the Mr for γ-globulin, HSA and adducts of the drug with low Mr molecules, respectively.78
Among blood proteins, transferrin (Tf), albumin (HSA) and hemoglobin (Hb) are important biomolecules that play indispensable roles related, for instance, with the transport of essential substances. The interaction of Pt-based drugs with these proteins has been studied in depth for decades.
The high stoichiometries of the Pt-drug–HSA adducts observed cannot be explained by the only free cysteine group present in the protein (Cys 34). Experiments using bovine serum albumin (BSA) demonstrated the cleavage of disulfide bonds as a result of their interaction with cisplatin82,83 and transplatin.84 This was probably followed by an intramolecular interaction and the consequent modification of the secondary structure and the biological functions of the protein. Even considering Cys 34 and the disulfide bonds cleavage, additional binding sites must be involved. Indeed, methionines, histidines or even tyrosines have also been suggested as reactive groups towards cisplatin.72,85,86 In that way, a very recent work based on multidimensional protein identification technology (MudPIT) methodology describes precisely some binding sites for the Cisplatin–HSA interaction,87 including cysteine residueCys 34, two methionine sites (Met 329 and Met 548) and the tyrosine and aspartateO-donor sites Tyr 150 and Asp 375.
Slight alterations in the bond environment or secondary structure modifications can produce conformational changes in the α-helix structure (intact HSA has a 67% of α-helix). One way to detect this conformational change is to measure the quenching of fluorescence of Trp.83 Depending on the drug![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :protein molar ratio, the fluorescence response can decrease between 30 and 75%. Other techniques like circular dichroism (CD),88UV spectroscopy83 or FTIR, verify changes in the α-helix structure that range between 15 and 45%. Disulfide bond cleavages were also observed in cisplatin–γ-globulin interactions.
:protein molar ratio, the fluorescence response can decrease between 30 and 75%. Other techniques like circular dichroism (CD),88UV spectroscopy83 or FTIR, verify changes in the α-helix structure that range between 15 and 45%. Disulfide bond cleavages were also observed in cisplatin–γ-globulin interactions.
Cisplatin may also lead to HSA dimerisation. SEC-UV experiments86 allowed detecting Pt-species of around 140 KDa and probably the disulfide bond cleavages could be related with this aggregation process. Similar results were reported by Einhäuser et al.,69 not finding Pt-species in molecular weight fractions lower than 100 KDa. The interaction of cisplatin with HSA may produce additional effects like a change in the affinity for other ligands. For instance, the affinity of HSA towards heme group is strongly reduced by the presence of the drug bound to HAS.89
Finally, it should be mentioned that carboplatin seems to enhance its chemotherapeutic action when it interacts with HSA.90 This conclusion was extracted from experiments where a greater accumulation of the drug was observed in several organs after a combined administration of the drug and a significant amount of HSA, suggesting a possible role in the transport and delivery of Pt drugs by albumin .
 | ||
| Fig. 4 ESI-Q-TOF deconvoluted spectra of apo-Tf incubated with cisplatin simulating physiological conditions. | ||
Additionally, bottom-up experiments, combining RP-ICP-MS and RP-ESI-Q-TOF have shown the presence of Pt in peptides by comparing the isotopic pattern of the detected Pt-peptides with the theoretically expected isotopic pattern. The successful application of tryptic digestions to analyse Pt-adducts could lead to new approaches in Pt-containing drug metallomics, avoiding the highly complex analysis of intact macromolecules. This strategy requires a high strength of the drug–Tf bond to ensure that the detected Pt-species come from the original adduct .45 Other successful application of the bottom-up strategy to study Pt-binding proteins has been performed by Will et al.87 Some cisplatin–Tf binding sites as Met 256 or O-donor sites as Glu 265, Tyr 314, Glu 385 and Thr 457 were proposed using 2D-LC-ESI-MS.
The interaction of cisplatin with iron-saturated apo-Tf is not influenced by the presence of the metal. Moreover, iron is not displaced by cisplatin, indicating different binding sites for cisplatin and iron, being the results for apo-Tf and holo-Tf comparable.35
Several authors have compared the cytotoxic behaviour of free cisplatin with that of cisplatin bound to Tf.95 These in vitro experiments with human epidermis cancer cells show that the cisplatin–Tf adduct decreases the drug cytotoxicity.
Oxaliplatin interaction with holo-Tf has also been studied.96HPLC-ICP-MS and nanoESI-Q-TOF analyses lead to the conclusion that the formed adduct may consist on an intact molecule of oxaliplatin bound to the protein.
Regarding the reactive chemical form of the Pt-based drug, Mandal et al.98 proposed for cisplatin that (Pt (NH3)2)2+ is the residue bound to the Hb. An increase in the cisplatin concentration and temperature enhances the adduct formation and the heme group loss. Clinical concentrations of about 0.05 μmol L−1 of cisplatin are enough to obtain the cisplatin–Hb adduct .99
Recent works in the study of the oxaliplatin–Hb adducts identified the forms [(αhβh)2-oxaliplatin] and [(αhβh)2-Pt-DACH] by nanoESI-Q-TOF.100
3.3 Pt-based drugs excretion
Urine is used to follow the time course of Pt excretion after the drugs administration and to evaluate their biological half-time. Begerow et al.101 obtained basal levels of Pt in urine within the range 0.48–7.65 ng L−1 employing the high sensitivity technique double focusing (DF)-ICP-MS. These low basal levels allow the detection of changes in the Pt content in urine. In this way, concentrations of 180 ng L−1 of Pt were detected by voltammetry in patients treated 17 years earlier with cisplatin.71 The chemical species and the levels of the excreted drug are related with its reactivity and toxicity. The fact that carboplatin is excreted faster is in accordance with its low reactivity.The comparison of the excretion data available is complicated because of the variety of treatments and dosages used. Some studies reported that, after a single injection or prolonged injection treatments of 40 to 140 mg m−2, between 10 and 40% of cisplatin is excreted in the urine during the first 24 h.102 After 5 days of prolonged treatment with dosages between 40 and 100 mg m−2, 35–51% of the administered Pt has been excreted. Despite being found in bile or in the large intestine, Pt excretion by faeces is not significant.
Pt-species in human urine have been separated and identified by RP-HPLC-ICP-MS, 1H and 13C NMR and FAB-MS103 after the administration of 50 mg m−2. Intact cisplatin, the monoaqua derivative, cis-[Pt (NH3)2Cl(Creatinine)]+ complex, cisplatin–urea and cisplatin–uric acidadducts were detected just after the administration, being the Pt concentration measured of 30–50 mg L−1. Three weeks later, between 50 and 100 μg L−1 of Pt were found in urine and transformations in all the initially detected Pt-species were also observed. The same Pt-species were detected in urine from rats with induced diabetes, despite the fact that diabetic rats are resistant to cisplatin nephrotoxicity.104
Urine has also been proposed as the main excretion route for carboplatin.105 Between 50 and 70% of the drug is excreted 6 h after the treatment, in a process that is faster than for cisplatin. Besides, about 32% of the drug remains intact in urine. Three weeks later, high concentrations of Pt are measured in urine, but carboplatin undergoes biotransformation processes to Pt-biospecies different from the original form. In vivo transformation of carboplatin to cisplatin106 is supported by HPLC-ICP-MS studies where a co-elution of cisplatin and its monoaqua derivative with some Pt-species formed from carboplatin is observed.
The high reactivity of oxaliplatin is shown both in the excretion rate of the drug and in the amount of Pt-species found in urine.68 After 5 days from the administration of single dosages of 130 mg m−2, about 54% of the administered drug was excreted by urine and only 2% by faeces.107 Similar results have been reported elsewhere.77,108 Up to 16 Pt-species have been found in urine from in vitro experiments.109 The use of mass spectrometry techniques allowed the identification of Pt(DACH) bound to dicreatinine (24%), methionine (6%), monochloro (2%), monochlorocreatinine (14%), dichloro (7%) monocreatinine (11%) and the complex Pt(DACH)(H2O)2. Parallel in vivo experiments showed similar results: Pt (DACH) bound to dicreatinine (1–4%), methionine (1–7%), monochloro (2%), monochlorocreatinine (1–20%) and monocreatinine (1–10%). Pt (DACH)-glutathione (2–18%) was also identified, confirming the possible detoxification role of the intracellularglutathione.
3.4 Accumulation and distribution in tissues of Pt-based drugs
The chemical form of the drug when administered, the design of the administration procedure and the use of protective agents have influence in the final behaviour of the drug. For instance, low concentration dosages, waiting several weeks between injections, minimise the accumulation of Pt in organs.34 A slow administration of each of the dosages is also beneficial for the patient.110 Other common practices are the previous hydration of the patient, the administration of low amounts of NaCl or NH4Cl to prevent cisplatin nephrotoxicity,111 or the use of sodium thiosulfate to avoid ototoxicity.112 Other way to prevent toxicities is the use of protective or rescue agents which produce easily excretable Pt complexes.113Prior to the speciation analyses, accumulation studies are basic to know the distribution and affinity of the Pt-based drugs in the organism. For several decades, studies measuring the Pt content in different target organs have been reported. Human tissues from patients with different tumours and subjected to different treatments based on cisplatin were analysed by X-Ray Fluorescence. Liver, kidney and prostate were the organs with the highest Pt levels. Lower concentrations were found in bladder, muscle, testicles, pancreas and spleen, being the lowest concentrations detected in bowel, adrenal gland, heart, lung, brain and cerebellum.114 Similar works, performed by NAA, point out liver, kidney and testicles as the organs that accumulate cisplatin the most.30 The gamma camera spectroscopy employed by Areberg et al.115 is an interesting contribution to visualise the cisplatin accumulation and distribution. Recent studies carried out by ICP-MS agree with the preferential Pt accumulation in kidney and liver from rats treated with cisplatin, carboplatin and oxaliplatin.35 Although the accumulation pattern in the studied organs is similar for the three drugs, oxaliplatin relative accumulation is higher than for cisplatin and both greater than for carboplatin. The analysis of inner ear and kidney tissues, both toxicologically affected by Pt drugs116,117 revealed that for inner ear, the accumulation of cisplatin was higher than that for oxaliplatin or carboplatin, while in kidney, oxaliplatin accumulation was the highest observed. These results suggest that the key factor in toxicity may be the drug structure itself rather than the total Pt content in the organ. Furthermore, a ten-fold higher accumulation of Pt in the cochlea than in brain was found, being the elimination rate of Pt higher in cerebral tissue.
The ratio of Pt in blood plasma versus Pt in rat tissues is similar during the treatment with cisplatin and carboplatin.29 About 4% of the administered Pt was accumulated in rats after 3 months and 5% in pigs and humans after 11 months.118
Tissue fractions and sub-fractions from rats treated with cisplatin, carboplatin and oxaliplatin have also been analysed.35 The distribution of Pt among the nuclear, mitochondrial and cytosolic fractions was evaluated and a fractionation study among cytosolic biomolecules was performed. Although the three drugs present different accumulation rates in the organs tested, once they are within the cell, the three drugs show a similar distribution. The cytosolic fraction and especially a low molecular weight sub-fraction were attractive samples for speciation studies due to their significant Pt content, 47–63% in the cytosol and 20–30% in the fraction with molecular weight lower than 50 kDa.
3.5 Interaction of Pt-based drugs within the cell
Cisplatin, carboplatin and oxaliplatin enter the cell as neutral molecules, mainly in a passive diffusion process,43 but some authors have proposed a mixed mechanism.119 In this sense, protein-mediated transport systems, such as the copper transporter Ctr1120–122 have been proposed. However, despite some biological and biochemical evidences supporting this latter hypothesis, it is still unclear how exactly the entrance process occurs. In this regard, some approaches have been made for the identification of protein targets in a whole cellular system, where membrane proteins were found in the electrophoretic band with the highest Pt content.37 Although a preferential interaction between cisplatin and membrane proteins with respect to phospholipids has been described,123 the weak and reversible binding of the drug to the latter seems to result in structural changes within the membrane, causing alterations in properties such as permeability.124000 and 1 per 300000 bases for 1,2-d(GpG) and 1,2-d(ApG), respectively.136Oxaliplatin and carboplatin produced in cells fewer DNA–Pt adducts compared to those formed by cisplatin.133,137 However, less DNA lesions are needed for cell growth inhibition when either, oxali- or carboplatin are used instead of cisplatin.137
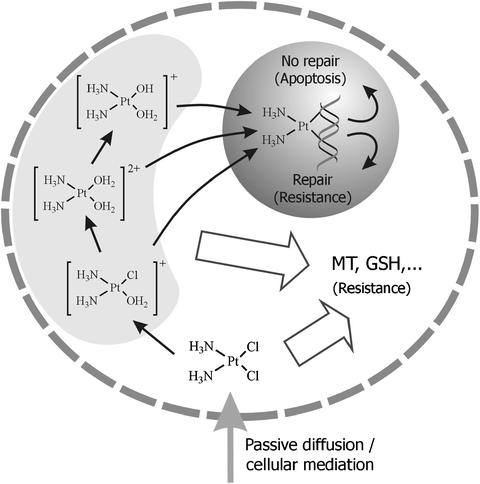 | ||
| Fig. 5 Scheme of the entrance and interactions of cisplatin in the cell. | ||
 | ||
| Fig. 6 Interaction of cisplatin with DNAnucleobases. | ||
The formation of DNA–drugadducts is a pharmacokinetic parameter to optimise in cancer therapy. Therefore, the quantification of DNA–drugadducts is of paramount importance. These adducts are indicators of the exposure to the drug, but they cannot be taken as direct and definitive proofs of genotoxic damage. In fact, a major clinical limitation for the use of cisplatin is the intrinsic or acquired resistance to the drug, mainly due to an increased level of repair of DNA–cisplatinadducts . There is a need for specific chemical methods to selectively detect those adducts formed in vivo at pharmacological concentrations.
The techniques traditionally used to study the interaction of these drugs with nucleobases have been, predominantly, X-ray crystallography,138NMR139 and immunoassay .133 However, they present serious disadvantages, namely that crystals of a suitable quality are required for crystallography, that a relatively high quantity of high-purity sample is required for NMR and that by using immunoassay techniques it is not possible to discern between different types of adducts . Tests used to evaluate Pt-drugs genotoxicity include those directed to study changes in DNA integrity such as the Comet assay , TUNEL methods, the number of micronuclei (MNi)140 and 32P post-labelling. The widely used Comet assay cannot differentiate among the many different types of DNA damage. Although 32P post-labelling provides excellent detection limits (it is possible to detect 1 adduct in 1010nucleotides), it is a complex assay (including radioactive phosphorus) and is not specific for cisplatinadducts .141
The highly sensitive and selective sector field ICP-MS technique was coupled to ion exchange chromatography and UV detector (HPLC-UV-SF-ICP-MS) for the investigation of the time-dependent reaction course of the cisplatin–5′ guanosine monophosphate (5′-GMP).142 Measuring the molar ratios of P and Pt provides direct stoichiometric information and facilitates the chromatographic identification of the major cis-[Pt (NH3)2(GMP)2]2−adduct . Parallel studies about binding kinetics, the nature of the adducts formed and the location of the binding site within the specifically designed double-stranded DNAoligonucleotides were carried out with the highly sensitive and selective Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR-MS)143 technique. It was demonstrated that binding to DNA takes places via a [Pt (NH3)2Cl]+ intermediate prior to the formation of bifunctional [Pt(NH3)2]2+adducts .
D. Garcia et al.144 performed in vitro incubations of individual DNAnucleotides with cisplatin. The cisplatin–nucleotideadducts (containing both Pt and P) were separated in a narrow-bore C8 column from the unreacted nucleotides (containing P but not Pt), and monitored by HPLC-ICP-MS. Incubations of cisplatin with either, a commercial oligonucleotide (dGMP) or Calf thymusDNA samples, followed by enzymatic hydrolysis, were prepared to ascertain the quantitative capabilities of this procedure. The successful quantitative analysis of the formed adduct : [Pt (NH3) (dGMP)2] confirmed the suitability of the method. Structural characterisation of the complex formed was performed by ESI-Q-TOF. These authors145 also proposed a sensitive and selective method by capHPLC-ICP-MS for the determination of 1,2-d(GpG) intra-strand cisplatin–DNAadducts . Somatic cells of Drosophila Melanogaster flies were exposed to cisplatin at biologically relevant concentrations of 0.5 and 1 mM (inducing damage but not death) and reaction products were cleaved by enzymatic hydrolysis of the DNA samples. These studies showed a direct correlation between the concentrations of cisplatinadducts , the induced genotoxic damage (measured as DNA strand breaks by Comet assay ) and the cisplatin concentration in flies whose response to DNA damage is known to be equivalent to that of mammals.
The amount of DNA–oxaliplatinadducts formation in white blood cells of patients with metastatic colorectal cancer was evaluated by Weber et al.146 using adsorptive stripping voltammetry (ASV). DNA was isolated by gradient density centrifugation and a four step solid phase extraction procedure and quantified by UV spectroscopy. Pt was determined after mineralisation of the DNA sample by ASV (formazone method). It was possible to determine Pt:nucleotide ratios in these samples down to five Pt atoms in 108nucleotides. With this method the time-dependent formation of oxaliplatin–DNAadducts can be monitored in clinical studies, which may help to understand inter-individual differences in the responses of patients to oxaliplatin-based therapy.
Using a linear ion trap ESI-MS, Kerr et al.147 demonstrated the formation of adducts between oxaliplatin and each of the DNAnucleobases in solutions containing them either individually or in mixtures. When the drug was incubated with the four bases together at close to equal molar concentrations, adducts containing predominantly adenine and guanine were found, cytosineadducts were formed to a lesser extent, whilst the yield of thymineadducts was minimal. A large excess of drug was required to ensure the formation of cytosine and thymineadducts in the presence of adenine and guanine. Even with a large excess of oxaliplatin, only mono-adducts of these nucleobases were observed when the four of them were present. This follows the expected pattern of mono-substitution being favoured over di-substitution and a reduction in successive formation constants. Consecutive losses of H2, ammonia and nucleobases from each of the cisplatin–nucleobaseadducts was demonstrated by multiple-stage tandem mass spectrometry (MSn), enabling the determination of the fragmentation pathways for each adduct and the elucidation of their respective structures. It was demonstrated that ESI-MSn is an ideal technique for studying these type of adducts , since structural information can be obtained in relatively short periods of time and the disadvantages of NMR and X-ray crystallography are avoided. These results provide the initial step towards a more complete understanding of the interaction and binding mode of the drug with intact DNA, where other factors such as steric hindrance and binding site geometry might have influence on the binding sites and the stability of the drug complex.
Le Pla et al.148 have developed a HPLC-ESI-MS/MS method that is capable of specifically measuring 1,2-d(GpG) and 1,2-d(ApG) oxaliplatin intra-strand crosslink adducts in DNA from liver, lung and kidney samples obtained from wild-type and GSTP1/2 null mice treated with the drug. Limits of detection for 1,2-d(GpG)-oxPt and 1,2-d(ApG)-oxPt were 23 and 19 adducts per 108nucleotides, respectively. The main advantages of this methodology over 32P post-labelling are the relatively quicker and easier sample preparation for the analysis, providing structural information that specifically identifies the adduct being analysed. No significant differences were observed in the levels of intra-strand crosslinks formed by oxaliplatin between the mouse strains in liver, kidney and lung DNA.
These secondary interactions may dramatically decrease the amount of free drug available to react with DNA, thus, reducing the cytotoxic effect in the cell. Moreover, those processes may be involved in the resistance observed in some tumours (Fig. 5) and the severe side-effects the treatment implies. For these reasons, the study of the interactions between Pt-drugs and cytoplasmic compounds has become a question of paramount importance. Among these, sulfur-containing MTs and GSH have been subjects of particular interest.149 When over-expressed, MTs have been proved to increase cell resistance to cisplatin and to reduce its nephrotoxicity.150 Moreover, in cancer cells basal MT levels are especially increased. In the case of GSH, a decrease in its cellular levels reduces the resistance to the drug and increases its cytotoxic effect.124 New generation Pt-based drugs have been designed to reduce the interaction with cytosolic sulfur-containing molecules and the resistance processes observed in cisplatin and carboplatin treatments. For instance, picoplatin, currently in phase III trials, presents a steric bulk around the Pt centre that leads to a decreased inactivation of the drug in presence of GSH151 or MT.152
3.5.2.1 Metallothioneins. Metallothioneins are low molecular weight, cysteine-rich (20 Cys residues) proteins known to bind several metals, such as Cd, Zn, Cu, Hg, with high affinity, producing metal–thiolate clusters. Each MT molecule is able to bind up to 7 atoms of Zn, Cd or Hg.153 In the case of Cu, the protein binds up to 12 atoms. Among those elements, Zn may be easily displaced by other metals, conferring on MT the ability to coordinate with potentially toxic metals present in the cell and consequently, leading to a detoxifying effect.
Coordination to Pt has also been proposed. Indeed, zinc′s displacement in the presence of cisplatin has been demonstrated by SEC-ICP-MS, while cadmium atoms remained bound to the protein.154 However, Hagrman et al.155 observed that not only Zn is displaced in MT in the presence of cisplatin but also Cd, making use of SEC with AAS detection. The key for these contradictory results may lay on the pH and the cisplatin/MT ratio assayed. In fact, the influence of the pH on the binding of Zn and Cd to MTs has been revealed by ESI-MS analyses.156 Simulating physiological conditions and pharmacological cisplatin levels, it was confirmed by SEC-ICP-MS that only Zn is rapidly displaced from MT by cisplatin.157 There is still scarce work directed to identify the cisplatin species that react with MT. Making use of X-Ray diffraction experiments, Pattanaik et al.158 suggested that cisplatin loses both amine and chloro ligands, coordinating to 4 sulfur atoms from MT. Another recent study by Knipp et al.,159 using simulated physiological conditions, confirmed by nESI-MS analyses that cis-[Pt(N-donor)2Cl2] compounds rapidly release all their ligands upon their reaction with Zn7MT-2, in contrast to trans-[Pt(N-donor)2Cl2] compounds, which tend to retain their N-donor ligands. In both cases, the previously observed substitution of Zn atoms for Pt was demonstrated.
The kinetics of the reaction between cisplatin and MT has also been studied. Hagrman et al.155 proposed reaction rates between first and second order in excess of cisplatin. Knipp et al.159 performed a kinetic study on the reaction of Zn7MT-2 with a 2-fold molar excess of several cis- and trans-[Pt (N-donor)2Cl2] complexes, obtaining kinetic constants of the same order of magnitude as those previously reported. Kinetics experiments prove that the nature of the ligands attached to the metallic centre exerts a great influence on the reaction parameters, due to the steric hindrance and the modulation of the Pt electrophilicity they may produce. Interestingly, trans-compounds reacted faster than their corresponding cis- isomers but to a similar extent.
Disagreement arises about cisplatin–MT adducts stoichiometry and binding sites. Cd4–Ptn-MT (n = 1–7) adducts were identified by nESI-MS, from incubations at neutral pH of MT in excess of the drug.154 On the other hand, up to 10 molecules of cisplatin were observed to bind to MT exposed to an excess of the drug.158 Furthermore, Knipp et al.159 observed Zn(7−n)Ptn-MT-2 (n = 0–3) and Zn4Pt4-MT-2 adducts after the incubation with a two-fold molar excess of cisplatin.
Recently, a top-down approach160 using nESI-Q-TOF allowed the identification of Cys5 and Cys7 as Pt-binding sites on MT incubated with cisplatin in a 1:5 molar ratio at neutral pH. However, apparently not only the highly reactive cysteine residues would be involved in the binding of Pt to MT. In fact, SEC-HPLC–UV155 experiments showed that sequential reactions take place during the interaction of the drug with MT, at the beginning not affecting the thiol groups absorbance.
In brief, there are still contradictory results in the few studies concerning the characterisation of the binding of cisplatin to MT, due to the diverse experimental conditions of the in vitro incubations used. It is therefore crucial to simulate physiological conditions and therapeutic concentrations of the drug to get information, which might be interesting from the biomedical point of view.
3.5.2.2 Glutathione . Glutathione, GSH (γGlu-Cys-Gly), is a peptide present at the cytosol at mmol L−1 concentration levels. GSH-Pt-drugs complexes can be excreted via an ATP-dependent pump able to transport outside the cell molecules with the structure GS-X, producing cellular resistance against Pt-drugs (cisplatin,161carboplatin, and to a lesser extent, new generation Pt drugs162).
The interaction of cisplatin, carboplatin and oxaliplatin with GSH has been extensively studied. Some approaches to the kinetics of the reaction reveal that, although cisplatin interacts more intensively with GSH than the other two drugs, the velocity of the reaction is, in the first stages, similar for cisplatin and oxaliplatin and up to five times faster than for carboplatin.163 These experiments, carried out under physiological conditions, using HPLC-UV to monitor the Pt–S bond, also reveal a first order reaction between all these drugs and GSH. Dabrowiak et al.164 performed similar studies by HPLC-UV and AAS, confirming the previously observed trend and concluding that the slow kinetics observed is in accordance with the efficient platination of DNA under mmol L−1 concentrations of GSH.
Structural elucidation of cisplatin–GSH adducts has also been performed. Miao et al.165 have suggested several structures for these adducts based on ESI-MS results. Two molecules of cisplatin could bind to one or two molecules of GSH through their thiol groups, when incubations were prepared at molar ratio 1:1 in aqueous media. However, the unrealistic conditions used in these experiments put into question the actual production of these species in vivo, considering the drug:peptide ratio used, and taking into account that in these experiments the reactant was the diaqua- complex and not cisplatin, something that is far from occurring in vivo. This may be the reason why Bernareggi et al.166 could not identify in real samples what they were able to characterise in vitro, incubating cisplatin and GSH (1:1) at pH 7.4. Furthermore, their results provided by HPLC-TQ MS/MS suggest a structure in which one cisplatin molecule binds to GSH, in contrast to the results by Miao et al.165 Contradictions often reside in the different experimental conditions used. In order to shed light on the real nature of the adducts generated in vivo, incubations performed in vitro at physiological conditions (pH 7.4, 37 °C, 4,6 mM NaCl, 500 μM GSH, 1 μM cisPt) were studied by Esteban-Fernández et al.157 Time-dependent evolution of the reaction, studied by SEC-ICP-MS, allowed the monitorisation of the incubation mixtures, finding out that two main species were produced. It has been shown that GSH is able to react with both, the cisplatin monoaqua- derivative and the dimer. Analysis by ESI-LIT-MS/MS also allowed the identification of one of the compounds, previously described by Miao et al.,165 in which two cisplatin molecules react with a GSH molecule. All these facts highlight again the need to simulate physiological conditions and therapeutic concentrations of the drug, in order to characterise the real adducts that may be present in clinical samples.
3.5.2.3 Other biomolecules. Some other potential ligands present at the cell have been object of study in an attempt to get a better understanding of the reactivity of Pt-drugs towards different cellular biomolecules, and to set the basis for the development of a reliable analytical methodology that may be useful for the characterisation of the Pt bio-compounds that may be formed in vivo in cells and tissues.
Although sulfur-containing groups in proteins, such as the aminoacids methionine or cysteine, have been considered as the main binding sites for Pt-based drugs, recent articles postulate O- and N-donors as other occurring binding sites. Bovine erythrocytesuperoxide dismutase was incubated with a 10-fold excess of cisplatin and analysed by X-Ray diffraction.167 Results showed His19 as the primary binding site for the drug, above Cys or Met residues. Similarly, the binding of cisplatin, transplatin, carboplatin or oxaliplatin to hen egg white lysozyme was studied at incubation ratios of 3:1 and analysed by ICP-OES,168 showing low platination levels for all the drugs tested. ESI-MS experiments revealed predominantly platinated mono-adducts, suggesting the presence of a preferential Pt binding site, which was identified by X-Ray diffraction analysis as the imidazole Nε of His15. Additionally, using ESI-MS, Miao et al.165 observed the participation of N-donors in the interaction of several sulfur-containing peptides with the cisplatin diaqua-complex. The adduct formed with Met-Arg-Phe-Ala involves peptide cyclation as a result of the interaction of the drug’s derivative with both the N-terminus and the sulfur in the methionine residue. On the other hand, the peptide (Ac)-Met-Ala-Ser coordinates to the diaqua-complex through the S in methionine and the deprotonated N in the amide adjacent to the acetate group.
An innovative approach for the elucidation of the binding sites of Pt-based drugs was proposed by Yang et al.169 on the study of the interaction of carboplatin with cytochrome C (Cyt C). A great excess of the drug was used in the incubations, but the observed stoichiometry carboplatin: Cyt C was 1:1 and 2:1 at pH 5.0 and 37 °C, while it was only 1:1 at pH 7.0 and 37 °C (closer to physiological conditions). However, the most relevant conclusion reached was that carboplatin induces the rupture of Cyt C on acidic conditions at high temperature (pH 2.5 and 50 °C), cleaving the peptide bond adjacent to the binding site. The ESI-MS and MS/MS analysis of the induced fragments allowed a straightforward identification of the binding sites of carboplatin, which turned out to be Met65 and to a lesser extent, Met80, thus, making unnecessary the use of an endopeptidase for this purpose.
The interaction of Pt-drugs with Cyt C has been object of further research, bearing in mind that this model protein presents several potential binding sites: Cys14, Cys17, Met65, Met80, His18, His26 and His33. Casini et al.170 studied different Pt–proteinadducts from incubations of Cyt C with either cisplatin, transplatin, carboplatin or oxaliplatin, in a drug molar excess ranging from three- to ten-fold, at pH 7.4, 37 °C, at different incubation times. Results obtained by ICP-OES indicated a significant degree of cytochrome C platination (1.2 to 1.5 mol of Pt per Cyt C) for all the drugs tested at a 3-fold excess, 72 h, while at a 10 fold-excess, 168 h, platination levels increased (6.7 and 5 moles per Cyt C, for cisplatin and carboplatin, respectively). Strikingly, the four Pt-drugs showed a similar pattern of reactivity with the protein, in contrast to the previously established different reactivity of these compounds. On the other hand, UV-visible spectra showed that the main visible bands on Cyt C and thus, the chromophore, remain unaltered in the presence of Pt-drugs. Furthermore, ESI-MS direct analysis of the samples revealed several adducts with Pt:protein stoichiometries of 1:1, 2:1, 3:1 or even 4:1, at the different incubation conditions, showing the survival of the adducts during the ionisation process. The authors predicted Met65 as the primary binding site and His26 and His33 as secondary binding sites.
In a subsequent article,171ESI-MS experiments were performed on incubations of carboplatin and Cyt C, prepared either in water (pH 5-6), in ammonium carbonate (pH 7.4), or in TMeAmAc (tetramethylammonium acetate) buffer (pH 7.4) with Pt:protein ratios of 3:1 or 10:1. Considering their results, the reactivity of carboplatin, which is considerably low in aqueous solution, is enhanced in the presence of certain compounds, such as Cyt C. Its binding to the protein occurs giving rise predominantly to 1:1 adducts , either by losing the cbdca (1,1-Cyclobutanedicarboxylate) ligand or through a ring-opening reaction, keeping the cbdca and releasing the amine. Moreover, the influence of the incubation conditions on the adducts generated was confirmed, showing that the carbonatebuffer induces an increase on the amount of cisplatin-like species bound to Cyt C. It was demonstrated, as well, that the platinated protein maintains its ability to react with 5′-GMP, producing ternary complexes. Finally, proteolysis of the incubation solutions with Asp-N, gave rise to platinated peptides, in which Met65 was the primary binding site and the histidines the secondary sites. Contrary to Yang et al.,169 these authors discarded Met80 as a possible binding site, due to its participation in the coordination to iron in the heme pocket. However, no MS/MS experiments and thus, no sequencing, were performed in order to prove these hypotheses.
Ubiquitin, which is relevant as a tag for protein degradation, has also been selected as a model protein to study protein interactions with Pt-based drugs. It is a small protein, 8565 Da, containing Met and His, both reactive aminoacids towards Pt. The utility and potential of mass spectrometry techniques for the study of platinated adducts have been demonstrated again on the aforementioned model. Gibson et al.172 analysed by ESI-MS aqueous incubations of ubiquitin with cisplatin, using different reaction conditions. Several monoadducts, and also less abundant biologically unrealistic diadducts and triadducts were observed after 15 days in ten-fold excess cisplatin incubation, with no unreacted protein remaining in solution. The characterisation of the adducts was based exclusively on the masses observed, on the coordination chemistry of Pt and on the parallel results provided by transplatin and cis-[Pt(en)Cl2]. In this way, the mechanism of the formation of monodentate, bidentate and tridentate adducts was proposed, discarding as an explanation the loss of ligands during the ionisation process. A kinetics study revealed the quick binding of the first Pt moiety giving rise to an initially monodentate adduct , and its conversion into bidentate or tridentate over time. Comparing the observed reaction kinetics with that of the protein containing an oxidized Met1, less reactive, Met1 was proposed as the main binding site for cisplatin, while His68 would remain as a secondary option. Interestingly, tryptic digests of adducts provided no Pt-containing fragments, being the authors explanation that the overnight treatment with NH4HCO3 may release the Pt moiety from the protein. Finally, experiments in non-denaturing conditions demonstrated that the bidentate monoadducts do not disrupt the folding of the protein, while tridentate adducts , which exist in two conformations, are able to unfold it.
An interesting comparative study on the use of MALDI-TOF, nESI-IT-MS and nESI-Q-TOF was performed by Hartinger et al.,173 on incubations, in water at 37 °C of cisplatin, transplatin or oxaliplatin with ubiquitin at a molar ratio of 2:1. Their results are in good agreement with those from Gibson et al.172 indicating that all the drugs tested give rise to the formation of mainly monoadducts and to a lesser extent, diadducts and even triadducts in the case of cisplatin. Cisplatin and transplatin form similar species with Ub, with differences laying on their different stability and hydrolysis pathways, being Ub-[Pt(NH3)2] and Ub-[Pt(NH3)2Cl] the most abundant adducts observed for cisplatin and transplatin, respectively. For oxaliplatin, the predominant adduct was Ub-[Pt (cyclohexane-1,2-diamine)], involving the release of the oxalate ligand. The study revealed subtle differences in the results depending on the ionisation technique and the mass analyser used. In this sense, nESI-QTOF allowed the characterisation of several mono- and di-adducts for cisplatin and transplatin. Using nESI-IT-MS the detection of diadducts of oxaliplatin was possible. Although the suitability of MALDI-TOF for this purpose was also demonstrated, the higher degree of fragmentation induced on the adducts , makes this technique a less attractive choice than ESI-based instruments, being the former only a real option for the study of small metalloproteins.
Recently, a top-down mass spectrometric approach, making use of ESI-FT-ICR-MS/MS,174 was applied on the study of the interaction of cisplatin, transplatin and oxaliplatin on ubiquitin. Results from the direct analyses of incubations of the drugs with the protein, indicate that both cisplatin and oxaliplatin bind to N-terminal fragments containing Met1 (1Met-Gln2 and 1Met-Gln-Ile-Phe4, respectively), whereas transplatin is bound within the sequence 19Pro-Ser-Asn-Thr-Ile-Glu24 (containing several potential donor atoms). These are in agreement with previous results based on indirect methods, but surprisingly, according to this, His68 would not to constitute a significant binding site for none of the compounds tested. This methodology allows a direct determination of the primary binding sites, avoiding the digestion involved in classical bottom-up approaches and its possible subsequent reactions.
Recently, Moreno-Gordaliza et al.175 demonstrated the capabilities of a top-down mass spectrometric approach using an affordable linear ion trap (LIT) for the full characterisation of small platinum-binding proteins. For this purpose, insulin, a serum protein that presents several histidines and cystines, which are potential binding-sites for platinum, was selected as a model. In vitro incubations were prepared under acidic and physiological conditions at 1:1 or 1:5 insulin:cisplatin molar ratios and different incubation times. SEC-ICP-MS analysis enabled the specific detection of Pt-containing species attributed to the binding of the drug to the protein. Further analysis through MALDI-TOF-MS and nESI-LIT-MS allowed the identification of platinated mono-, di-, and even triadducts in the incubations. The zoom scan mode provided by the LIT offered enough resolution to discern the isotopic pattern of ions, allowing the differentiation of Pt-containing ions as can be seen in Fig. 7. Pt binding sites were identified by CID-MSn as: B Chain N-terminus, His5, and His10 residues, finding also evidence on the binding of Pt to B Chain Cys7. Although the main adduct observed for acidic incubations ([insulin+Pt(NH3)2Cl]) differs from the main adducts observed for physiological conditions ([insulin+Pt(NH3)2] and [insulin+Pt(NH3)2H2O]), the Pt binding sites turned out to be the same in all the cases.
![Ultrazoom scan nESI-LIT spectra of a mixture of insulin : cisplatin 1 : 5 incubated for 96 h in 0.1% TFA at 37 °C showing (a) the insulin ion with charge +5 at m/z 1146.93 and (b) a platinated insulin ion at m/z 1199.27 corresponding to [insulin+Pt(NH3)2Cl+4H]5+. Their respective theoretical isotopic patterns are included as insets for comparison.](/image/article/2010/MT/b911438f/b911438f-f7.gif) | ||
| Fig. 7 Ultrazoom scan nESI-LIT spectra of a mixture of insulin:cisplatin 1:5 incubated for 96 h in 0.1% TFA at 37 °C showing (a) the insulin ion with charge +5 at m/z 1146.93 and (b) a platinated insulin ion at m/z 1199.27 corresponding to [insulin+Pt(NH3)2Cl+4H]5+. Their respective theoretical isotopic patterns are included as insets for comparison. | ||
Speciation studies were performed on kidney and inner ear cytosols from rats treated with cisplatin, carboplatin and oxaliplatin, using two dimensional liquid chromatography separations and serving as a starting point to develop a methodology for the separation of Pt-containing biomolecules present in tissues. Analysis by SEC-ICP-MS and further analysis by SAX-ICP-MS with a previous head-column focusing of selected fractions, obtained in the first dimension separation, were carried out. Results demonstrated that Pt-drugs present at the cytosol are completely bound to biomolecules, being predominantly bound to compounds of 25–65 kDa and around 12 kDa for the inner ear, while in the case of kidney and liver, the main Pt-compounds comprised masses of 50–60 kDa and around 20 kDa.116,157
The stability and strength of the drug–biomolecule adducts should be studied in order to explore high resolution separation strategies that involve more aggressive sample treatments. In this way, the cytosolic fractions of kidney and ear impacted by cisplatin were subjected to several treatments common in gel electrophoresis, employing SDS, BME (β-mercaptoethanol), heating and sonication. Analysis by SEC-ICP-MS revealed that cisplatin–biomolecule interactions resisted denaturing conditions, basing on the absence of the peak corresponding to free cisplatin in the treated samples and the non significant chromatographic differences observed between treated samples and the original ones.35
Indeed, gel electrophoresis has already been successfully applied for the separation of Pt-containing proteins. In particular, Allardyce et al.37 administered cisplatin to Escherichia Coli cells, and proteins were partially separated by SDS-PAGE, followed by a Pt total content analysis by laser ablation ICP-MS. Membrane proteins were identified in a Pt-rich electrophoretic band by RP-ESI-Q-TOF, after an in-gel tryptic digestion. In this case, the outer membrane protein A (ompA) was identified and pointed out to be involved in the cellular cisplatin intake.
Moreover, a highly interesting study was recently presented by Will et al.,176 in which MudPIT was used to elucidate cisplatin binding sites in a whole cell system. MudPIT was applied on tryptic digests of extracts from Escherichia Coli cells treated with cisplatin at pH 7.0. MS/MSspectra were analysed by SEQUEST, making use of a search file where Pt-modified aminoacids had also been included. 31 protein targets were found for cisplatin, including DNA- and RNA-binding proteins, stress regulated proteins, enzymes and resistance efflux proteins. The main Pt coordination sites in the proteins were successfully identified as Asp, Glu, Ser, Thr, Tyr and Met. The level of detection of proteins with this technique is at 100 copies per cell, with a background of an abundant protein present at 106 copies. Therefore, at least 10% of the less abundant proteins must be platinated at a single site in order to be detected. In this case, some platinated peptides from those proteins were only probably be detectable because a high amount of cisplatin was used in the incubations. This is the main drawback concerning the applicability of this technique on real samples.
On the other hand, Coling et al.177 demonstrated the changes in the expression of 22 cochlear proteins after the administration of cisplatin on rats, using 2D-DIGE (two dimensional difference gel electrophoresis) and MALDI-TOF analysis. Five proteins underwent an increase in their expression while seventeen proteins decreased significantly after the cisplatin administration. Seven additional cochlear proteins were identified with less significant drug-induced changes. Those results were interpreted considering the functions of the identified proteins. Therefore, this work constitutes a pioneer proteomics study related to the ototoxicity produced by cisplatin.
4. Conclusions
Pt-based drugs efficacy is limited by their interactions with different biomolecules along their way to the therapeutic target. Metallomics studies clearly offer a way to improve the knowledge about the behaviour of these drugs in the organism.Some disagreements in the different studies performed pointed out the complicated interpretation of in vitro experiments, where small differences could result in contradictory conclusions. Furthermore, the low content of Pt-adducts and the complexity of real samples pose new challenges. To deal with these difficulties, refined analytical methodologies must be used.
Special attention must be paid to avoid bio-transformation or cleavage of the Pt-adducts along the sample preparation and analytical procedures. Even the original structure of the Pt-drug administered should be controlled to avoid dealing with its derivatives. Unfortunately, few papers discuss the stability of the sample.
Different separation and detection techniques may be combined to improve analytical results and/or to obtain more specific information. However, the high complexity of real samples makes recommendable the complementary use of both a molecular and elemental mass spectrometric detection technique. Pt and other elements can be highly sensitively monitored by LC-ICP-MS to follow Pt-species along the separation processes. Further structural information of the preconcentrated and purified Pt-adducts may be obtained by ESI-MS (Table 1).
| Ref. | Technique | Drug | Sample | Research topic |
|---|---|---|---|---|
| 55 | HPLC(RP)-ICP-MS, HPLC(RP)-UV | Cisplatin | Drug aqueous solution | Cisplatin interaction with mobile phases |
| 57 | HPLC(RP)-ESI-MS | Cisplatin | Drug aqueous solution | Evolution of the drug |
| 58 | HPLC-ICP-MS | Cisplatin, Carboplatin, Oxaliplatin | Wastewater from hospital | Separation of cisplatin, carboplatin, oxaliplatin and their derivatives |
| 60 | HPLC(RP)-UV, RMN | Cisplatin | Drug aqueous solution | Conditions for the analysis of cisplatin and its derivatives |
| 63 | HPLC(RP)-UV, ESI-MS | Carboplatin | Drug aqueous solution | Evolution of the drug |
| 64 | HPLC(RP)-UV-ESI-MS | Carboplatin | Drug aqueous solution | Evolution of the drug |
| 65 | HPLC-ESI-MS | Oxaliplatin | Drug aqueous solution | Evolution of the drug |
| 33 | HPLC, GFAAS | Cisplatin | Plasma | Evolution of the drug |
| 34 | ICP-MS, ICP-AES | Cisplatin | Plasma | Free drug quantification |
| 45 | HPLC(RP)-ICP-MS, HPLC(RP)-ESI-Q-TOF | Cisplatin | Human serum | Interaction of Tf and HSA with the drug |
| 52 | HPLC(SCX-SAX)-UV | Cisplatin | Plasma | Evolution of the drug |
| 54 | HPLC-UV | Cisplatin | Blood, plasma, ultrafiltered plasma (UFP) | Evolution of the drug |
| 53 | HPLC-UV | Cisplatin | Blood | Evolution of the drug |
| 56 | RMN | Cisplatin | Drug aqueous solution, UFP | Evolution of the drug |
| 69 | ICP-AES, ET-AAS | Cisplatin and other Pt-based drugs | UFP | Quantification of the drug bound to proteins |
| 70 | SEC-ICP-MS | Cisplatin | Serum | Separation of free drug and drug-binding proteins |
| 71 | Voltammetry | Cisplatin | Serum, urine | Drug quantification |
| 73 | HPLC(RP)-ESI-MS | Carboplatin | UFP, tumoral tissue | Free drug quantification |
| 74 | SEC-ICP-MS | Carboplatin | Plasma | Drug–protein interactions |
| 75 | HPLC(RP), FAAS | Cisplatin, carboplatin, oxaliplatin and others | Blood, red cells, plasma, cellular lines | Drug distribution, biotransformation , citotoxicity |
| 78 | SEC-ICP-MS, SEC-ESI-MS | Oxaliplatin | UFP, red cells, urine | Drug biotransformation |
| 65 | HPLC-ICP-MS | Oxaliplatin | UFP, GSH, Met, Cys | Interaction of sulfur-containing biomolecules with the drug |
| 80 | CE-ICP-MS | Cisplatin | HSA | HSA-Drug interaction |
| 81 | CE-UV | Cisplatin, oxaliplatin | HSA, Tf | Interaction of blood serum proteins with the drugs |
| 84 | GFAAS, UV, CD, Fluorescence | Transplatin | HSA | HSA-Drug interaction |
| 86 | NMR | Cisplatin | HSA | Binding sites to HSA |
| 87 | 2D-LC-ESI-MS | Cisplatin | Blood serum/Drug incubations | Binding sites to HSA and Tf |
| 91 | ESI-Q-TOF, UV/Vis | Cisplatin | Tf | Tf-Drug interaction |
| 92 | ESI-TOF-TOF | Cisplatin | Tf | Tf-Drug interaction |
| 94 | HPLC-UV, CE-UV NMR | Cisplatin | Tf | Tf-Drug interaction |
| 96 | SEC-ICP-MS, ESI-Q-TOF | Oxaliplatin | holoTf | holoTf-Drug interaction |
| 97 | SEC-ICP-MS, ESI-Q-TOF | Cisplatin, carboplatin, oxaliplatin | Red cells, Hb | Hb-Drug interaction |
| 98 | SEC-ICP-MS, ESI-Q-TOF | Cisplatin | Hb | Hb-Drug interaction |
| 99 | SEC-ICP-MS, ESI-Q-TOF | Cisplatin | Hb | Hb-Drug interaction |
| 100 | ESI-Q-TOF | Oxaliplatin | Red cells, Hb | Hb-Drug interaction |
| 101 | DF-ICP-MS | — | Urine | Quantification of physiological Pt levels |
| 103 | HPLC(RP)-ICP-MS, FAB-TQ, NMR | Cisplatin, carboplatin | Urine, creatinine, urea, uric acid | Drug biotransformation in urine |
| 30 | NAA | Cisplatin | Fluids, tissues | Pt quantification in fluids and tissues |
| 35 | FIA-ICP-MS, SEC-ICP-MS | Cisplatin, carboplatin, oxaliplatin | Rat tissues | Drug distribution among cellular fractions. Accumulation/elimination in tissues |
| 114 | X-Ray Fluorescence | Cisplatin | Fluids, tissues | Pt quantification in fluids and tissues |
| 115 | Gamma spectroscopy | Cisplatin | Human body | Pt distribution in tissues |
| 116 | SEC-ICP-MS, AE-ICP-MS | Cisplatin, carboplatin, oxaliplatin | Rat tissues | Separation of Pt-adducts |
| 118 | ICP-MS | Cisplatin, carboplatin | Blood, red cells, urine, tissues | Pt quantification in fluids and tissues |
| 43 | ICP-MS | Cisplatin, carboplatin, oxaliplatin and others | Cellular culture | Drug uptake by the cell |
| 144 | HPLC(RP)-ICP-MS, ESI-Q-TOF | Cisplatin | Nucleotides, DNA | Speciation of DNAnucleotides–Drugadducts |
| 145 | HPLC(RP)-ICP-MS | Cisplatin | Drosophila Melanogaster DNA treated with the drug | Detection of DNA–Drugadducts and correlation with genotoxic damage |
| 132 | nESI-FT-ICR | Cisplatin | Oligonucleotides | Characterisation of Pt-binding sites, kinetics studies |
| 133 | AAS, ELISA, immunocytochemistry | Carboplatin | DNA from rat tissues | Identification and quantification of the drug–DNAadducts |
| 142 | HPLC(IC)-UV-ICP-SFMS | Cisplatin | 5′-GMP | Identification of 5′-GMP–cisplatin adducts |
| 143 | ESI-FT-ICR | Cisplatin | DNA oligonucleotides | Characterisation of oligonucleotides–drugadducts , kinetics studies |
| 146 | Adsoptive Stripping Voltammetry | Oxaliplatin | White blood cells from oncological patients | Time-dependent formation of oxaliplatin–DNAadducts |
| 147 | ESI-LIT | Oxaliplatin | DNA nucleobases | DNA nucleobases–drug interaction |
| 148 | HPLC(RP)-ESI-TQ | Oxaliplatin | DNA from mouse tissues | Determination of GG- and AG- drug intrastrand crosslinks |
| 154 | SEC-ICP-MS, ESI-Q-TOF | Cisplatin | MT | Binding of Zn, Cd and Pt to MT, stoichiometry |
| 155 | HPLC-AAS, RP-HPLC-UV | Cisplatin | MT | Binding of Zn, Cd and Pt to MT, binding kinetics |
| 157 | SEC-ICP-MS, ESI-LIT | Cisplatin | Rat tissues, GSH, MT | GSH and MT-Drug interactions |
| 158 | X-ray diffraction | Cisplatin | MT | Binding of cisplatin to MT |
| 159 | nESI-Q-TOF, AAS | Pt(II) complexes | MT | Reaction and kinetics of MT-Pt complexes |
| 160 | nESI-Q-TOF | Cisplatin | MT | Structural study of the drug–proteinadducts |
| 163 | HPLC(RP)-UV, AAS | Cisplatin, carboplatin, oxaliplatin | GSH | Kinetics of the Drug-GSH interaction |
| 164 | HPLC(RP)-UV, AAS | Cisplatin | GSH, WR-1065, 2-mercaptoethanesulfonate | Kinetics of the Drug-Thiols interaction |
| 165 | ESI-IT | Pt(II) complexes | GSH, MAS, AcMAS, MRFA | Sulfur-peptides interaction with Pt(II) complexes |
| 166 | HPLC(RP)-tQ, RP-HPLC-UV, AAS | Cisplatin | GSH, human and rat plasma, UFP | Drug–GSH adducts characterisation |
| 167 | X-Ray Diffraction | Cisplatin | Superoxodismutase | Structural study of the drug–proteinadducts |
| 168 | ESI-LIT, ICP-OES, X-ray Diffraction | Cisplatin, transplatin, carboplatin, oxaliplatin | Hen egg lysozyme | Lysozyme-drug interaction |
| 169 | ESI-IT | Carboplatin | Cytocrome-C | Cytocrome C-Drug interaction |
| 170 | ESI-LIT, ICP-OES, UV | Cisplatin, carboplatin, oxaliplatin, transplatin | Cytocrome-C | Cytocrome C-Drug interaction |
| 171 | ESI-LIT | Carboplatin, others | Cytocrome-C | Cytocrome C-Drug interaction-binding sites |
| 172 | ESI-tQ | Cisplatin, others | Ubiquitin | Ubiquitin-Drug interaction |
| 173 | nESI-Q-TOF, nESI-IT, MALDI-TOF | Cisplatin, transplatin, oxaliplatin | Ubiquitin | Ubiquitin-Drug interaction |
| 174 | ESI-FT-ICR | Cisplatin, transplatin, oxaliplatin | Ubiquitin | Structural study of the drug–proteinadducts |
| 175 | SEC-ICP-MS, nESI-LIT, MALDI-TOF | Cisplatin | Insulin | Characterisation of cisPt–insulinadducts |
| 37 | SDS-PAGE, LA-ICP-MS, RP-ESI-Q-TOF | Cisplatin | E. Coli cells | Identification of Pt-containing proteins |
| 176 | 2D-SCX-RP-ESI-LIT | Cisplatin | E. Coli cells | Identification and characterisation of Pt-containing proteins |
| 177 | 2D-DIGE, MALDI-TOF | Cisplatin | Cochleas from rats | Quantitative proteomics on cochleas after drug administration |
| 117 | ICP-MS | Cisplatin | Brain and cochlea from rats | Pt accumulation in organs |
Acknowledgements
This work was financially supported by the Spanish CICYT projects CTQ-2005-08593 and CTQ-2008-04873. D.E. gratefully acknowledges the Spanish Ministerio de Ciencia e Innovación and the European Union for the post-doctoral fellowship and the Marie Curie contract, respectively.References
- X. Sun, C.-N. Tsang and H. Sun, Metallomics, 2009, 1, 25–31 RSC.
- S. Mandel, T. Amit, O. Bar-Am and M. B. H. Youdim, Prog. Neurobiol., 2007, 82, 348–360 CrossRef CAS.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- Z. Guo and P. J. Sadler, Angew. Chem., Int. Ed., 1999, 38, 1512–1531 CrossRef CAS.
- C. F. Shaw III, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS.
- S. Nobili, E. Mini, I. Landini, C. Gabbiani, A. Casini and L. Messori, Med. Res. Rev., 2009 DOI:10.1002/med.20168.
- M. A. Jakupec and B. K. Keppler, Curr. Top. Med. Chem., 2004, 4(15), 1575–1583 CrossRef CAS.
- M. J. Clarke, Coord. Chem. Rev., 2003, 236, 209–233 CrossRef CAS.
- F. Pruchnik and D. Dus, J. Inorg. Biochem., 1996, 61, 55–61 CrossRef CAS.
- A. Alama, B. Tasso, F. Novelli and F. Sparatore, Drug Discovery Today, 2009, 14, 500 CrossRef CAS.
- A. M. Ramos, C. Fernández, D. Amrán, D. Esteban, E. de Blas, M. A. Palacios and P. Aller, J. Cell. Physiol., 2006, 209, 1006–1015 CrossRef CAS.
- B. Rosenberg, L. van Camp and T. Krigas, Nature, 1965, 205, 698–699 CAS.
- P. M. Takahara, A. C. Rosenzweig, C. A. Frederick and S. J. Lippard, Nature, 1995, 377, 649–652 CrossRef CAS.
- R. C. Todd and S. J. Lippard, Metallomics, 2009, 1, 280–291 RSC.
- C. L. Litterst, Toxicol. Appl. Pharmacol., 1981, 61, 99–108 CrossRef CAS.
- C. Schaake-Koning, W. Van den Bogaert and O. Dalesio, et al., N. Engl. J. Med., 1992, 326, 524–530 CAS.
- J. Cassidy, Int. J. Clin. Pract., 2000, 54, 399–402 Search PubMed.
- F. I. Raynaud, P. Mistry, A. Donaghue, G. K. Poon, L. R. Kelland, C. F. J. Barnard, B. A. Murrer and K. R. Harrap, Cancer Chemother. Pharmacol., 1996, 38, 155–162 CrossRef CAS.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS.
- N. Shah and D. S. Dizon, Future Oncol., 2009, 5, 33–42 Search PubMed.
- M. J. Cleare and J. D. Hoeschele, Bioinorg. Chem., 1973, 2, 187 CrossRef CAS .17.
- H. Haraguchi, J. Anal. At. Spectrom., 2004, 19, 5–14 RSC.
- D. Templeton, F. Ariese, R. Cornelis, L. G. Danielsson, H. Muntau, H. P. van Leeven and R. Lobinski, Pure Appl. Chem., 2000, 72, 1453–1470 CrossRef CAS.
- J. Szpunar, Anal. Bioanal. Chem., 2004, 378, 54–56 CAS.
- T. G. Appleton, J. R. Hall, S. F. Ralph and C. S. M. Thompson, Inorg. Chem., 1989, 28, 1989–1993 CrossRef.
- K. Wang, J. Lu and R. Li, Coord. Chem. Rev., 1996, 151, 53–58 CrossRef CAS.
- A. L. Rosen and G. M. Hieftje, Spectrochim. Acta, Part B, 2004, 59, 135–146 CrossRef.
- J. Szpunar, Analyst, 2005, 130, 442–465 RSC.
- Z. H. Siddik, M. Jones, F. E. Boxall and K. R. Harrap, Cancer Chemother. Pharmacol., 1988, 21, 19–24 CAS.
- K. Heydorn, B. Rietz and A. J. Krarup-Hansen, J. Trace Elem. Exp. Med., 1998, 11, 37–43 CrossRef CAS.
- Z. Yang, X. Hou and B. T. Jones, Appl. Spectrosc. Rev., 2002, 37, 57–88 CAS.
- O. Nygren, G. T. Vaughan, T. M. Florence, G. M. P. Morrison, I. M. Warner and L. S. Dale, Anal. Chem., 1990, 62, 1637–1640 CrossRef CAS.
- M. Verschraagen, K. van der Born, T. H. Ursula Zwiers and W. J. F. van der Vijgh, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2002, 772, 273–281 CrossRef CAS.
- F. Morazzoni, C. Canevali, I. Moschetti, R. Todeschini, S. Caroli, A. Alimonti, F. Petrucci, G. Ravasi, A. V. Bedini, F. Milani, M. Palazzi, S. Villa and G. Giudice, Cancer Chemoter. Pharmacol., 1995, 35, 529–532 Search PubMed.
- D. Esteban-Fernández, J. M. Verdaguer, R. Ramírez-Camacho, M. A. Palacios and M. M. Gómez-Gómez, J. Anal. Toxicol., 2008, 32, 140–147 CAS.
- J. Spzunar and R. Lobinski, Anal. Bioanal. Chem., 2002, 373, 404–411 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, F. R. Abou-Shakra, H. Birtwistle and J. Coffey, Chem. Commun., 2001, 2708–2709 RSC.
- I. Pizarro, M. Gómez, C. Cámara, M. A. Palacios and D. A. Román, J. Anal. At. Spectrom., 2004, 19, 292–296 RSC.
- A. R. Timerbaev, A. Küng and B. K. Keppler, J. Chromatogr., A, 2002, 945, 25–44 CrossRef CAS.
- B. Cañas, D. López-Ferrer, A. Ramos-Fernández, E. Camafeita and E. Calvo, Briefings Funct. Genomics Proteomics, 2006, 4, 295–320 Search PubMed.
- A. Sanz-Medel, M. Montes-Bayón, M. R. Fernández de la Campa, J. Ruiz Encinar and J. Bettmer, Anal. Bioanal. Chem., 2008, 390, 3–16 CrossRef CAS.
- A. Sanz-Medel, Anal. Bioanal. Chem., 2008, 391, 885–894 CrossRef CAS.
- A. R. Ghezzi, M. Aceto, C. Cassino, E. Gabano and D. Osella, J. Inorg. Biochem., 2004, 98, 73–78 CrossRef CAS.
- B. Cañas, C. Piñeiro, E. Calvo, D. López-Ferrer and J. M. Gallardo, J. Chromatogr., A, 2007, 1153, 235–258 CrossRef CAS.
- D. Esteban-Fernández, M. Montes-Bayón, E. Blanco González, M. M. Gómez-Gómez, M. A. Palacios and A. Sanz-Medel, J. Anal. At. Spectrom., 2008, 23, 378–384 RSC.
- K. Weidt, C. Logan Mackay, P. R. R. Langridge-Smith and P. J. Sadler, Chem. Commun., 2007, 1719–1721 RSC.
- C. J. Boreham, J. A. Broomhead and D. P. Fairlie, Aust. J. Chem., 1981, 34, 659–664 CAS.
- T. G. Appleton, R. D. Berry, C. A. Davis, J. R. Hall and H. A. Kimlin, Inorg. Chem., 1984, 23, 3514–3521 CrossRef CAS.
- S. E. Miller and D. A. House, Inorg. Chim. Acta, 1989, 161, 131–137 CrossRef CAS.
- S. E. Miller and D. A. House, Inorg. Chim. Acta, 1989, 166, 189–197 CrossRef CAS.
- S. E. Miller and D. A. House, Inorg. Chim. Acta, 1990, 173, 53–60 CrossRef CAS.
- A. Andersson and H. Ehrsson, J. Chromatogr., B: Biomed. Sci. Appl., 1994, 652, 203–210 CrossRef CAS.
- A. Andersson, J. Fagerberg, R. Lewensohn and H. Ehrsson, J. Pharm. Sci., 1996, 85, 824–827 CrossRef CAS.
- A. Andersson and H. Ehrsson, J. Pharm. Biomed. Anal., 1995, 13, 639–644 CrossRef CAS.
- O. Heudi, A. Cailleux and P. Allain, Chromatographia, 1997, 44, 19–24 CAS.
- K. H. Briere, R. Goel, F. H. Shirazi, D. J. Stewart and C. P. Smith, Cancer Chemother. Pharmacol., 1996, 37, 518–524 CrossRef CAS.
- M. Cui and Z. Mester, Rapid Commun. Mass Spectrom., 2003, 17, 1517–1527 CrossRef CAS.
- S. Hann, Z. Stefanka, K. Lenz and G. Stingeder, Anal. Bioanal. Chem., 2005, 381, 405–412 CrossRef CAS.
- M. Cui, L. Ding and Z. Mester, Anal. Chem., 2003, 75, 5847–5853 CrossRef CAS.
- M. El-Khateeb, T. G. Appleton, B. G. Charles and L. R. Gahan, J. Pharm. Sci., 1999, 88, 319–326 CrossRef CAS.
- R. J. Knox, F. Friedlos, D. A. Lydall and J. J. Roberts, Cancer Res., 1986, 46, 1972–1979 CAS.
- K. Aabo, M. Adams, P. Adnitt, D. S. Alberts, A. Athanazziou, V. Barley, D. R. Bell, U. Bianchi, G. Bolis, M. F. Brady, H. S. Brodovsky, H. Bruckner, M. Buyse, R. Canetta, V. Chylak, C. J. Cohen, N. Colombo, P. F. Conte, D. Crowther, J. H. Edmonson, C. Gennatas, E. Gilbey, M. Gore, D. Guthrie and B. Y. Yeap, et al., Br. J. Cancer, 1998, 78, 1479–1487 CAS.
- K. Vivekanandan, M. G. Swamy, S. Prasad, G. C. Maikap, R. Mukherjee and A. C. Burman, Int. J. Pharm., 2006, 313, 214–221 CrossRef CAS.
- R. B. Burns, R. W. Burton, S. P. Albon and L. Embree, J. Pharm. Biomed. Anal., 1996, 14, 367–372 CrossRef CAS.
- E. Jerremalm, M. Hedeland, I. Wallin, U. Bondesson and H. Ehrsson, Pharm. Res., 2004, 21, 891–894 CrossRef CAS.
- M. A. Graham, G. F. Lockwood, D. Greenslade, S. Brienza, M. Bayssas and E. Gamelin, Clin. Cancer Res., 2000, 6, 1205–1218 CAS.
- J. T. Reardon, A. Vaisman, S. G. Chaney and A. Sancar, Cancer Res., 1999, 59, 3968–3971 CAS.
- A. R. Timerbaev, C. G. Hartinger, S. S. Aleksenko and B. K. Keppler, Chem. Rev., 2006, 106, 2224–2248 CrossRef CAS.
- T. J. Einhäuser, M. Galanski and B. K. Keppler, J. Anal. At. Spectrom., 1996, 11, 747–750 RSC.
- J. Szpunar, A. Makarov, T. Pieper, B. K. Keppler and R. Lobinski, Anal. Chim. Acta, 1999, 387, 135–144 CrossRef CAS.
- A. Gerl and R. Schierl, Acta Oncol., 2000, 39, 519–522 Search PubMed.
- J. Reedijk, Chem. Rev., 1999, 99, 2499–2510 CrossRef CAS.
- P. Guo, S. Li and J. M. Gallo, J. Chromatogr. A, 2003, 783, 43–52 CrossRef CAS.
- R. Xie, W. Johnson, L. Rodríguez, M. Gounder, G. S. Hall and B. Buckley, Anal. Bioanal. Chem., 2007, 387, 2815–2822 CrossRef CAS.
- L. Pendyala and P. J. Creaven, Cancer. Res., 1993, 53, 5970–5976 CAS.
- M. A. Graham, E. Gamelin, J. L. Misset, S. Brienza, P. Allain, M. Boisdron-Celle, A. Krikorian, D. Greenslade and M. Bayssas, Proc. Am. Assoc. Cancer Res., 1998, 39, 159.
- M. A. Graham, S. Brienza, J. L. Misset, E. Cupissol, E. Gamelin and P. Allain, Sanofi Research Report VAR3149, 1998 Search PubMed.
- P. Allain, O. Heudi, A. Cailleux, A. Le Bouil, F. Larra, M. Boisdron-Celle and E. Gamelin, Drug Metab. Dispos., 2000, 28, 1379–1384 CAS.
- B. P. Espósito and R. Najjar, Coord. Chem. Rev., 2002, 232, 137–149 CrossRef CAS.
- A. R. Timerbaev, S. S. Aleksenko, K. Polec-Pawlak, R. Ruzik, O. Semenova, C. G. Hartinger, S. Oszwaldowski, M. Galanski, M. Jarosz and B. K. Keppler, Electrophoresis, 2004, 25, 1988–1995 CrossRef CAS.
- A. V. Rudnev, S. S. Aleksenko, O. Semenova, C. G. Hartinger, A. R. Timerbaev and B. K. Keppler, J. Sep. Sci., 2005, 28, 121–127 CrossRef CAS.
- A. Sugii, K. Nishimura, K. Harada, M. Nakayama and S. Masuda, Chem. Pharm. Bull., 1991, 39, 408–410 CAS.
- N. Ohta, T. Yotsuyanagi, D. Chen, R. Ono, S. Ito and K. Ikeda, Int. J. Pharm., 1992, 85, 39–44 CrossRef CAS.
- N. Ohta, D. Chen, S. Ito, T. Futo, T. Yotsuyanagi and K. Ikeda, Int. J. Pharm., 1995, 118, 85–93 CrossRef CAS.
- J. Reedijk, Inorg. Chim. Acta, 1992, 198–200, 873–881 CrossRef CAS.
- A. I. Ivanov, J. Christodoulou, J. A. Parkinson, K. J. Barnham, A. Tucker, J. Woodrow and P. J. Sadler, J. Biol. Chem., 1998, 273, 14721–14730 CrossRef CAS.
- J. Will, D. A. Wolters and W. S. Sheldrick, ChemMedChem, 2008, 3, 1696–1707 CrossRef CAS.
- E. N. Zhmareva, G. D. Zegzhda, G. B. Kasain and O. A. Livanskaya, Ukr. Biokhim. Zh., 1996, 68, 74–79 Search PubMed.
- L. Trynda-Lemiesz, H. Kozlowski and B. K. Keppler, J. Inorg. Biochem., 1999, 77, 141–146 CrossRef CAS.
- J. Ni, Y. Wang, Q. Wang, L. Lu and Q. Zheng, Zhongguo Yiyuan Yaoxue Zazhi, 1996, 16, 246 Search PubMed.
- I. Khalaila, C. S. Allardyce, C. S. Verma and P. J. Dyson, ChemBioChem, 2005, 6, 1788–1795 CrossRef CAS.
- C. S. Allardyce, P. J. Dyson, J. Coffey and N. Johnson, Rapid Commun. Mass Spectrom., 2002, 16, 933–935 CrossRef CAS.
- M. R. Schlabach and G. W. Bates, J. Biol. Chem., 1975, 250, 2182–2188 CAS.
- M. C. Cox, K. J. Barnham, T. A. Frenkiel, J. D. Hoeschele, A. B. Mason, Q.-Y. He, R. C. Woodworth and P. J. Sadler, JBIC, J. Biol. Inorg. Chem., 1999, 4, 621–631 CrossRef CAS.
- T. Hoshino, M. Misaki, M. Yamamoto, H. Shimizu, Y. Ogawa and H. Toguchi, J. Controlled Release, 1995, 37, 75–81 CrossRef CAS.
- Y.-Y. Zhao, R. Mandal and X.-F. Li, Rapid Commun. Mass Spectrom., 2005, 19, 1956–1962 CrossRef CAS.
- R. Mandal, R. Kalke and X.-F. Li, Chem. Res. Toxicol., 2004, 17, 1391–1397 CrossRef CAS.
- R. Mandal, C. Teixeira and X.-F. Li, Analyst, 2003, 128, 629–634 RSC.
- R. Mandal, R. Kalke and X.-F. Li, Rapid Commun. Mass Spectrom., 2003, 17, 2748–2754 CrossRef CAS.
- J. Peng, R. Mandal, M. Sawyer and X.-F. Li, Clin. Chem., 2005, 51, 2274–2281 CrossRef CAS.
- J. Begerow, M. Turfeld and L. Dunemann, J. Anal. At. Spectrom., 1996, 11, 913–916 RSC.
- Prospect from Platinol-AQ drug (cisplatin), Bristol Laboratories, Princeton, New Jersey 08543 USA, 1998 Search PubMed.
- X. Tang, J. W. Hayes II, L. Schroder, W. Cacini, J. Dorsey, R. C. Elder and K. Tepperman, Met.-Based Drugs, 1997, 4, 97–108 Search PubMed.
- W. Cacini and Y. Singh, Proc. Soc. Exp. Biol. Med., 1991, 197, 285–289 Search PubMed.
- Prospect from Carboplatin drug, Pfizer Laboratories, Auckland, New Zealand, 2005 Search PubMed.
- N. D. Tinker, H. L. Sharma and C. A. McAuliffe, in Platinum and other metal coordination compounds in cancer chemotherapy: Procedings of the fifth international symposium, ed. M. Nicolini and M. Nijhoff, Boston, 1987, p. 144 Search PubMed.
- J. Allen, M. A. Graham, J. Firth, S. Woolfrey, D. Greenslade, J. G. Morrison, S. McDougall, P. Ross and D. Cunningham, Proc. Am. Assoc. Cancer Res., 1998, 39, 159.
- J. L. Misset and P. Allain, Pharmacokinetics, urinary, and fecal excretion of oxaliplatin in cancer patients, Debiopharm/Sanofi Report TDR3500, 1995 Search PubMed.
- S. McDougall and J. Allen, In vitro stability of 3H-oxaliplatin in human plasma ultrafiltrate and urine, Sanofi Research Report SPP0111, 1998 Search PubMed.
- E. E. Cohen, M. W. Lingen and E. E. Vokes, J. Clin. Oncol., 2004, 22, 1743–1752 CrossRef.
- P. T. Daley-Yates and D. C. H. McBrien, Biochem. Pharmacol., 1985, 34, 2363–2369 CrossRef CAS.
- N. D. Doolittle, L. L. Muldoon, R. E. Brummett, R. M. Tyson, C. Lacy, J. S. Bubalo, D. F. Kraemer, M. C. Heinrich, J. A. Henry and E. A. Neuwelt, Clin. Cancer Res., 2001, 7, 493–500 CAS.
- L. M. Schuchter, Eur. J. Cancer, 1996, 32, S40–42 CrossRef.
- D. J. Stewart, R. S. Benjamin, M. Luna, L. Feun, R. Caprioli, W. Seifert and T. L. Loo, Cancer Chemother. Pharmacol., 1982, 10, 51–54 CAS.
- J. Areberg, S. Bjorkman, L. Einarsson, B. Frankenberg, H. Lundqvist, S. Mattsson, K. Norrgren, O. Scheike and R. Wallin, Acta Oncol., 1999, 38, 221–228 Search PubMed.
- D. Esteban-Fernández, M. M. Gómez-Gómez, B. Cañas, J. M. Verdaguer, R. Ramírez and M. A. Palacios, Talanta, 2007, 72, 768–773 CrossRef CAS.
- R. Ramírez-Camacho, D. Esteban-Fernández, J. M. Verdaguer, M. M. Gómez-Gómez, A. Trinidad, J. R. García-Berrocal and M. A. Palacios-Corvillo, Acta Oto-Laryngol., 2008, 128, 505–509 CrossRef CAS.
- P. Tothill, H. S. Klys, L. M. Matheson, K. McKay and J. F. Smith, Eur. J. Cancer, 1992, 28, 1358–1361 CrossRef.
- D. P. Gately and S. B. Howell, Br. J. Cancer, 1993, 67, 1171–1176 CAS.
- S. Ishida, J. Lee, D. J. Thiele and I. Herskowitz, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14298–14302 CrossRef CAS.
- N. Pabla, R. F. Murphy, K. Liu and Z. Dong, Am. J. Physiol., 2009, 296, F505–F511 CAS.
- R. Safaei, Cancer Lett., 2006, 234, 34–39 CrossRef CAS.
- G. Speelmans, W. M. Rutger, M. Staffhorts, K. Versluis, J. Reedijk and B. De Kruijff, Biochemistry, 1997, 36, 10545–10550 CrossRef CAS.
- F. Lu, K. Wang, X.-Z. Sun, F. Xing, P.-D. An, Z.-H. Yang and J.-J. Yin, Met.-Based Drugs, 1995, 2, 73–80 Search PubMed.
- S. J. Lippard and J. M. Berg, in Principles of Bioinorganic Chemistry, ed. University Science Books, California, 1994, p. 113 Search PubMed.
- J. J. Roberts and J. M. Pascoe, Nature, 1972, 235, 282–284 CrossRef CAS.
- J. J. Roberts and A. J. Thomson, Prog. Nucleic Acid Res. Mol. Biol., 1979, 22, 71–133 Search PubMed.
- J. J. Roberts, Adv. Inorg. Biochem., 1981, 3, 274.
- J. P. Caradona, S. J. Lippard, M. J. Gait and M. Singh, J. Am. Chem. Soc., 1982, 104, 5793–5795 CrossRef CAS.
- J. R. Yachnin, I. Tallin, R. Lewensohn, F. Sirzén and H. Ehrsson, Cancer Lett., 1998, 132, 175–180 CrossRef CAS.
- A. Rodger, K. K. Patel, K. J. Sanders, M. Datt, C. Sacht and M. J. Hannon, J. Chem. Soc., Dalton Trans., 2002, 3656–3663 RSC.
- A. E. Egger, C. G. Hartinger, H. B. Hamidane, Y. O. Tsybin, B. K. Keppler and P. J. Dyson, Inorg. Chem., 2008, 47, 10626–10633 CrossRef CAS.
- F. A. Blommaert, C. Michael, H. C. M. van Dijk-Knijnenburg, J. H. Schornagel, L. des Engelse and A. M. J. Fichtinger-Schepman, Cancer Chemother. Pharmacol., 1996, 38, 273–280 CrossRef CAS.
- D. Pluim, M. Maliepaard, R. C. A. M. van Waardenburg, J. H. Beijnen and J. H. M. Schellens, Anal. Biochem., 1999, 275, 30–38 CrossRef CAS.
- R. Gupta, J. L. Beck, M. M. Sheil and S. F. Ralph, J. Inorg. Chem., 2005, 99, 552–559 CAS.
- M. J. P. Weleters, M. Maliepaard, A. J. Jacobs-Bergmans, R. A. Baan, J. H. M. Schellens, J. G. Ma, W. J. F. van der Vijgh, B. J. M. Braakhuis and A. M. J. Fichtinger-Schepman, Carcinogenesis, 1997, 18, 1767–1774 CrossRef.
- J. M. Woynarowski, S. Faivre, M. C. S. Herzig, B. Arnett, W. G. Chapman, A. V. Treviño, E. Raymond, S. G. Chaney, A. Vaisman, M. Varchenko and P. E. Juniewiez, Mol. Pharmacol., 2000, 58, 920–927 CAS.
- B. Spingler, D. A. Whittington and S. J. Lippard, Inorg. Chem., 2001, 40, 5596–5602 CrossRef CAS.
- S. G. Chaney, S. L. Campbell, B. Temple, E. Bassett, Y. Wu and M. Faldu, J. Inorg. Biochem., 2004, 98, 1551–1559 CrossRef.
- K. Jirsova, V. Mandys, W. H. Gispen and P. R. Bar, Neurosci. Lett., 2006, 392, 22–26 CrossRef CAS.
- D. H. Phillips, Mutat Res., 1997, 378, 1–12 CrossRef CAS.
- S. Hann, A. Zenker, M. Galanski, T. L. Bereuter, G. Stingeder and B. K. Keppler, Fresenius J. Anal. Chem., 2001, 370, 581–586 CrossRef CAS.
- E. L. Meczes, A. Azim-Araghi, C. J. Ottley, D. G. Pearson and M. J. Tilby, Biochem. Pharmacol., 2005, 70, 1717–1725 CrossRef CAS.
- D. García Sar, M. Montes-Bayón, L. Aguado Ortiz, E. Blanco-González, L. M. Sierra and A. Sanz-Medel, J. Anal. At. Spectrom., 2006, 21, 861–868 RSC.
- D. García Sar, M. Montes-Bayón, L. Aguado Ortiz, E. Blanco-González, L. M. Sierra and A. Sanz-Medel, Anal. Bioanal. Chem., 2008, 390, 37–44 CrossRef CAS.
- G. Weber, J. Messerschmidt, A. C. Pieck, A. M. Junker, A. Wehmeier and U. Jaehde, Anal. Bioanal. Chem., 2004, 380, 54–58 CrossRef CAS.
- S. M. Kerr, T. Shoeib and B. L. Sharp, Anal. Bioanal. Chem., 2008, 391, 2339–2348 CrossRef CAS.
- R. C. Le Pla, K. J. Ritchie, C. J. Henderson, C. R. Wolf, C. F. Harrington and P. B. Farmer, Chem. Res. Toxicol., 2007, 20, 1177–1182 CrossRef CAS.
- J. Reedik and J. M. Teuben, in Cisplatin chemistry and biochemistry of a leading anticancer drug, ed. B. Lipperd, Wiley-VCH, Weinheim, Zürich, 1999, pp. 339–362 Search PubMed.
- M. Saroh, D. M. Kloth, S. A. Kadhim, J. L. Chin, A. Naganuma, N. Imura and M. G. Cherian, Cancer Res., 1993, 53, 1829–1832 CAS.
- J. Holford, F. Raynaud, B. A. Murrer, K. Grimaldi, J. A. Hartley, M. Abrams and L. R. Kelland, Anticancer Drug Design, 1998, 13, 1–18 CAS.
- J. Holford, P. J. Beale, F. E. Boxall, S. Y. Sharp and L. R. Kelland, Eur. J. Cancer, 2000, 36, 1984–1990 CrossRef CAS.
- S. J. Lippard and J. M. Berg, in Principles of Bioinorganic Chemistry, ed. University Science Books, California, 1994, p. 23 Search PubMed.
- R. Mandal, G. Jiang and X. F. Li, Appl. Organomet. Chem., 2003, 17, 675–681 CrossRef CAS.
- D. Hagrman, J. Goodisman, J. C. Dabrowiak and A. Souid, Drug Metab. Dispos., 2003, 31, 916–923 CrossRef CAS.
- G. van Vyncht, G. Bordin and A. R. Rodríguez, Chromatographia, 2000, 52, 745–752 CAS.
- D. Esteban-Fernández, B. Cañas, I. Pizarro, M. A. Palacios and M. M. Gómez-Gómez, J. Anal. At. Spectrom., 2007, 22, 1113–1121 RSC.
- A. Pattanaik, G. Bachowski, J. Laib, D. Lemkuil, C. F. Shaw III, D. H. Petering, A. Hitchcock and L. Saryan, J. Biol. Chem., 1992, 267, 16121–16128 CAS.
- M. Knipp, A. V. Karotki, S. Chesnov, G. Natile, P. J. Sadler, V. Brabec and M. Vasak, J. Med. Chem., 2007, 50, 4075–4086 CrossRef CAS.
- R. Mandal and X. F. Li, Rapid Commun. Mass Spectrom., 2006, 20, 48–52 CrossRef CAS.
- P. D. Sadowitz, B. A. Hubbard, J. C. Dabrowiak, J. Goodisman, K. A. Tacka, M. K. Aktas, M. J. Cunningham, R. L. Dubowy and A. H. Souid, Drug Metab. Dispos., 2002, 30, 183–190 CrossRef CAS.
- B. A. J. Jansen, J. Brouwer and J. Reedijk, J. Inorg. Biochem., 2002, 89, 197–202 CrossRef CAS.
- D. Hagrman, J. Goodisman and A. K. Souid, J. Pharmacol. Exp. Ther., 2003, 308, 658–666 CrossRef.
- J. C. Dabrowiak, J. Goodisman and A. K. Souid, Drug Metab. Dispos., 2002, 30, 1378–1384 CrossRef CAS.
- R. Miao, G. Yang, Y. Miao, Y. Mei, J. Hong, C. Zhao and L. Zhu, Rapid Commun. Mass Spectrom., 2005, 19, 1031–1040 CrossRef CAS.
- A. Bernareggi, L. Torti, R. M. Facino, M. Carini, G. Depta, B. Casetta, N. Farrell, S. Spadacini, R. Ceserani and S. Tognella, J. Chromatogr., B: Biomed. Sci. Appl., 1995, 669, 247–263 CrossRef CAS.
- V. Calderone, A. Casini, S. Mangani, L. Messori and P. L. Orioli, Angew. Chem., Int. Ed., 2006, 45, 1267–1269 CrossRef CAS.
- A. Casini, G. Mastrobuoni, C. Temperini, C. Gabbiani, S. Francesc, G. Moneti, C. T. Supuran, A. Scozzafava and L. Messori, Chem. Commun., 2007, 156–158 RSC.
- G. Yang, R. Miao, C. Jin, Y. Mei, H. Tang, J. Hong, Z. Guo and L. Zhu, J. Mass Spectrom., 2005, 40, 1005–1016 CrossRef CAS.
- A. Casini, C. Gabbiani, G. Mastrobuoni, L. Messori, G. Moneti and G. Pieraccini, ChemMedChem, 2006, 1, 413–417 CrossRef CAS.
- C. Gabbiani, A. Casini, G. Mastrobuoni, N. Kirshenbaum, O. Moshel, G. Pieraccini, G. Moneti, L. Messori and D. Gibson, JBIC, J. Biol. Inorg. Chem., 2008, 13, 755–764 CrossRef CAS.
- D. Gibson and C. E. Costello, Eur. J. Mass Spectrom., 1999, 5, 501–510 CrossRef CAS.
- C. G. Hartinger, W. H. Ang, A. Casini, L. Messori, B. K. Keppler and P. J. Dyson, J. Anal. At. Spectrom., 2007, 22, 960–967 RSC.
- C. G. Hartinger, Y. O. Tsybin, J. Fuchser and P. J. Dyson, Inorg. Chem., 2008, 47, 17–19 CrossRef CAS.
- E. Moreno-Gordaliza, B. Cañas, M. A. Palacios and M. M. Gómez-Gómez, Anal. Chem., 2009, 81(9), 3507–3516 CrossRef CAS.
- J. Will, W. S. Sheldrick and D. Wolters, JBIC, J. Biol. Inorg. Chem., 2008, 13, 421–434 CrossRef CAS.
- D. E. Coling, D. Ding, R. Young, M. Lis, E. Stofko, K. M. Blumenthal and R. J. Salvi, Hear. Res., 2007, 226, 140–157 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |