A novel target-based de novo ligand design by use of pseudomolecular probe
Kinya
Toda
a,
Junichi
Goto
a and
Noriaki
Hirayama
*b
aComputational Science Department, Ryoka Systems Inc., 1-28-38 Shinkawa, Chuo-ku, Tokyo 104-0033, Japan
bTokai University School of Medicine, 143 Shimokasuya, Isehara, Kanagawa 259-1143, Japan. E-mail: hirayama@is.icc.u-tokai.ac.jp; Tel: +81 463 93 1121
First published on 14th October 2010
Abstract
Three-dimensional structures of drug target molecules are extremely useful for designing novel ligands. We have developed a novel structure-based de novo design method by the combination of a novel concept of pseudomolecular probe(PMP) and alpha spheres aiming to make the utmost use of the three-dimensional structures of the target molecules. Alpha spheres generated at the binding site give good clues to place molecular fragments at the site. A PMP consisting of a functional group and a supporting group can determine the most appropriate position of the functional group at the binding site. The results have indicated that the strategy works reasonably well in reproducing the structures of the original ligands at the binding sites and have proved that this method can become one of the target-based de novo design methods.
Introduction
Nowadays, it is common that reliable X-ray structures of complexes between certain small molecules and a target protein are available at the start of drug discovery project. The structural information is quite helpful to discover molecules with higher affinity and specificity to the target molecule. If the structural information is in our hand, in silico approaches can now replace the traditional high-throughput screening to minimize the resources required in the screening. Docking simulation between the target molecule and small molecules from libraries of commercially available compounds is one of the most important in silico approaches. Although docking simulations have been applied successfully to find hit compounds from the chemical libraries, hit compounds extracted from the chemical libraries are usually not suitable enough for lead compounds. To proceed with the drug design project further, it is highly required to find novel and patentable compounds with higher activities and lower toxicities starting from the hit compounds. For this purpose it is common that medicinal chemists synthesize various derivatives starting from the hits. However, it is not straightforward to take full advantage of the three-dimensional information of the target-ligand complexes in this designing process.Structure-based de novo molecule-design software has emerged to help medicinal chemists to take advantage of structural information of target molecules to design promising molecules. Several dozen de novo design programs have been published1 and some of them have been successfully applied in generating novel compounds. Most of the currently available de novo design algorithms are based on the construction of novel molecules from multiple molecular fragments. The first and most important step of assembling of fragments is placing suitable molecular fragments at each interaction center in the binding site.
Mainly two strategies are taken for this purpose, i.e. the link/grow and lattice strategies. For the link/grow strategies employed in software such as LUDI,2 mostly rule-based methods are applied to place chemical groups at the biding site. Since they are based on the empirical rules, the results are heavily dependent on the rules regarded as allowable. The performance of grid strategy3 employed in software such as LUDI crucially depends on the resolution of the grid. In addition, many meaningless grids must be taken in account.
A concave portion at the surface of a target molecule where a ligand is supposed to bind can be identified as a collection of spheres by use of the modified Delaunay triangulation.4 The sphere is designated as ‘alpha sphere.’ We have already confirmed that a set of the alpha spheres generated at the binding site is a very useful clue to place docking poses.5 As the set of the alpha spheres can represent physico-chemical properties and shape of the binding site, it can be used to place molecular fragments at the binding site. In this study we have applied the alpha spheres to find appropriate positions of the molecular fragments at the binding site. Since the alpha-sphere-based approach is not based on the empirical rules and does not depend on the resolution of the grid it is expected to be reasonably accurate and fast.
One of the most important steps to build novel molecules is disposition of functional groups at appropriate sites in a concavity of the target protein. We have introduced a novel concept of pseudomolecular probe (PMP) for this purpose. A PMP consists of a functional group and a supporting group. A supporting group helps the functional group properly position at the binding site. The supporting group should not be too large and should be rigid enough in order to determine the position of the functional group at the binding site ambiguously and efficiently. We found 3-methylcyclopropene (Fig. 1) is a suitable supporting group to anchor the functional group at the proper position of the binding site.
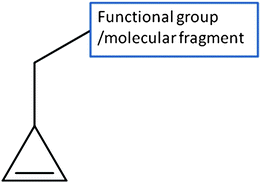 | ||
| Fig. 1 The structure of a pseudomolecular probe. | ||
De novo design software in general is inherently confronted with a virtually infinite search space. An essential problem underlying is combinatorial explosion. From the practical point of view, the search space for structure generation should be reduced in some way to avoid this NP-hard problem. Since X-ray structure of a ligand bound in a target molecule is crucially important to start the optimization or de novo design procedure, we make utmost use of this structural information in this study to avoid this problem. Ten target systems for which X-ray structures of protein-ligand complexes are available were used in the validation of this method. The results have indicated that the strategy works reasonably well in reproducing the original ligands at the binding sites of the target molecules and have proved that this method can become one of the target-based de novo design methods.
Methods
Pseudomolecular probe
Alpha spheres which are used to determine the position of a functional group at the binding site were generated by a program named Alpha Site Finder implemented in MOE.6 In the present method, optimum position of the functional group in the binding site are determined by docking procedure by use of a docking software ASEDock.7 Since functional groups are usually small in size, they tend to be trapped at various trivial crevices in the target molecule which are too small for ligand binding. Therefore we attached a support group to the functional group as shown in Fig. 1. Such a virtual molecule is designated as a pseudomolecular probe (PMP). Ideally the supporting group should function as a substitute for the remaining moiety of the ligand. Various small, rigid and hydrophobic groups were tested whether the relevant PMP can reproduce the position of the functional group of ligands in the crystal structures of protein-ligand complexes. The supporting group should not be too large and should be rigid enough in order to determine the position of the functional group at the binding site ambiguously and efficiently. After trial-and-error, it was found that cyclopropenylmethyl group is the best one that we came across so far. As an example binding modes of two different PMP's at the binding site are compared in Fig. 2. The functional group of the ligand determined by X-ray analysis is also shown for comparison. If we use cyclobutenylmethyl group as a supporting group, the functional group cannot approach near enough to the binding site. If we increase the size of the supporting group, this tendency increases as expected. We have used the PMP with the cyclopropenylmethyl group in this study.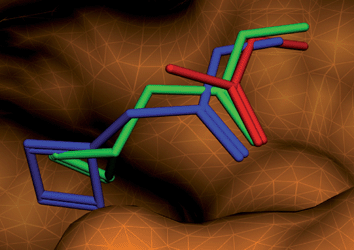 | ||
| Fig. 2 Comparison of pseudomolecular probes with cyclopentenyl (green) and cyclobutenyl (blue) groups. The functional group of the ligand determined by X-ray analysis is shown in red. | ||
Linking the PMP's by a Linker
In order to minimize the efforts and maximize the chance to obtain drug-like molecules, we selected the linker structures that connect the functional groups from ca. 1440 drugs used in Japan now. By applying a filter of drug-likeness defined by Oprea,8 we have further selected 831 molecules. This process eliminated molecules which are used for diagnosis, contrast study, transfusion, disinfection, etc. From these 831 molecules, the druggable linker fragments were selected. Although all possible molecules are not considered, we think that most linker fragments of interest are covered in this way. After determination of the structures and the locations of the PMP's at the binding site by the docking procedure, suitable linker fragments and their structures were determined. In order to link multiple PMP's with a linker, we set linking points both in PMP's and a linker as shown in Fig. 3.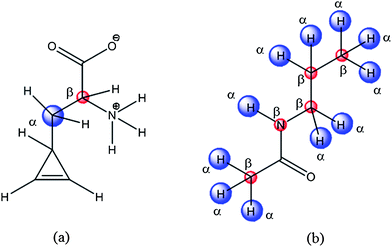 | ||
| Fig. 3 Anchoring points in a pseudo molecular probe (a) and a linker (b). | ||
Two points of α and β in PMP's are superposed to the corresponding α and β points in a linker. Here, the α and β points are pseudo-atoms with the distance between α and β points being set to the typical Csp3–Csp3 bond length of 1.54 Å. In the linking procedures, the allowance errors of superposition at the anchoring points were set to 0.2 and 0.3 Å, for α and β points, respectively. Since taking all 831 linkers into account is not practical and certain linkers are inherently unsuitable to link the particular PMP's, a simple selection rule is applied. The van der Waals volume (VL) of the linker moiety of the X-ray structure of the ligand is used to determine the size of the linkers to be selected. In this study, the linkers whose volumes range between 0.9VL and 1.1VL were selected. Through this procedure we could eliminate unsuitable linkers preliminarily.
Optimization of the structure and locations of de novo molecules at the binding site
De novo molecules constructed by connecting PMP's and a linker were docked at the binding site by use of software ASEDock. The previously reported docking protocol7 was used in this study. Alpha spheres generated at the concavity usually span a wider range than the area where the ligand is actually bound. From the point of making utmost use of the ligand information in the complex structure, alpha spheres distributed far from the ligand binding site are useless and should be count out. Therefore only the alpha spheres located within 3.5 Å from non-hydrogen atom of the ligand were selected, and the docking calculations were performed using these alpha spheres.Results and discussion
For the validation of this de novo ligand design method, we selected ten crystal structures of ligand-target complexes randomly. These structures meet the following selection criteria: the ligand is a drug or drug-like molecule which contains at least two functional groups/molecular fragments connected by a linker structure. The Protein Data Bank9 codes of the crystal structures are 1EKX, 1EVE, 1G98, 1KE5,1KI2, 1OTH, 1X70, 2PRG, 2WD9 and 3B7E. Six drug molecules currently used clinically are included. The chemical structures of the ligands are shown in Fig. 4 and their chemical names are given in Table 1. In the validation study, two functional groups in the original ligand were selected, and novel structures with different linkers were constructed. These functional groups/molecular fragments are indicated in Fig. 4. We paid close attention to whether the original ligands can be regenerated by this method. The predicted binding affinity of the de novo molecules is judged by the docking energy of Udock defined as follows:| Udock = Uele + Uvdw + Ustrain. |
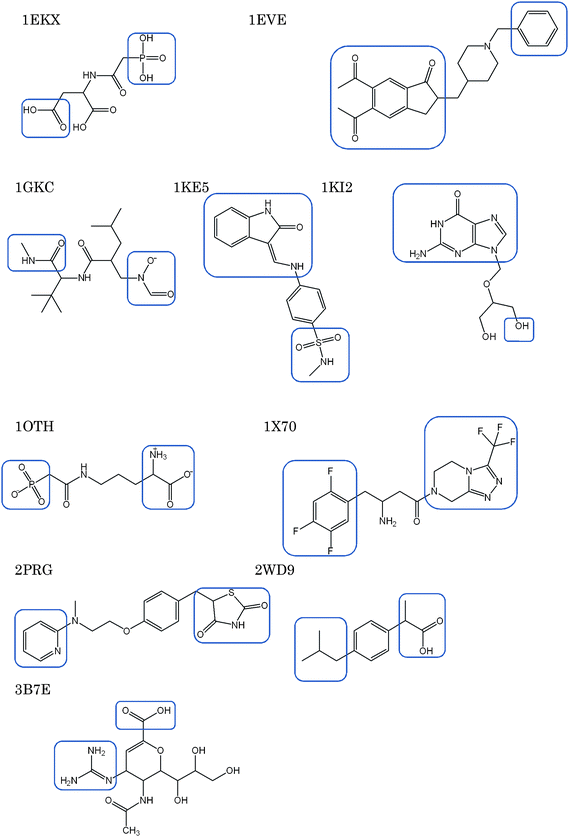 | ||
| Fig. 4 Chemical structures of the ligands used for the validation. Squares are put around the functional groups which were used for de novo design. | ||
| PDB code | Ligand identifier | Number of linkers | Number of generated molecules (number of conformations) | U dock/kcal mol−1 of the original ligand | The ranking of the original ligand | rmsd/Å | V m/Udock |
|---|---|---|---|---|---|---|---|
| a N-(phosphonacetyl)-L-aspartic acid. b Donepedil. c Reverse hydroxamate inhibitor. d N-methyl-4-{[(2-oxo-1,2-dihydro-3H-indol-3-ylidene)methyl] amino} benzenesulfonamide. e Ganciclovir. f N-(phosphonoacetyl)-L-ornithine. g Sitagliptin. h Rosiglitazon. i Ibuprofen. j Zanamivir. | |||||||
| 1EKX | PALa | 11 | 76(284) | −548.3 | 3 | 1.348 | 2 |
| 1EVE | E20b | 17 | 411(2123) | −36.2 | 990 | 2.611 | 498 |
| 1GKC | BUM + STNc | 59 | 1953(9627) | −150.0 | 139 | 0.232 | 22 |
| 1KE5 | LS1d | 19 | 136(313) | −48.5 | 52 | 0.496 | 27 |
| 1KI2 | GA2e | 12 | 158(1338) | −62.4 | 13 | 1.161 | 13 |
| 1OTH | PAOf | 20 | 108(255) | −377 | 1 | 0.349 | 1 |
| 1X70 | 715g | 61 | 1184(4943) | −92.9 | 34 | 2.055 | 26 |
| 2PRG | BRLh | 186 | 6202(24602) | −34.5 | 3320 | 2.000 | 1777 |
| 2WD9 | IBPi | 17 | 222(1208) | −37.2 | 589 | 1.727 | 214 |
| 3B7E | ZMRj | 60 | 5462(81496) | −199.5 | 176 | 2.042 | 13 |
Here, Uele and Uvdw mean electrostatic and van der Waals interaction energies, respectively, between the target and the ligand molecules. Ustrain refers to the difference between the conformation energy of a docked ligand and the conformation energy of the energy minimum conformation nearest to that of the docked ligand. The results are summarized in Table 1. As the linkers similar in size to the original linker are used to connect the PMP's, the number of linkers is different for each system. Since the original ligand was reconstructed, the number of generated molecules minus one is the number of the novel molecules. Multiple conformations were generated for each de novo molecule and the number of conformations given in the parenthesis means the total number of generated conformations for all de novo molecules. The total number of conformations depends on the number of linkers used and the flexibility of the generated molecule. The ranking of the original ligand means the ranking of the regenerated original molecule in the order of Udock values. The rmsd (Å) means the root mean square deviation between the non-hydrogen atoms of the original ligand molecule and those of the regenerated molecule by docking simulation. From the standpoint of making maximum use of the X-ray structure of the complex, the space occupied by the ligand at the binding site should be taken into account. The volume (Vcom) commonly occupied by the alpha spheres and the original ligand can be a simple index to judge how well a generated molecule corresponds to the original ligand. Suppose the common volume between the alpha spheres and a generated molecule is V, we can select the utmost similar molecules to the ligand by picking up the molecules whose V values range between 0.9Vcom and 1.1Vcom. The rankings of the regenerated ligands by Udock after this filtering are generally higher as shown in the last column (Vm/Udock) of Table 1. From the practical point, this filtering may be useful. Although the original ligands are highly ranked in all systems, the results indicate some de novo molecules with higher predicted binding affinity than the original ligand were generated. The exception is 1OTH. In this case, the original ligand came in first.
Although the present study proposes a novel method of de novo design, it is interesting to quick sketch of a few de novo molecules generated by this method. Human matrix metalloproteinase 9(MMP9) is one of important cardiovascular disease targets. Peptidic reverse hydroxamates such as shown in Fig. 4 are known to inhibit MMP9 and expected to be starting point for drug design. By use of the crystal structure of 1GKC,10 9,627 conformations of 1,952 novel molecules were generated. In this case 59 linkers were used. The original ligand ranked 139th in terms of the Udock value. The significant low rmsd of 0.232 Å has proven that the present de novo design method has succeeded in reconstruction and redocking of the original ligand. The ranking advanced from 139th to 22nd when the Vcom is taken into account. The reconstructed and original ligands are superposed in Fig. 5.
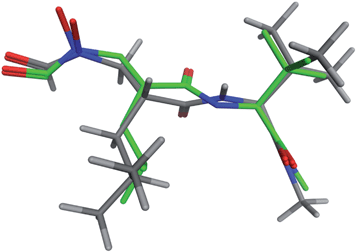 | ||
| Fig. 5 The X-ray (grey) and the docked (green) ligand of 1GKC are superimposed. | ||
The chemical structure of the molecule with the lowest Udock value is shown in Fig. 6(a). In this novel molecule, the linker contains a relatively long hydrophobic chain with a carboxyl group at the end. The docked structure of the original and the novel molecules are superposed at the binding site in Fig. 7. The molecules with carbon atoms shown in green and yellow are the novel molecule and the original ligand, respectively. The hydroxamate groups, shown on the right side in this figure, share the same position at the binding site. The terminal carboxylic acid of the novel molecule which is located at the left side in Fig. 7 binds to the backbone nitrogen atom of Gly77. The N atom of the N-methylamide group in the original ligand is hydrogen-bonded to the backbone oxygen atom of Gly77. The corresponding N atom in the novel molecule is, however, bonded to the backbone oxygen atom of Tyr136.
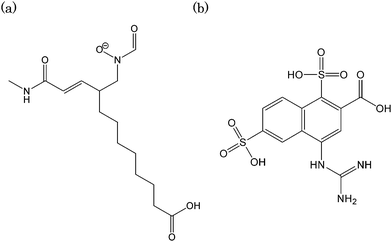 | ||
| Fig. 6 The chemical structures of the novel molecule with the lowest Udock values. (a) 1GKC (b) 3B7E. | ||
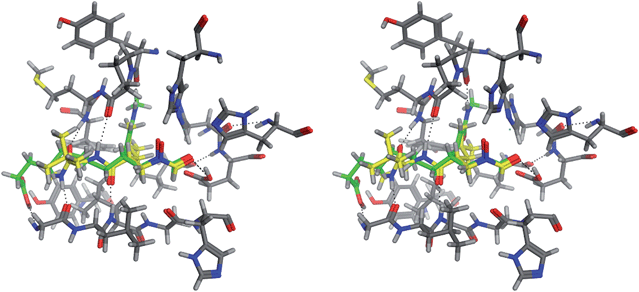 | ||
| Fig. 7 Comparison of the binding modes of the X-ray ligand and the novel molecule shown in Fig. 6(a) at the binding site of 1GKC. | ||
The influenza virus neuraminidase is one of the important targets of antiinfluenza drugs. Based on the crystal structure of the complex between a neuraminidase and an antiinfluenza drug of zanamivir(3B7E),11 81,496 conformations of 5,462 novel molecules were generated. In this case, 60 linkers were used. The original ligand ranked 176th in terms of the Udock value. The rmsd of non-hydrogen atoms is 2.042 Å. If the Vcom is considered to judge the results, the original ligand ranks 13th. The chemical structure of the novel molecule with the lowest Udock value is shown in Fig. 6(b). In the novel molecule, the ring system of 3,4-dihydro-2H-pyran in zanamivir is replaced by a naphthalene ring. In addition, two sulfonate groups are attached to the naphthalene ring. The superposition of zanamivir and the novel molecule at the binding site of neuraminidase is shown in Fig. 8. The molecules with carbon atoms shown in green and yellow are the novel molecule and the original ligand, respectively. The sulfonate groups are hydrogen bonded to the side chains of Asp151 and Glu227. In the complex structure of zanamivir, no corresponding hydrogen bonds are observed.
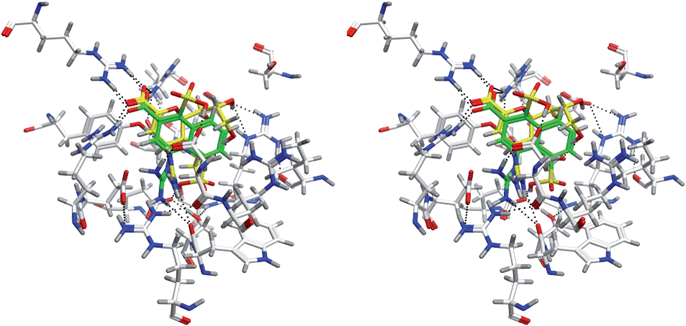 | ||
| Fig. 8 Comparison of the binding modes of zanamivir and the novel molecule shown in Fig. 6(b) at the binding site of 3B7E. | ||
As these two examples have demonstrated, the de novo design method proposed in this study can generate novel molecules which are significantly different from the original ligands and possibly bind to the target molecules stronger than the original ligands. Therefore, the present method is expected to give medicinal chemists important clues to facilitate lead optimization.
Conclusions
The present study has demonstrated that the combination of alpha sphere and pseudomolecular probe is useful in generating novel molecules based on the crystal structure of a complex between a target and a small molecule. Although the number of the complexes used for validation is limited in this paper, this method is expected to be applicable to other systems in general.The results of this method obviously depend on the libraries of linkers and functional groups. In this study, we have selected linkers and functional groups only from the database of drugs which are currently applied clinically in Japan. We understand the number of linkers and functional groups which can be applicable to drugs is strictly limited, but incorporation of novel chemical groups in generating novel molecules will be interesting. On the other hand, for specific targets the number of applicable linkers and functional groups may be more restrictive.
The present study has clearly indicated that a new and powerful strategy of de novo design has deployed in the methodology for in silico drug discovery.
Acknowledgements
This work was partly supported by Grant-in-Aid for Scientific Research (A) (19200024) from MEXT (Ministry of Education, Culture, Sports, Science and Technology) for N.H.References
- G. Schneider and U. Fechner, Nat. Rev. Drug Discovery, 2005, 4, 649–663 CrossRef CAS.
- H. J. Böhm, J. Comput.-Aided Mol. Des., 1992, 6, 593–606 CAS.
- P. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS.
- H. Edelsbrunner, M. Facello, R. Fu and J. Liang, Proceedings of the 28th Hawaii International Conference on System Sciences, 1995, 5, 256–264 Search PubMed.
- J. Goto, R. Kataoka and N. Hirayama, J. Med. Chem., 2004, 47, 6804–6811 CrossRef CAS.
- MOE (Molecular Operating Environment), version 2006.0801; Chemical Computing Group Inc.: Montreal, Quebec, Canada, 2006 Search PubMed.
- J. Goto, R. Kataoka, H. Muta and N. Hirayama, J. Chem. Inf. Model., 2008, 48, 583–590 CrossRef CAS.
- T. I. Oprea, J. Comput.-Aided Mol. Des., 2000, 14, 251–264 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Fenz, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS.
- S. Rowsell, P. Hawtin, C. A. Minshull, H. Jepson, S. Brockbank, D. Barratt, A. M. SlaterW.Mcpheat, D. Waterson, A. Henney and R. A. Pauptit, J. Mol. Biol., 2002, 319, 173–181 CrossRef CAS.
- X. Xu, X. Xhu, R. A. Dwek, J. Stevens and I. A. Wilson, J. Virol., 2008, 82, 10493–10501 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |