The kinetics of bulk hydration of the disaccharides α-lactose and trehalose by in situ neutron powder diffraction†
Valeska P.
Ting
a,
Marc
Schmidtmann
b,
Paul F.
Henry
c,
Sandie E.
Dann
d,
Jenna L.
Crisp
d,
Chick C.
Wilson
b and
Mark T.
Weller
*a
aSchool of Chemistry, University of Southampton, U.K.. E-mail: mtw@soton.ac.uk; Fax: +44 23 8059 33781; Tel: +44 23 8059 3592
bWestCHEM, Department of Chemistry, University of Glasgow, U.K.. E-mail: c.c.wilson@chem.gla.ac.uk; Tel: +44 141 330 8522
cMI-1, Helmholtz Zentrum Berlin für Materialien und Energie, Berlin, Germany. E-mail: paul.henry@helmholtz-berlin.de; Tel: +49 30 8062 2686
dDepartment of Chemistry, Loughborough University, U.K.. E-mail: s.e.dann@lboro.ac.uk
First published on 11th October 2010
Abstract
The hydrations of the excipients stable α-lactose (Lαs), with two particle sizes, and trehalose have been studied by neutron powder diffraction; analysis of the Avrami plots, derived from the bulk polycrystalline phase compositions, allows elucidation of the kinetics and mechanisms involved in these hydration processes.
Introduction
Many saccharides, including di-, tri- and poly-saccharide forms and their hydrated derivatives, are important and widely used excipients for the stabilisation and convenient delivery of active pharmaceutical ingredients.1 As a consequence, understanding the hydration behaviours of these saccharides is highly relevant to their application as filler-binders in tablets and solid form inhalers where hydration and dissolution factors control tablet and dosing rates. Two important excipients are the disaccharides lactose and trehalose. Lactose (β-D-galactopyranosyl-(1 → 4)-D-glucose) consists of galactose and glucose fragments bonded through a β-1 → 4 glycosidic linkage; in α-lactose the glucose is in the α-pyranose form.2 α-Lactose also exists as a monohydrate, (Lα·H2O), whose crystal structure has recently been redetermined.3 Lα·H2O is widely used in the pharmaceutical industry in dry powder inhaler (DPI) formulations. Recently, Lαs within a narrow particle size range of less than one micron in ethanolic suspension has been shown to give improved consistency of dose.4 Beta-lactose is mainly used as a filler-binder in the formulations of tablets formed by direct compression, so controlling the physical properties of these materials in relation to their compaction and dissolution behaviours is an area of considerable current interest. For example, recent work has shown that modified anhydrous α-lactoses with high surface areas have improved compaction and dissolution behaviours.5,6 Trehalose, α-D-glucopyranosyl-(1 → 1)-α-D-glucopyranoside, is formed from two glucose units joined by a 1-1 alpha bond.7,8 and frequently crystallizes as a dihydrate.9,10Trehalose is used as a protein-stabilizing agent and is used in several biopharmaceutical monoclonal antibody formulations, in organ protection solutions and as a moisturizer in cosmetics and dry eye treatments. It is also described as a potential excipient for use in powder inhalers. The significance of studying the behaviours of (bio)pharmaceutical compounds, particularly in situ during thermal or humidity changes, has been recently discussed by a number of authors.11–14 Information on phase stability and phase transformations as a result of heating, cooling, or water or solvent molecule uptake is relevant due to ambient weather conditions in various countries where DPI units are used. However, the changes in relative humidity can be obtained using powder X-ray diffraction, including structural changes that occur. Powder X-ray diffraction has inherent drawbacks, as it samples mainly the surface of a polycrystalline mixture so that it is only structural changes occurring on the surface of a crystallite that are probed. This limitation is of particular concern when investigating the phase and reaction chemistry of a pharma-compound as a function of particle size – a key parameter in controlling dissolution rates.15–18 Similar limitations apply to other commonly-used techniques for studying pharmaceuticals in situ, such as infrared and Raman spectroscopy and optical microscopy, in that these methods are also confined to monitoring the surface reaction chemistry. Therefore, to determine changes in bulk and overall structure, and also to investigate transformation kinetics as a function of particle size, a method that probes the entire bulk sample is required. To this end we have used neutron diffraction, which is well known as a non-destructive technique for in situ investigations of bulk systems and non-surface structures, for example, of archaeological artefacts and engineering structures. Problems with incoherent scattering from the 1H isotope, which may have precluded application of this method to hydrogenous pharmaceutical compounds previously, have recently been overcome through use of modern high-flux instrumentation.19 Here, we describe the in situ hydration of α-lactose (Lαs) samples of different particle sizes (∼50 μm and >1 μm) and trehalose followed by neutron powder diffraction (NPD), demonstrating the type of information that can be extracted on the kinetics of hydration processes for bulk polycrystalline materials.
Experimental
Neutron diffraction experiments were performed at the ILL in Grenoble, using the high-flux powder diffraction instrument D20, operating in its high take-off angle configuration at a fixed wavelength of 1.86 Å.20 High quality powder diffraction data were initially obtained from anhydrous stable Lαs (>99.9%, particle size ∼50 μm, hereafter denoted HCα- lactose) with the sample mounted in 9 mm diameter vanadium cans (∼5 g) and data were collected over a period of 4 h at 298 K. After normalisation and reduction including fitting and removal of the high incoherent scattering background, data were analysed via Rietveld refinement using the GSAS21 structure refinement program in conjunction with the EXPGUI22 user interface using the published crystallographic structure as a starting model.3 Full details of the refinement process are given in the ESI†; such analysis has the benefit of producing a structural model with nuclear positions, rather than the electron distributions probed by X-rays, and provides a fit to the NPD data. The structures refined to provide final χ2 values of 11.1 and 7.47 and RF2 values of 9.79 and 4.59 for the HCα-lactose and the monohydrate samples, respectively. The resulting extracted unit cell dimensions and atomic coordinates were subsequently input as initial values for the refinement of the shorter data sets collected under conditions of 100% relative humidity (RH). The profile fits and atomic coordinates where refined/modified have been deposited in the ESI.†For the in situ hydration experiments, powdered samples of stable α-lactose (two samples, the highly crystalline form described above, and a small particle size form (particle size 0.1–1μm), hereafter denoted SPSα-lactose or trehalose (>99%) were loaded into 5 mm diameter containers formed from a heavily perforated vanadium-foil. The can size was kept to a minimum to reduce the possibility of moisture gradients between powder at the surface and the core of the can while ensuring that diffraction data collected in 2.5 min intervals had sufficient peak-to-background ratios to permit profile refinement with extraction of phase fractions. The sample environment consisted of heated aluminium vessel with an atmosphere of controlled-humidity modified from that described previously.22 A regulated flow of humid gas was achieved through the use of a RH-200 humidity generator unit, which combined a stream of dry nitrogen gas with a humidified N2 stream in a mixing ratio that could be set and remotely controlled via a computer program. The humidity of the output gas stream was directly measured within the generator unit using a dew-point sensor and in the sample chamber using an in-cell hygrometer. Temperature control was achieved over the temperature range 30–200 °C through heating tapes mounted around the aluminium chamber and a remote PID interface; pre–experiment tests showed that 100% humidity could be achieved in the cell at temperatures up to 55 °C and lower humidity values at the higher temperatures.
The background contribution from the humid gas flow through the sample environment was found to be negligible by measurements on a non-hygroscopic powder at various humidity levels, dispelling concerns of significant beam attenuation or incoherent scattering by the H2O vapour.
Full powder diffraction patterns were collected at 2.5 min intervals during the hydration of the HCα-lactose, SPSα-lactose and trehalose, under conditions of 100% RH at 25 °C. Diffraction profiles were modelled via Rietveld refinement as stated above; for the trehalose pairs of diffraction data sets were summed to give the equivalent of 5 min data sets. Analysis was undertaken using the extracted crystallographic models for stable α-lactose and α-lactose monohydrate (ESI†) and the literature data for, trehalose and trehalose dihydrate.2,3,7,9 During profile fitting only the profile parameters and the phase fractions of the anhydrous and hydrated disaccharides were refined. Sequential GSAS refinements (SEQGSAS)19 was used to analyse the full set of diffraction profiles for each system and the phase fraction ratio was extracted in each case.
Results
Fig. 1 shows the diffraction profiles for HCα-lactose collected as a function of time. Strong peaks characteristic of the monohydrate phase, occurring between 49 and 52°, start to appear after 30 min and become dominant after 120 min. It has been previously reported that the reaction progress and kinetics of hydration reactions in a neutron beam can be monitored through the increase in total counts scattered into the detectors which increases monotonically with the level of hydrogen in the sample.23,24 From comparisons of both the increase in the normalised background counts for each sample (see ESI Fig. S1†) as well as the extracted phase percentages of the monohydrate in the sample as a function of time, (as plotted in Fig. 2 for the samples of stable α-lactose with two different particle sizes), it is clear that the conversion to the monohydrate occurred at a significantly faster rate for the sample of SPSα-lactose. The rate of growth of the monohydrate phase for SPSα-lactose is approximately twice as fast as for HCα-lactose, as can be seen from the gradient of the plots in Fig. 2. The Avrami equation can be used to describe the transformation between two solid crystalline phases and also provide some information on the likely mechanism involved,25 using the expression| X = 1 − e−(kt)n |
| ln[−ln(1 − X)] = nlnk + nlnt |
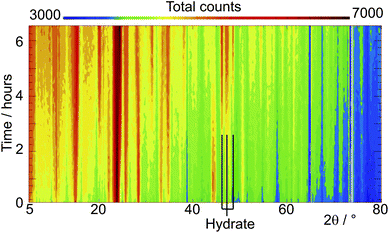 | ||
| Fig. 1 Stacked plot viewed down the intensity direction showing the evolution of the diffraction patterns of standard stable α-lactose as a function of time in 100% relative humidity at 45 °C; the positions of three strong reflections from α-lactose monohydrate are shown. | ||
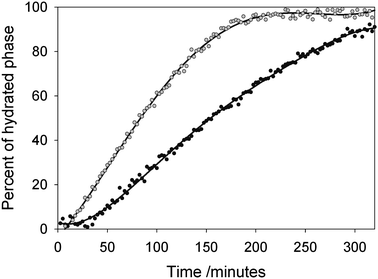 | ||
| Fig. 2 Phase percentage of α-lactose monohydrate in HCα-lactose (black filled circles) and SPSα-lactose (grey filled circles) exposed to 100% RH at 25 °C, as refined from sequential NPD data sets as a function of time. | ||
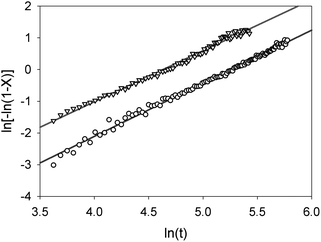 | ||
| Fig. 3 Avrami plots for the formation of α-lactose monohydrate from HCα-lactose (circles) and SPSα-lactose (triangles) exposed to a humid gas flow, as a function of time. | ||
For trehalose a similar analysis shows slightly more complex behaviour, Fig. 4 and 5. The phase fraction of dihydrate does not present a simple sigmoid shape but rather an initial induction period of around 60 min followed a more rapid growth of dihydrate finally a slower rate of formation of the product phase. A straight line fit to the Avrami plot is much poorer (r2 = 0.984) though it yields a similar value for n (1.645) as was found for stable α-lactose. A better fit to this plot can be achieved using two straight lines corresponding to a reaction time up to 150 min (n = 2.35, r2 = 0.973) and thereafter (n = 1.45, r2 = 0.995. This behaviour might be explained by the fact that the process of hydration of trehalose eventually leads to trehalose dihydrate and probably involves a two stage mechanism with an intermediate partially hydrated phase. As no additional unfitted reflection was observed at intermediate hydration times it seems likely that only small levels of this intermediate phase are formed at the reaction boundary or that it is amorphous. The formation of this intermediate phase may account for the induction time before the dihydrate phase is observed. The values of n for the two reaction processes, between 2–3 and 1–2 respectively, are consistent with an initial 3-dimensional bulk growth process for the formation of the initial intermediate phase followed by a lower dimensionality (i.e. surface or linear) growth for crystallisation of the dihydrate.
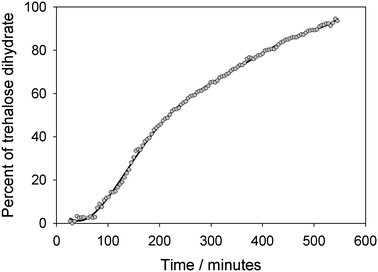 | ||
| Fig. 4 Phase percent of trehalose dihydrate as a function of time, as refined from sequential NPD data from anhydrous trehalose exposed to 100% RH. at 25 °C. | ||
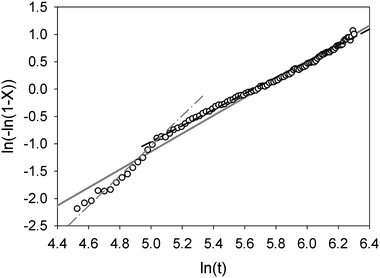 | ||
| Fig. 5 Avrami plot for the hydration of trehalose; linear regression fitted to whole data range is shown as solid grey line. Fits to data at short reaction times and long reaction times are shown as grey dash-dot and black dashed lines respectively. | ||
Conclusions
It has been demonstrated here that neutron powder diffraction can be used to observe the bulk hydration or solvation of pharma-compounds in situ. Information on the rates of such processes and their mechanisms can also be gleaned. For α-lactose a simple hydration process occurs which is consistent with the simple growth of the monohydrate from the crystallite surface and this process moves more rapidly to completion for smaller crystallite sizes. In trehalose, a more complex mechanism seems to be involved in the formation of an intermediate hydrated phase on route to its dihydrate.The ability to observe the structures of pharmaceutical compounds in situ in controlled humidity and temperature conditions and study the bulk material rather than simply the surface structures is of considerable relevance to production and the storage of many active pharmaceutical compounds. The technique could also readily be applied to complex formulations and those in pelletized forms.
Acknowledgements
This work was supported by EPSRC grants EP/E051049 and EP/E050859. The authors thank Dr Thomas Hansen for technical advice and the ILL for awards of beam time through LTP-5A-2 for the development of the sample environment. We also acknowledge 3M Health Care (Loughborough) for providing samples of smaller particle anhydrous stable α-lactose (Lαs) and the studentship for JLCNotes and references
- Handbook of Pharmaceutical Excipients, Fifth edition. Edited by R. C. Rowe, P. J. Sheskey and S. C. Owen. 2005, Pharmaceutical PressLondon Search PubMed.
- C. Platteau, J. Lefebvre, F. Affouard and P. Derollez, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 453 CrossRef.
- J. H. Smith, S. E. Dann, M. R. J. Elsegood, S. H. Dale and C. G. Blatchford, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61(8), o2499 CrossRef.
- P. A. Jinks, Proc. DDL XIV, 2003 Search PubMed.
- S. Ziffelsa and H. Steckel, Int. J. Pharm., 2010, 387, 71–78 CrossRef.
- E. A. Schmitt, D. Law and G. Q. Zhang, J. Pharm. Sci., 1999, 88, 291 CrossRef CAS.
- T. Taga, M. Senma and K. Osaki, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3258 CrossRef CAS.
- H. Nagase, T. Endoa, H. Uedaa and M. Nakagakib, Carbohydr. Res., 2002, 337, 167 CrossRef CAS.
- G. M. Brown, D. C. Rohrer, B. Berking, C. A. Beevers, R. O. Gould and R. Simpson, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 3145 CrossRef CAS.
- G. A. Jeffrey and R. Nanni, Carbohydr. Res., 1985, 137, 21 CrossRef CAS.
- K. D. M. Harris, American Pharmaceutical Review, 2004, 7, 86–91 Search PubMed.
- M. L. MacCalman, K. J. Roberts, C. Kerr and B. Hendriksen, J. Appl. Crystallogr., 1995, 28, 620 CrossRef CAS.
- T. D. Davis, K. R. Morris, H. Huang, G. E. Peck, J. G. Stowell, B. J. Eisenhauer, J. L. Hilden, D. Gibson and S. R. Byrn, Pharm. Res., 2003, 20(11), 1851 CrossRef CAS.
- D. Giron, Ch. Goldbronn, M. Mutz, S. Pfeffer, Ph. Piechon and Ph. Schwab, J. Therm. Anal. Calorim., 2002, 68, 453 CrossRef CAS.
- M. Charoenchaitrakool, F. Dehghani, N. R. Foster and H. K. Chan, Ind. Eng. Chem. Res., 2000, 39, 4794 CrossRef CAS.
- N. Rasenacka, H. Hartenhauerb and B. W. Müller, Int. J. Pharm., 2003, 254, 137 CrossRef.
- J. L. Crisp, S. E. Dann, M. Edgar and C. G. Blatchford, Int. J. Pharm., 2010, 391, 38 CrossRef CAS.
- D. Traini, P. M. Young, F. Thielmann and M. Acharya, Drug Dev. Ind. Pharm., 2008, 34, 992 CrossRef CAS.
- M. T. Weller, P. F. Henry, V. P. Ting and C. C. Wilson, Chem. Commun., 2009, 2973 RSC.
- T. C. Hansen, P. F. Henry, H. E. Fischer, J. Torregrossa and P. Convert, Meas. Sci. Technol., 2008, 19, 034001 CrossRef.
- A. C. Larson and R. B. V. Dreele, Los Alamos National Laboratory Report, 1994, 86–748 Search PubMed.
- B. H. Toby, J. Appl. Crystallogr., 2001, 34, 210 CrossRef CAS.
- V. P. Ting, P. F. Henry, M. Schmidtmann, C. C. Wilson and M. T. Weller, Chem. Commun., 2009, 7527 RSC.
- X. Turrillas, P. Barnes, D. Gascoigne, J. Z. Turner, S. L. Jones, C. J. Norman, C. F. Pygall and A. J. Dent, Radiat. Phys. Chem., 1995, 45, 491 CrossRef CAS.
- A. P. Wilkenson, J. S. Speck, A. K. Cheetham, S. Natarajan and J. M. Thomas, Chem. Mater., 1994, 6, 750 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary figures and final crystallographic model. See DOI: 10.1039/c0md00093k |
| This journal is © The Royal Society of Chemistry 2010 |