Antivascular and anticancer activity of dihalogenated A-ring analogues of combretastatin A-4†
Thomas M.
Beale
a,
Rebecca M.
Myers
*a,
James W.
Shearman
a,
D. Stephen
Charnock-Jones
bc,
James D.
Brenton
d,
Fanni V.
Gergely
d and
Steven V.
Ley
a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, United Kingdom CB2 1EW. E-mail: rmm32@cam.ac.uk.; Fax: +44 1223 336442
bDepartment of Obstetrics and Gynaecology, University of Cambridge, The Rosie Hospital, Cambridge, United Kingdom CB2 0SW
cNational Institute for Health Research, Cambridge Comprehensive Biomedical Research Centre, Cambridge, UK
dCancer Research UK Cambridge Research Institute, Li Ka Shing Centre, Robinson Way, Cambridge, United Kingdom, CB2 0RE
First published on 23rd August 2010
Abstract
The generally accepted view is that the 3,4,5-trimethoxy-substituted aromatic A-ring of combretastatin A-4 (CA-4) and its analogues should be conserved in order to maintain biological activity through enforcing an active molecular conformation. Contrary to this, we have found that substituting the larger meta-methoxy groups of CA-4 with smaller halogen atoms results in compounds that are equipotent or more potent than CA-4 itself in vitro.
Introduction
Microtubules (MTs), formed from α- and β-tubulin subunits, are key participants in cellular activities including mitosis. Due to this intimate involvement in cell division, chemical agents that act on the tubulin/MT system are first-line therapies for treating cancer e.g. the natural product-derived drugs Velban™ (vinblastine, 1), Taxol™ (paclitaxel, 2) and Ixempra™ (epothilone B, 3) (Fig. 1).1 However, despite their widespread use in the clinic, drug resistance and dose-limiting adverse effects are major issues.2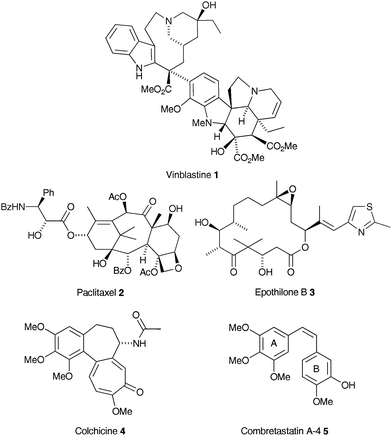 | ||
| Fig. 1 | ||
Antitubulin agents change the extent of tubulin polymer assembly or disassembly through binding to one of three currently well-defined binding sites.3 These changes include MT-stabilisation (polymerisation) elicited by drug binding through the so-called paclitaxel binding site on β-tubulin, and MT-destabilisation (depolymerisation) in response to drug binding at either the Vinca or the colchicine binding site. A more complex, concentration-dependent binding mode leads to suppression of MT dynamics.4 Such is the unabated interest in these agents there are at least 28 tubulin targeting agents in clinical development.5
The small natural product combretastatin A-4 (CA-4) (5)6 is highly effective at binding to the colchicine site on β-tubulin.7 At micromolar concentrations, despite its simple structure, 5 inhibits MT polymerisation, whilst at nanomolar concentrations it stabilises MT dynamics, arresting the cell cycle in metaphase and triggering apoptosis.8 Furthermore, unlike 2 and the Vinca alkaloid drugs, 5 is not a substrate for the multi-drug resistance efflux pump, P-glycoprotein.9 CA-4 (5) also has antivascular activity in vivo at doses less than 10% of the MTD.10 However, an additional mode of action related to the endothelial effect has been suggested which could account for this activity.11
Antivascular therapy is an attractive strategy for the treatment of some cancers. Tumour vasculature is abnormal—the branching pattern is not hierarchical—leading to avascular hypoxic voids and vessels that are leaky and tortuous. These abnormalities are due to peturbation of the normal balance of pro- and antiangiogenic regulators. A consequence of this is that local capillary sprouting is enhanced. This process of neovascularisation is generally uncommon in healthy adult tissues. Strategies for targeting tumour vasculature involve antiangiogenic agents and vascular disrupting agents (VDAs).12 The antiangiogenic agents (such as those targeting the VEGF system) normalise abnormal tumour blood vessels which blocks further endothelial proliferation, reduces leakiness and improves perfusion allowing more effective treatment with traditional cytotoxic drugs.13,14 Despite success in some cancers, on-going clinical trials have found competing production of other proangiogenic factors by some tumour types to be a problem. In contrast, direct targeting of endothelial cells with VDAs causes the rapid collapse of abnormal blood vessels. The remaining viable rim of tumour cells at the periphery responds well to traditional chemotherapy when given before VDA treatment, cutting off the supply of oxygen and nutrients and leading to the rapid induction of tumour necrosis.15
The appeal of 5 is its simple stilbenic structure coupled with its remarkable anticancer activity, especially when these attributes are compared to highly complex structures such as 1 or 2. Although these latter two drugs target tubulin binding in dividing cancer cells, it is the fact that 5 has its most pronounced effect on the tumour endothelium that sets it apart from the other tubulin targeting agents. Analogues of 5 with improved or additional activity against proliferating endothelial cells giving faster blood vessel shut down at lower minimum concentrations are attractive targets.
The syntheses of CA-4 analogues and structure activity relationships for the binding of colchicine (4) to tubulin were reviewed recently.16 Linking these published structures, almost without exception, is a trimethoxyaryl ring (referred to as the A-ring) which is widely considered to be essential for biological activity. Superficially, the rationale for this appears to revolve around the preponderance of trimethoxyaryl motifs in nature, perhaps best illustrated by the vast family of plant lignan natural products.17 Its presence in other natural product-derived anticancer drugs e.g. the topoisomerase II inhibitor etoposide and other colchicine binding site antitubulin agents e.g. steganacin and podophyllotoxin, have also been cited as compelling evidence. Indeed, it has been suggested that the trimethoxyaryl unit may make an additive contribution to the strength of binding to tubulin, serving as an anchor that maintains the B-ring portion of the molecule in the optimum orientation within the binding locus.18 Thus far, and possibly as a consequence of this, the number of A-ring substitution studies has been limited, with most effort having focussed on analogues with alternative bridging groups and B-rings.19 In one study, all three methoxy groups of 5 were replaced by methyl groups, which resulted in an approximate 10-fold loss in cytotoxicity (EC50 = 0.001 μM and EC50 = 0.02 μM in K562 cells respectively).18 Removal of a single meta-methyl group resulted in a further drop in activity to 0.07 μM. Removal of all three methyl groups (to an unsubstituted aryl ring) resulted in a much larger drop in activity to 0.5 μM. This pattern of activity has also been shown for the removal of methoxy groups from colchicine analogues, with the most significant contribution to this loss arising from the 4-methoxy substituent.20 It has, therefore, been suggested that the 4-methoxy group is essential for binding to tubulin with the meta-methoxy groups required to enforce an active confirmation.21 It has also been reported that replacement of a single meta-methoxy group with a halogen atom (F, Cl, Br) resulted in little difference in human cancer cell line growth inhibition and antitubulin activity in comparison to 5,21 which seems to reinforce this hypothesis. However, the preferred conformation of these halogenated compounds in the crystal phase or when bound to tubulin is not known. As a semi-empirical study has suggested, the energy barrier to torsional rotation of 5 is negligible.22 Thus, it could be considered that the meta-halogen atoms are not merely enforcing an active conformation, but rather providing an electronic interaction with the protein arising from halogen bonding.
Although a familiar concept in the area of supramolecular chemistry,23 the importance of halogen bonding in biological systems has remained uninvestigated until relatively recently. The first extensive treatment of this phenomenon involved targeted data processing of the Protein Data Bank (PDB, July 2004 release). This resulted in the discovery of 113 distinct halogen-protein interactions (defined by distances less than the sum of the Van der Waal's radii of the atoms involved and a bonding angle of greater than 120°).24 A second study using the PDB (December 2008 release) found 248 halogen-protein interactions using the same definition but with a 140° bonding angle threshold.25 Following ab initio modelling of electrostatic potentials in different systems, it was postulated that this interaction arises between electronegative portions of protein backbone or amino acid side-chain and the electropositive cap found at the end of the halogen atoms along the C–X axis for X = Cl, Br, I. It was found that aromatic carbon-halogen bonds produce a larger electropositive cap and that the strength of interaction followed the series F≪Cl<Br<I, mirroring their trend of polarisability.25 Most commonly used molecular modelling platforms are presently inadequate for predicting this type of interaction.24,25
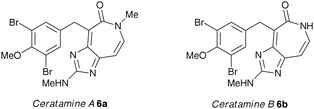
We were, therefore, interested in the activity of a series of 3,5-dihalogenated analogues of 5, maintaining the required 4-methoxy unit, to investigate whether two halogen atoms would maintain or possibly improve the activity of the parent compound. The 3,5-dibromo-4-methoxy motif is found in many marine natural products,26 including the ceratamines (6a and 6b, Fig. 2).27a The ceratamines have been reported to have an antimitotic effect through the stabilisation of MTs,27b and the syntheses and biological activity of these and a number of other dibromotyrosine-derived natural products are currently under investigation by our group.28 Since both 5 and 6 interact with tubulin, it was envisaged that compounds with structural features from both families may exhibit interesting or enhanced antimitotic and, potentially, antivascular activity. Furthermore, in the light of recent reports into the effects of halogen bonding on ligand-biomolecule interaction,24,25,29 we felt that halogenated A-ring analogues of 5 warranted further investigation.
 | ||
| Fig. 2 | ||
Results and discussion
Chemical synthesis
A series of CA-4 analogues were prepared using the Wittig olefination, adopting the procedure of Pettit et al.,21 which required access to substituted benzaldehydes 7–11 (Fig. 3).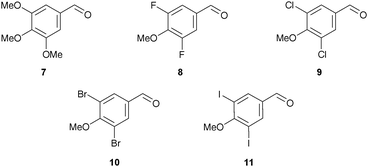 | ||
| Fig. 3 | ||
3,4,5-Trimethoxybenzaldehyde (7) and 3,5-difluoro-4-methoxybenzaldehyde (8) are commercially available and aldehydes 9, 10 and 11 were readily synthesised (Scheme 1). After some experimentation, we found that treatment of 4-hydroxybenzaldehyde (12) with sulfuryl chloride under microwave irradiation gave the best ratio of the desired dichlorinated (13) to monochlorinated product (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Without purification, methylation of 13 gave 9 in 69% yield over two steps. 3,5-Dibromo-4-methoxybenzaldehyde (10) was obtained in 90% yield via methylation of phenol 14. Iodination of 12 was achieved using potassium iodide, sodium chloride and sodium periodate which gave 15 in excellent yield (97%).30 Subsequent methylation produced 3,5-diiodo-4-methoxybenzaldehyde (11) in 90% yield.
:1). Without purification, methylation of 13 gave 9 in 69% yield over two steps. 3,5-Dibromo-4-methoxybenzaldehyde (10) was obtained in 90% yield via methylation of phenol 14. Iodination of 12 was achieved using potassium iodide, sodium chloride and sodium periodate which gave 15 in excellent yield (97%).30 Subsequent methylation produced 3,5-diiodo-4-methoxybenzaldehyde (11) in 90% yield.
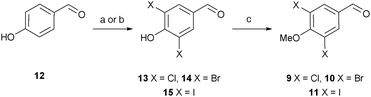 | ||
| Scheme 1 Reagents and conditions: (a) for 13, SO2Cl2, CH2Cl2, μW, 80 °C, 10 h; (b) for 15, KI, NaCl, NaIO4, AcOH/H2O (9:1), rt, 96 h, 97%; (c) K2CO3, MeI, acetone, 60 °C, 9 69% over 2 steps, 10 91%, 11 90%. | ||
n-Butyllithium was used to generate the corresponding ylide from phosphonium salt 16,31 required for the Wittig olefinations. This was followed by addition of the requisite aldehyde as a solution in THF (Table 1).32 As expected, the Wittig reactions gave 17–21 as approximately 1:1 mixtures of the (E)- and (Z)-isomers which were either separated at this stage or after TBDMS deprotection. A similar ratio of alkene isomers of 17 was obtained when potassium hexamethyldisilazide (KHMDS) was used as base to form the ylide at −78 °C.
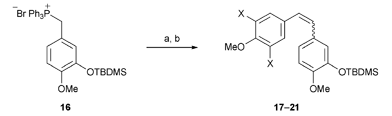
| Compound | X | (E):(Z)b | Isolated (E):(Z):mixture/% | Yield/%c |
|---|---|---|---|---|
| a Reagents and conditions: (a) n-BuLi, THF, −20 °C to rt, 10 min; (b) aldehyde, THF, rt, 30 min. b Ratio determined by 1H NMR spectroscopy. c Total isolated yield. d Isomers separated after deprotection. | ||||
| 17 | OMe | 53:47 |
40:35:2 |
77 |
| 18 | F | 53:47 |
36:37:11 |
84 |
| 19 | Cl | 57:43 |
90 | |
| 20 | Br | 60:40 |
96 | |
| 21 | I | 58:42 |
97 |
Interestingly, inverting the sense of Wittig coupling; i.e. reaction of the ylide derived from 22 using KHMDS as the base with TBDMS-protected isovanillin, increased the ratio of isomers towards desired (Z)–17 (E:Z ratio 22:78) when compared with the Pettit procedure above (E:Z ratio 53:47) without a reduction in overall yield (Scheme 2). This represents an improvement to a similar reported procedure.33 In contrast, when applied to the synthesis of 20 there was no improvement in the ratio of isomers, presumably due to electronic effects operating in the initial coupling event.
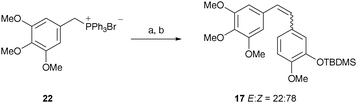 | ||
| Scheme 2 Reagents and conditions: (a) KHMDS, THF, −78 °C; (b) TBDMS-isovanillin, THF, −78 °C to rt, 77%. | ||
Deprotection of the desired (Z)-isomers of 17 and 18 with TBAF in THF proceeded smoothly to give 5 and 23 in 73% and 92% yield respectively. Separation of the (E)- and (Z)-isomers for 24, 25 and 26 was performed following deprotection (Table 2). In deuterated chloroform (Z)-26 isomerised to afford exclusively the (E)-isomer over the course of a few days; this was not observed for 5 or 23, 24 or 25.
xCELLigence cytotoxicity assays on human umbical vein endothelial cells (HUVECs)
Human umbilical vein endothelial cells (HUVECs) are a model of tumour endothelium and can be used to evaluate antivascular effects of bioactive compounds. The HUVECs were isolated from umbilical cords using the method described in the Supplementary Information and the xCELLigence system was used to measure cell growth.34 This equipment measures impedance recorded from the base of 96-well plates to give a real-time indication of changes in cell number. The data obtained was used to calculate EC50 values for 5 and 23–26 at two timepoints and the assay was repeated on four independent HUVEC isolates (Table 3).| HUVEC Isolate | Time/h | Compound EC50/nM | ||||
|---|---|---|---|---|---|---|
| 5 (CA-4) | 23 (F) | 24 (Cl) | 25 (Br) | 26 (I) | ||
| a HUVECs were seeded at 7.5 × 103 cells per well and incubated for 5 h prior to drug addition. Real-time monitoring every 30 min for 45 h allowed extraction of EC50 data. | ||||||
| 1 | 24 | 1.9 | 193.3 | 3.8 | 2.2 | 0.9 |
| 45 | 1.8 | 224.4 | 2.3 | 2.2 | 1.0 | |
| 2 | 24 | 2.2 | 263.1 | 2.6 | 2.3 | 1.7 |
| 45 | 0.9 | 318.0 | 2.4 | 1.1 | 0.9 | |
| 3 | 24 | 2.3 | 448.6 | 11.0 | 3.0 | 2.6 |
| 45 | 3.0 | 563.6 | 15.8 | 3.2 | 1.9 | |
| 4 | 24 | 2.2 | 1088.0 | 28.3 | 4.5 | 2.5 |
| 45 | 3.0 | 1276.0 | 49.1 | 5.5 | 2.4 | |
In this assay, CA-4 (5) was highly potent in the low nanomolar range with mean EC50 values across all four independent HUVEC isolates of 2.2 nM at both 24 h and 45 h (Table 3). Interestingly, 26 (I) proved to be marginally more potent than 5 with mean EC50 values of 1.9 nM at 24 h and 1.6 nM at 45 h. The activity of 26 (I) agrees with that previously reported (EC50 = 0.6 nM, HUVECs).3525 (Br) was slightly less potent than 5 with mean EC50 values across all isolates of 3.0 nM at both 24 and 45 h with very little variance across the HUVEC isolates. The pattern became disrupted with 24 (Cl) which retained good potency against isolates 1 and 2 (EC50 = 1.1–3.8 nM). However, a considerable drop was observed for isolates 3 and 4 (EC50 = 11.0–49.1 nM). The mean EC50 values for 24 (Cl) across all four isolates were 22.9 nM at 24 h and 17.4 nM at 45 h. The most striking loss of potency was observed for 23 (F) across all four HUVEC isolates (mean EC50 = 498.3 nM at 24 h and 595.9 nM at 45 h) with the most significant loss again seen with isolates 3 and 4. Therefore, the order of activity of dihalogenated CA-4 analogues against HUVECs can be described as F≪Cl<Br<OMe<I.
MTS assays on HUVECs
Given that we found 26 (I) to be prone to isomerisation,3625 (Br) was used for further investigation. Using the colourimetric MTS assay on HUVECs the ability of 25 to inhibit cell proliferation relative to 5 was determined (Table 4). Under these conditions 25 was active in the picomolar range (EC50 = 0.2–0.4 nM) when compared to 5 (EC50 = 2.0–3.7 nM) in their action on HUVECs.MTS assays on human carcinoma cell lines and peripheral blood mononuclear cells
CA-4 (5) and 25 were assayed for activity on three cancer cell lines: human epithelial cervical cancer (HeLa), ovarian cancer (SK-OV-3) and its taxol-resistant variant (SK-OV-3TR) (Table 5). The MTS assays confirmed 25 to be approximately equipotent with 5 after 48 h drug treatment. Furthermore, neither compound was affected by the mechanism of taxol resistance, as shown by the low nanomolar EC50 data for the SK-OV-3TR cell line (5 EC50 = 3.1 nM, 25 EC50 = 1.1 nM). To check for non-specific toxicity, 5 and 25 were assayed against non-proliferating peripheral blood mononuclear cells consisting of monocytes and lymphocytes. We found neither compound reduced cell viability at concentrations of up to 10 μM (data not shown).FACS analysis
The effect of 5 and 23–26 on the SK-OV-3 cell cycle was investigated using flow cytometry. After treatment with compound for 24 h the cells were fixed, stained with propidium iodide and counted (Fig. 4). The profile of untreated cells showed that 54.4% were in the cell growth (G1) phase with 31.2% in DNA synthesis (S) phase and 13.6% entering into or undergoing cell division (G2/M) (Fig. 4a).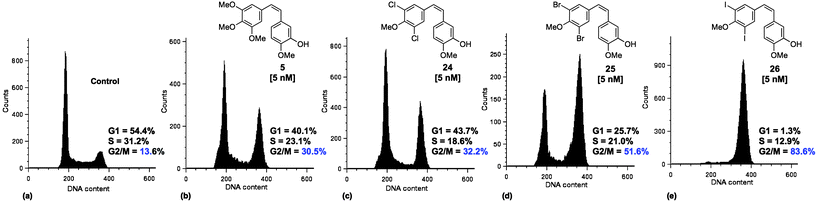 | ||
| Fig. 4 FACS analysis of SK-OV-3 (ovarian cancer) cells stained with propidium iodide after 24 h exposure to control, 5 and 24–26 at 5 nM. (a) Control, (b) 5, (c) 24, (d) 25, (e) 26. Data is representative of three independent experiments. | ||
Treatment with CA-4 (5) at a concentration of 5 nM increased the amount of cells in G2/M to 30.5% (Fig. 4b). Treatment with 24 (Cl) at 5 nM increased the percentage further to 32.2% (Fig. 4c) and levels of mitotic block increased significantly on treatment with 25 (Br) at 5 nM (51.6%, Fig. 4d), reaching a maximum of 83.6% for 26 (I) at the same concentration (Fig. 4e). Treatment with 23 (F) resulted in a large percentage of G2/M cells (73.7%), however, this required a concentration of 200 nM (see ESI,† Fig. S1). Therefore, the ability of these analogues to induce mitotic block was shown to increase in the order F≪Cl≈OMe<Br<I. This order fits with the proposed strength of halogen bonding24 and correlates with halogen Van der Waal's radii. For all of these compounds the population increase in G2/M is consistent with the hypothesis that they are acting as disruptants to the action of MTs in cell division.
Confocal microscopy
To determine if these antimitotic effects were due to interaction with tubulin, cancer cell lines were treated with 5 or 25 for 16 h, fixed, antibody stained for α-tubulin (Dm1a antibody), and visualised using confocal microscopy. Untreated HeLa cells included interphase cells possessing well-defined MT networks (Fig. 5a, panel 1 and Fig. 5b, panel 1). Mitotic cells characterised by condensed chromatin and dense mitotic spindles were also present. Treatment with 25 at 1 μM (Fig. 5a, panel 2) and 100 nM (Fig. 5a, panel 3) resulted in depolymerisation of the interphase MT network and membrane blebbing. Furthermore, a striking redistribution of MTs around the nuclear envelope of a number of the cells were seen (marked with arrows, Fig. 5a, panel 3, enlarged as Fig. 6); a phenomenon also observed in cells treated with 20 μM of the marine alkaloid ceratamine A (6a).27b Treatment with 25 at 10 nM (Fig. 5a, panel 4) resulted in a normal interphase MT network, however, a disproportionately large number of mitotic cells were present. Also observed were MT asters (marked with an asterisk, Fig. 5a, panel 4).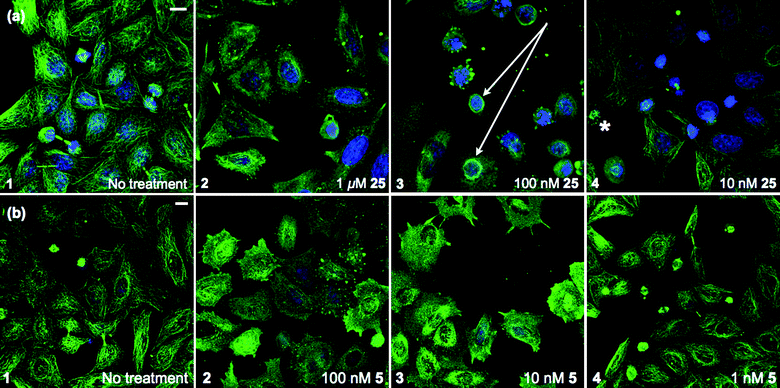 | ||
| Fig. 5 Confocal images of HeLa (cervical cancer) cells. (a) Treatment with 25. Accumulations of MTs encircling some nuclei were seen at 100 nM (arrow, panel 3) along with asters 10 nM (asterisk, panel 4). (b) Treatment with CA-4 (5). Staining: anti-tubulin, Dm1a (green), DNA, DAPI (blue). Representative images shown, scale bar 10 μm. | ||
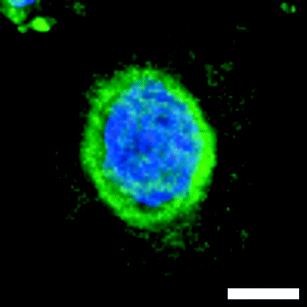 | ||
| Fig. 6 Redistribution of MTs around the nuclear envelope of HeLa cells treated with 25 at 100 nM, scale bar 5 μm. | ||
Treatment with 5 produced similar phenotypic effects at 10-fold lower concentrations i.e. MT depolymerisation in interphase cells was observed at 100 nM (Fig. 5b, panel 2) and 10 nM (Fig. 5b, panel 3) with interphase MT networks intact at 1 nM (Fig. 5b, panel 4). Consistent with a mitotic block phenotype, a large number of mitotic cells were observed at 1 nM, however, circular accumulations of MTs around nuclei were not seen. These data suggest that 25 induces mitotic block at 10 nM as cells had a similar appearance to those treated with 5 at 1 nM. Photomicrographs were also obtained for SK-OV-3 and SK-OV-3TR cell lines that reflect the observations in the HeLa cells and their relative EC50 values in the MTS assay (ESI,† Fig. S2 and S3).
Live-cell microscopy on HeLa cells
To investigate these antitubulin effects in more detail, live-cell microscopy was performed with HeLa cells expressing green-fluorescent protein-tagged tubulin using spinning-disk confocal microscopy, which is a fast and sensitive technique for time-lapse imaging. We first observed the response of interphase and mitotic MTs to treatment with 25 at 100 nM (ESI,† Fig. S4 and S5). On first exposure interphase MTs were clearly visible but between 31 and 34 min cell membranes began to bleb (ESI,† Fig. S4). Mitotic spindles formed during metaphase began to disappear from 21 min and had disappeared entirely within 36 min (ESI,† Fig. S5).On treatment with a 10 nM concentration of 25 this effect on mitotic cells was still evident. The cell in late G2 at t = 0 min (Fig. 7, panel 1) successfully entered prophase at t = 11 min, whereupon the nuclear envelope broke down and MT spindles began to form (Fig. 7, panel 2). At t = 21 min bipolar MT spindles could be seen in the mitotic cell (Fig. 7, panel 3). This cell would normally progress through anaphase but treatment with 25 resulted in collapse of the mitotic spindle and the cell failed to exit mitosis after t = 31 min (Fig. 7, panels 4–6). Interphase cells were unaffected by drug treatment until t = 51 min, where the cell denoted by an asterisk began to bleb, whether this is a direct response to 25 is unclear (Fig. 7, panel 6). These responses were not due to photobleaching or phototoxicity as control cells progressed normally through mitosis under the same conditions (Fig. 8).
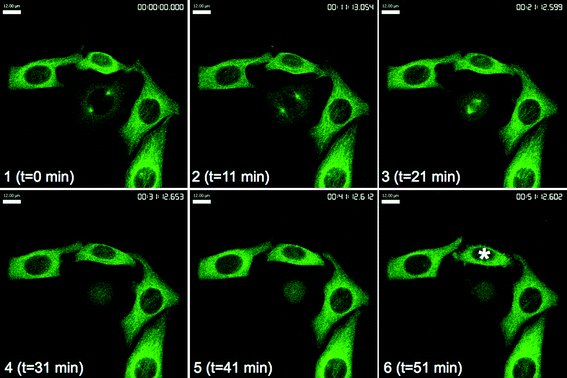 | ||
| Fig. 7 GFP-tubulin HeLa (cervical cancer) cells after treatment with 25 at 10 nM. The asterisk denotes blebbing interphase cell at t = 51 min. Images taken at 10 min intervals immediately after drug addition. Scale bar, 12 μm. | ||
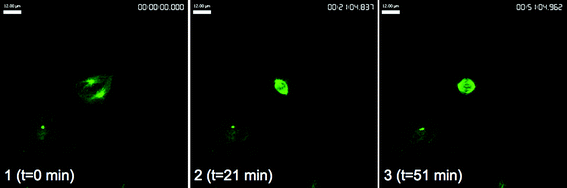 | ||
| Fig. 8 GFP-tubulin HeLa (cervical cancer) cells after treatment with control. Scale bar, 12 μm. | ||
The data from the FACS analysis and confocal microscopy demonstrates that 25 induces significant G2/M arrest through an interaction with tubulin. At high concentrations (>100 nM) 25 has a depolymerising effect on MTs. At concentrations closer to its EC50 value (1–10 nM) 25 has a larger effect on mitotic over interphase MTs. It is thought that at these concentrations 25 is influencing MT dynamics and in doing so induces mitotic block in dividing cells, whilst interphase cells are unaffected.
In summary, we have shown that replacement of two meta-methoxy groups of CA-4 (5) with F, Cl, Br or I atoms results in conservation of the antitubulin and antimitotic effects of the parent compound. Difluorination, however, results in a significant drop in potency over those substituted with Cl, Br or I. A similar finding was reported for trifluorinated derivatives,18,37 whereas mono-fluorination gave no significant reduction in efficacy.21 Dichlorination resulted in a moderate loss of activity, which is significant since mono-chlorination was reported21 to conserve the activity of 5. We have demonstrated that dibromination or diiodination results in compounds (25 and 26) that have equal or superior potency to 5 in both cell viability and cell cycle analysis assays. This seems to undermine the hypothesis that the steric bulk of the meta-groups results in a more favourable binding geometry, given that bromine (1.85 Å)24 and iodine (1.98 Å)24 have smaller van der Waal's radii than a methoxy (2.60 Å)38 group. This raises the possibility that an increased ability to interact with protein through a halogen bonding mechanism could be responsible for the activity of the dibrominated and diiodinated analogues, as these atoms are those expected to have a halogen bond with the greatest strength. This challenges the established structure-activity relationship formulated for analogues of 5, in which the 3,4,5-trimethoxyaryl group is considered optimal for tubulin binding, and other meta-substitutions merely maintain the same conformation.
Acknowledgements
TMB and JWS are supported by The Cambridge Cancer Research UK PhD Training Programme in Medicinal Chemistry. Richard Farndale (Department of Biochemistry) and Tai-Ping Fan (Department of Pharmacology) at the University of Cambridge for their assistance with biological assays.References and notes
- (a) K.-H. Altmann, Curr. Opin. Chem. Biol., 2001, 5, 424–431 CrossRef CAS; (b) M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253–265 CrossRef CAS; (c) B. Weaver and D. Cleveland, Cancer Cell, 2005, 8, 7–12 CrossRef CAS; (d) M. Schmidt and H. Bastians, Drug Resist. Updates, 2007, 10, 162–181 CrossRef CAS; (e) M. Steinberg, Clin. Ther., 2008, 30, 1590–1617 CrossRef CAS; (f) A. Canta, A. Chiorazzi and G. Cavaletti, Curr. Med. Chem., 2009, 16, 1315–1324 CrossRef CAS.
- D. Schiff, P. Y. Wen and M. J. van den Bent, Nat. Rev. Clin. Oncol., 2009, 6, 596–603 Search PubMed.
- M. Mahindroo, J.-P. Liou, J.-Y. Chang and H.-P. Hsieh, Expert Opin. Ther. Pat., 2006, 16, 647–691 Search PubMed.
- A. L. Risinger, F. J. Giles and S. L. Mooberry, Cancer Treat. Rev., 2009, 35, 255–261 CrossRef CAS.
- R. O. Carlson, Expert Opin. Invest. Drugs, 2008, 17, 707–722 Search PubMed.
- G. R. Pettit, S. B. Singh, E. Hamel, C. M. Lin, D. S. Alberts and D. Garcia-Kendall, Experientia, 1989, 45, 209–211 CAS.
- C. M. Lin, H. H. Ho, G. R. Pettit and E. Hamel, Biochemistry, 1989, 28, 6984–6991 CrossRef CAS.
- M. Cushman, D. Nagarathnam, D. Gopal, H. M. He, C. M. Lin and E. Hamel, J. Med. Chem., 1992, 35, 2293–2306 CrossRef CAS.
- L. Lee, L. M. Robb, M. Lee, R. Davis, H. Mackay, S. Chavda, B. Babu, E. L. O'Brien, A. L. Risinger, S. L. Mooberry and M. Lee, J. Med. Chem., 2010, 53, 325–334 CrossRef CAS.
- (a) G. G. Dark, S. A. Hill, V. E. Prise, G. M. Tozer, G. R. Pettit and D. J. Chaplin, Cancer Res., 1997, 57, 1829–1834 CAS; (b) J. Griggs, J. C. Metcalfe and R. Hesketh, Lancet Oncol., 2001, 2, 82–87 CrossRef CAS.
- E. L. Schwartz, Clin. Cancer Res., 2009, 15, 2594–2601 CrossRef CAS.
- (a) V. L. Heath and R. Bicknell, Nature Reviews Clinical Oncology, 2009, 6, 395–404 Search PubMed; (b) D. W. Siemann and M. R. Horsman, Cell Tissue Res., 2009, 335, 241–248 CrossRef CAS; (c) M. M. Cooney, W. van Heeckeren, S. Bhakta, J. Ortiz and S. C. Remick, Nat. Clin. Pract. Oncol., 2006, 3, 682–692 Search PubMed.
- S. P. Ivy, J. Y. Wick and B. M. Kaufman, Nature Reviews Clinical Oncology, 2009, 6, 569–579 Search PubMed.
- (a) R. K. Jain, Science, 2005, 307, 58–62 CrossRef CAS; (b) C. Rüegg, M. Hasmim, F. J. Lejeune and G. C. Alghisi, Biochim. Biophys. Acta, 2006, 1765, 155–177.
- G. M. Tozer, C. Kanthou and B. C. Baguley, Nat. Rev. Cancer, 2005, 5, 423–435 CrossRef CAS.
- (a) S. R. Singh and H. Kaur, Synthesis, 2009, 2471–2491 CrossRef; (b) B. Bhattacharyya, D. Panda, S. Gupta and M. Banerjee, Med. Res. Rev., 2008, 28, 155–183 CrossRef CAS.
- (a) J.-Y. Pan, S.-L. Chen, M.-H. Yang, J. Wu, J. Sinkkonen and K. Zoud, Nat. Prod. Rep., 2009, 26, 1251–1292 RSC; (b) S. Apers, A. Vlietinck and L. Peters, Phytochem. Rev., 2003, 2, 201–217 CrossRef CAS.
- K. Gaukroger, J. A. Hadfield, N. J. Lawrence, S. Nolana and A. T. McGown, Org. Biomol. Chem., 2003, 1, 3033–3037 RSC.
- G. C. Tron, T. Pirali, G. Sorba, F. Pagliai, S. Busacca and A. A. Genazzani, J. Med. Chem., 2006, 49, 3033–3044 CrossRef CAS.
- J. M. Andreu, B. Perez-Ramirez, M. J. Gorbunoff, D. Ayala and S. N. Timasheff, Biochemistry, 1998, 37, 8356–8368 CrossRef CAS.
- G. R. Pettit, M. D. Minardi, H. J. Rosenberg, E. Hamel, M. C. Bibby, S. W. Martin, M. K. Jung, R. K. Pettit, T. J. Cuthbertson and J.-C. Chapuis, J. Nat. Prod., 2005, 68, 1450–1458 CrossRef CAS.
- R. R. Monaco, W. C. Gardiner and S. Kirschner, Int. J. Quantum Chem., 1999, 71, 57–62 CrossRef CAS.
- P. Metrangolo, F. Meyer, T. Pilati, G. Resnati and G. Terraneo, Angew. Chem., Int. Ed., 2008, 47, 6114–6127 CrossRef CAS.
- P. Auffinger, F. A. Hays, E. Westhof and P. Shing Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS.
- Y. Lu, T. Shi, Y. Wang, H. Yang, X. Yan, X. Luo, H. Jiang and W. Zhu, J. Med. Chem., 2009, 52, 2854–2862 CrossRef CAS.
- F. Hentschel and T. Lindel, Synthesis, 2010, 42, 181–204.
- (a) E. Manzo, R. van Soest, L. Matainaho, M. Roberge and R. J. Andersen, Org. Lett., 2003, 5, 4591–4594 CrossRef CAS; (b) G. Karjala, Q. Chan, E. Manzo, R. J. Andersen and M. Roberge, Cancer Res., 2005, 65, 3040–3043 CAS.
- J. W. Shearman, R. M. Myers, T. M. Beale, J. D. Brenton and S. V. Ley, Tetrahedron Lett., 2010 DOI:10.1016/j.tetlet.2010.07.016.
- P. Metrangolo and G. Resnati, Science, 2008, 321, 918–919 CrossRef CAS.
- L. Emmanuvel, R. K. Shukla, A. Sudalai, S. Gurunath and S. Sivaram, Tetrahedron Lett., 2006, 47, 4793–4796 CrossRef CAS.
- S. B. Singh and G. R. Pettit, J. Org. Chem., 1989, 54, 4105–4114 CrossRef CAS.
- G. R. Pettit, S. B. Singh, M. R. Boyd, E. Hamel, R. K. Pettit, J. M. Schmidt and F. Hogan, J. Med. Chem., 1995, 38, 1666–1672 CrossRef CAS.
- F. Orsini, F. Pelizzoni, B. Bellini and G. Miglierini, Carbohydr. Res., 1997, 301, 95–109 CrossRef CAS.
- The xCELLigence system (Roche) for measuring cell growth was used for these assays, as described in J. Z. Xing, L. Zhu, J. A. Jackson, S. Gabos, X.-J. Sun, X. Wang and X. Xu, Chem. Res. Toxicol., 2005, 18, 154–161 Search PubMed.
- US Pat., 7 507 851, 2009.
- The 3,5-diiodo-4-methoxy analogue, (Z)–26, used in these assays was isomerically pure as determined by 1H NMR spectroscopy.
- J. J. Hall, M. Sriram, T. E. Strecker, J. K. Tidmore, C. J. Jelinek, G. D. K. Kumar, M. B. Hadimani, G. R. Pettit, D. J. Chaplin, M. L. Trawick and K. G. Pinney, Bioorg. Med. Chem. Lett., 2008, 18, 5146–5149 CrossRef CAS.
- D. F. Paris, N. L. Wolfe, W. C. Steen and G. L. Baughman, Appl. Environ. Microbiol., 1983, 45, 1153–1155 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of 5 and 23–26 with full spectral data, along with procedures for HUVEC isolation, biological assays and microscopy methods. See DOI: 10.1039/c0md00095g |
| This journal is © The Royal Society of Chemistry 2010 |