DOI:
10.1039/C0MD00027B
(Concise Article)
Med. Chem. Commun., 2010,
1, 209-211
Biofunctional TiO2 nanoparticle-mediated photokilling of cancer cells using UV irradiation†
Received
12th March 2010
, Accepted 2nd April 2010
First published on
11th May 2010
Abstract
Cancer cell-specific photokilling was successfully demonstrated using antibody-immobilized TiO2 nanoparticles with only 1 J cm−2 UV irradiation.
Introduction
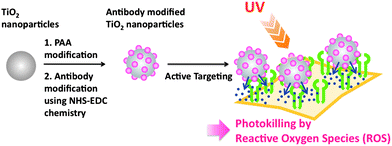
In the context of cancer therapy, nanoparticles (NPs) such as platinum,1 gold,2 quantum dots and liposomes have attracted great interest.3 Compared with organic nanoparticles such as liposomes or chitosan, metal nanoparticles have the advantage that due to their photodynamic characteristics, their therapeutic effect is autonomous and, thus, does not require encapsulation of therapeutic agents. Among various metal nanoparticles, titanium dioxide (TiO2) has a great generation property of reactive oxygen species (ROS) under ultraviolet (UV) light excitation depending on the sizes and morphologies of the particles.4 In spite of its high potential for photodynamic cancer therapy (PDT), the use of these particles has been hampered because of the difficulty of size control, aggregation in aqueous solution and nanotoxicity itself5,6, and so far only studies on their antibacterial effect have been successful.7 Few studies on cancer cell-specific photokilling by TiO2 are reported;8 however, they only focus on the immobilization method of the biomolecule or mechanical delivery method. To control this “friends and foes” photocharacteristic of TiO2 under UV irradiation, a fundamental study about the relationship between TiO2 concentration and UV irradiation strength is necessary. In addition, cancer cell-specific molecules have to be immobilized to reduce the side effect on normal cells around the cancer.
In this study, to expand the versatility of TiO2 NP-mediated cell-specific photokilling method, we evaluated the cytotoxicity of TiO2 NPs according to UV irradiation strength, and measured photokilling of target cells via active cell targeting using antibody-immobilized TiO2 NPs.
Experimental
Two kinds of TiO2 NPs were used in this study, with sizes of about 30 nm and 10 nm, as measured by dynamic light scattering. For surface modification of TiO2 NPs, polyacrylic acids (PAA) were used to avoid TiO2 NP aggregation under physiological conditions. PAA modification of each of the TiO2 NPs was carried out according to previous reports.9 UV-driven hydroxyl radical generation from various concentrations of NPs was measured using aminophenyl fluorescein (APF), which has a fluorescent response to various ROS. Chemical modifications of biofunctional molecules on the PAA-modified TiO2 NPs were made with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundN-hydroxysuccinimide and COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compound1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride chemistries.9 The variable domain of a camelid heavy chain-only antibody (la), which has high affinity to EGFR and is prepared easily using Escherichia coli as a host,11 was immobilized as a cell-specific homing molecule on TiO2 NPs. Detailed experimental procedures are shown in ESI.†
Preparation and evaluation of antibody-immobilized TiO2 nanoparticles
HeLa cells were incubated with 0.03 wt% PAA-TiO2/la for 10 min and then directly irradiated with UV (3 J cm−2 or 1 J cm−2). Cell toxicity was evaluated immediately or 24 h later using calcein-AM (live cells) and ethidium homodimer-1 (EthD-1, dead cells) fluorescent dye. Detailed experimental procedure are shown in ESI.†
Results and discussion
Our strategy is shown in Fig. 1. First, we investigated the radical generation ability of TiO2 and PAA-modified TiO2 NPs with UV irradiation using fluorescence derived from APF as an indicator. TiO2 NPs (30 nm) generated the highest fluorescence level when 0.3 and 0.03 wt% NPs were irradiated with 1.5 J cm−2 (Fig. 2A), suggesting the appropriate conditions to generate high amount of radicals. Interestingly, the fluorescence intensity decreased at higher levels of UV exposure. For NPs of lower concentration (0.003 wt%), more UV exposure (5 J cm−2) was required to generate nearly the same fluorescence levels. After PAA modification, the highest fluorescence intensity was detected with 3 J cm−2 of exposure with 0.03 wt% NPs (Fig. 2C). We found that UV irradiation and pH did not affect APF fluorescence. PAA modification slightly decreased the amount of generated radicals, because produced radicals or UV radiation may be blocked by PAA. Similar results were obtained for TiO2 NPs of 10 nm, although lower fluorescence (i.e. a smaller amount of radicals) was observed (Fig. 2B). However, after PAA modification, the amount of radicals decreased, and a maximum peak was not reached even after UV irradiation up to 10 J cm−2 (Fig. 2D). From these results, there was an optimal size for NPs for ROS generation with UV irradiation. TiO2 NPs of 30 nm possessed better ROS generation potential whether the NPs were modified or not, which corresponds to a previous report.12 For target cell-specific photokilling, a large amount of radicals should be produced with a lower NP concentration and with lower UV exposure levels. We assumed that PAA-modified TiO2 NPs of 30 nm (hereafter named PAA-TiO2) were optimal for protein modification and cell photokilling in further experiments.
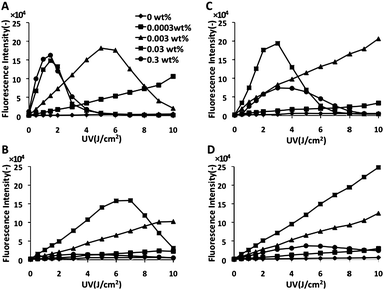 |
| | Fig. 2 The amount of radicals from TiO2 exposed to UV. (A) TiO2 (30 nm). (B) TiO2 (10 nm). (C) PAA-TiO2 (30 nm). (D) PAA-TiO2 (10 nm). | |
We modified PAA-TiO2 by anti-epidermal growth factor receptor (EGFR) antibody because EGFR is over-expressed on the surface of 20–30% of cancer cell lines.10 The anti-EGFR antibody modification of TiO2 NPs, named as PAA-TiO2/la, was analyzed by SDS-PAGE. The band corresponding to PAA-TiO2/la was observed at a much higher molecular weight, suggesting successful modification (ESI, Fig. S1†). We also confirmed the antigen-binding ability of PAA-TiO2/la by ELISA (ESI, Fig. S2†). Anti-EGFR antibody (la) and PAA-TiO2/la efficiently bound the HeLa cells, although PAA-TiO2 did not bind the cell.
Cell-specific photokilling using antibody-immobilized TiO2 NPs with UV irradiation
Encouraged by these findings, we next carried out cell-specific photokilling using PAA-TiO2/la. HeLa cells were employed as a model of EGFR-expressing cells. The addition of PAA-TiO2 and/or anti-EGFR antibody without UV exposure did not affect cell viability (Fig. 3, left column), suggesting low toxicity of TiO2 NPs itself on cultured cells at this concentration. HeLa cells treated with PAA-TiO2/la showed extensive cell death (about 50% estimated by cell counting) after UV irradiation (Fig. 3D). In contrast, there was no cell death after treatment with only PAA-TiO2 and anti-EGFR antibody mixtures (Fig. 3B, C), or BSA-modified TiO2 NPs after UV exposure (Fig. 3E, F). When MCF-7, which is a low EGFR expression cell line, was treated with PAA-TiO2/la, cell toxicity was not observed even under UV exposure (Fig. 3G, H). This clearly shows that PAA-TiO2/la NPs allow EGFR expressing cell-specific TiO2 NP accumulation and cell-specific killing by radicals generated with UV irradiation. However, after further cultivation for 24 h following UV irradiation (3 J cm−2), cells without TiO2 NPs were detected to be dead in the end. It suggests apoptosis might be induced with large amount of UV irradiation. Therefore, to optimize the UV irradiation level, we applied UV irradiation at 1 J cm−2 followed by a further 24 h incubation, and then cell viability was evaluated. HeLa cells treated with PAA-TiO2/la under UV exposure (1 J cm−2) showed major cell death after 24 h incubation (Fig. 4D). In contrast, only a few dead cells under other conditions were observed (Fig. 4A–C). We also evaluated cell death depending on the amount of TiO2 NPs with 1 J cm−2 UV exposure. HeLa cells were efficiently damaged (about 80% estimated by cell counting) with 0.03 and 0.003 wt% PAA-TiO2/la (Fig. 4B, C), suggesting that accumulation around the cell surface enhanced cell toxicity caused by radical generation with UV exposure, even at a low TiO2 NP concentration. Effective photokilling with a low TiO2 concentration demonstrates the advantage of active targeting, and is also a desirable property in therapy.
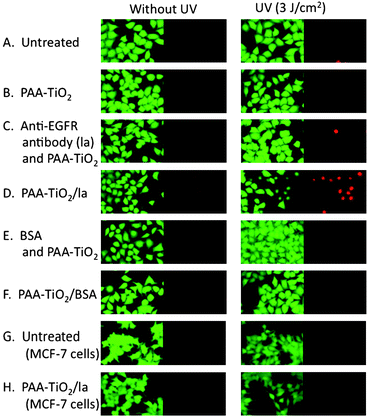 |
| | Fig. 3 HeLa cell viability after treatment with anti-EGFR antibody-immobilized TiO2 NPs followed by UV irradiation (3 J cm−2). Cell viability was examined immediately after UV irradiation by calcein AM/EthD-1 fluorescence staining, and representative images from each sample are shown. Cells with green fluorescence are considered alive, whereas those with red are considered dead. (A) Untreated (B) PAA-TiO2 (C) anti-EGFR antibody (la) and PAATiO2 mixtures. (D) PAA-TiO2/la. (E) la and BSA mixture; (F) PAA-TiO2/BSA (G) Untreated MCF-7 cells (COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundH) MCF-7 cells treated with PAA-TiO2/la. | |
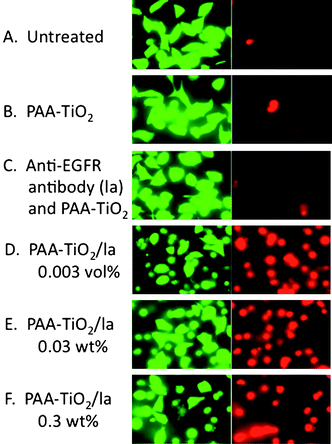 |
| | Fig. 4 HeLa cell viability after treatment with PAA-TiO2/la followed by UV irradiation (1 J cm−2) and further incubation for 24 h. (A) Untreated (B) PAA-TiO2 (C) anti-EGFR antibody (la) and PAATiO2 mixtures. (D) 0.003 wt% of PAA-TiO2/la. (E) 0.03 wt% of PAA-TiO2/la. (F) 0.3 wt% of PAA-TiO2/la. | |
Conclusions
We established a cell-specific photokilling method using antibody immobilized TiO2 NPs. It allows target cell-specific killing with low TiO2 concentrations and low UV exposure levels. Our strategy is applicable for other targeting molecules such as various antibodies or bioactive ligands, as well as photoactivating nanoparticles. Although our system can be applicable for tumors near the skin surface, one possible disadvantage is that UV irradiation cannot be exposed to deeper tissues such as the liver and pancreas. Hence, novel nanoparticles which generate ROS with some radial ray, such as X-rays or gamma-rays, should be developed in the future.
Acknowledgements
This research was supported in part by Special Coordination Funds for Promoting Science and Technology, Creation of Innovation Centers for Advanced Interdisciplinary Research Areas (Innovative Bioproduction Kobe), MEXT, Japan.
Notes and references
- N. G. Blanco, C. R. Maldonado and J. C. Marreque-Rivas, Chem. Commun., 2009, 5257–5259 RSC.
- Y. T. Lim, M. Y. Cho, B. S. Choi, J. M. Lee and B. H. Chung, Chem. Commun., 2008, 4930–4932 RSC.
-
(a) D. J. Bharali, M. Khalil, M. Gurbuz, T. M. Simon and S. A. Mousa, Nanomedicine, 2009, 4, 1–7 CrossRef CAS;
(b) M. Ferrari, Nat. Rev. Cancer, 2005, 5, 161–171 CrossRef CAS;
(c) M. E. Davis, Z. G. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS;
(d) D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS;
(e) N. G. Blanco, C. R. Maldonado and J. C. Mareque-Rivas, Chem. Commun., 2009, 5257–5259 RSC.
-
(a) U. M. K. Shahed, Al-S. Mofareh and B. I. William Jr., Science, 2002, 297, 2243–2245 CrossRef CAS;
(b) R. Huang, A. Wallqvist and D. G. Covell, Biochem. Pharmacol., 2005, 69, 1009–1039 CrossRef CAS;
(c) R. Cai, Y. Kubota, T. Shuin, H. Sakai, K. Hashimoto and A. Fujishima, Cancer Res, 1992, 52, 2346–2348 CAS;
(d) Y. Kubota, T. Shuin, C. Kawasaki, M. Hosaka, H. Kitamura, R. Cai, H. Sakai, K. Hashimoto and A. Fujishima, Br. J. Cancer, 1994, 70, 1107–1111 CAS;
(e) J. W. Seo, H. Chung, M. Kim, J. Lee, I. Choi and J. Cheon, Small, 2007, 3, 850–853 CrossRef CAS;
(f) N. K. Shrestha, J. M. Macak, F. Schmidt-Stein, R. Hahn, C. T. Mierke, B. Fabry and P. Schmuki, Angew. Chem., Int. Ed., 2009, 48, 969–972 CrossRef CAS;
(g) Y. Luo, Y. Tian and Q. Rui, Chem. Commun., 2009, 3014–6 RSC.
-
(a) A. Nel, T. Xia, L. Mädler and N. Li, Science, 2006, 311, 622–627 CrossRef CAS;
(b) T. Xia, M. Kovochich, J. Brant, M. Hotze, J. Sempf, T. Oberley, C. Sioutas, J. I. Yeh, M. R. Wiesner and A. E. Nel, Nano Lett., 2006, 6, 1794–1807 CrossRef CAS;
(c) S. Singh, T. Shi, R. Duffin, C. Albrecht, D. van Berlo, D. Hoehr, B. Fubini, G. Martra, I. Fenoglio, P. J. A. Borm and R. P. F. Schins, Toxicol. Appl. Pharmacol., 2007, 222, 141–151 CrossRef CAS.
- S. M. Hussain, K. L. Hess, J. M. Gearhart and K. T. Geiss, Toxicol. in Vitro, 2005, 19, 975–983 CrossRef CAS.
-
(a) F. R. Marciano, D. A. Lima-Oliveira, N. S. Da-Silva, A. V. Diniz, E. J. Corat and V. J. Trava-Airoldi, J. Colloid Interface Sci., 2009, 340, 87–92 CrossRef CAS;
(b) D. S. Kim and S. Y Kwak, Environ. Sci. Technol., 2009, 43, 148–151 CrossRef CAS;
(c) B. K. Erdural, A. Yurum, U. Bakir and G. Karakas, J. Nanosci. Nanotechnol., 2008, 8, 878–886 CrossRef CAS;
(d) C. Hu, J. Guo, J. Qu and X. Hu, Langmuir, 2007, 23, 4982–4987 CrossRef CAS.
-
(a) A. R. Elena, U. Ilya, L. Barry, M. D. Nada, S. L. Maciej and R. Tijana, Nano Lett., 2009, 9, 3337–3342 CrossRef;
(b) J. Xu, Y. Sun, J. Huang, C. Chen, G. Liu, Y. Jiang, Y. Zhao and Z. Jiang, Bioelectrochemistry, 2007, 71, 217–222 CrossRef CAS.
-
(a) K. Kanehira, T. Banzai, C. Ogino, N. Shimizu, Y. Kubota and S. Sonezaki, Colloids Surf., B, 2008, 64, 10–15 CrossRef CAS;
(b) C. Ogino, K. Kanehira, R. Sasai, S. Sonezaki and N. Shimizu, J. Biosci. Bioeng., 2007, 104, 339–342 CrossRef CAS.
-
(a) N. E. Hynes and D. F. Stern, Biochim. Biophys. Acta, 1994, 1198, 165–184;
(b) V. P. Collins, Semin. Cancer Biol, 1993, 4, 27–32 CAS.
- R. C. Roovers, T. Laeremans, L. Huang, S. De Taeye, A. J. Verkleij, H. Revets, H. J. de Haard and P. M. P. van Bergen en Henegouwen, Cancer Immunol. Immunother., 2007, 56, 303–317 CAS.
- J. Jiang, G. Oberdoumlrster, A. Elder, R. Gelein, P. Mercer and P. Biswas, Nanotoxicology, 2008, 2, 33–42 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure, and Figure S1 and S2. See DOI: 10.1039/c0md00027b |
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.