Discovery of the highly potent PI3K/mTOR dual inhibitor PF-04691502 through structure based drug design†
Hengmiao
Cheng
*a,
Shubha
Bagrodia
b,
Simon
Bailey
a,
Martin
Edwards
a,
Jacqui
Hoffman
a,
Qiyue
Hu
a,
Robert
Kania
a,
Daniel R.
Knighton
a,
Matthew A.
Marx
a,
Sacha
Ninkovic
a,
Shaoxian
Sun
b and
Eric
Zhang
c
aCancer Chemistry, Pfizer Worldwide Research and Development, La Jolla Laboratories, 10770 Science Center Drive, San Diego, CA 92121, U.S.A. E-mail: henry.cheng@pfizer.com; Tel: +1 (858) 622-3208
bOncology Research Unit, Pfizer Worldwide Research and Development, La Jolla Laboratories, 10770 Science Center Drive, San Diego, CA 92121, U.S.A.
cPDM, Pfizer Worldwide Research and Development, La Jolla Laboratories, 10770 Science Center Drive, San Diego, CA 92121, U.S.A.
First published on 23rd July 2010
Abstract
The phosphatidylinositol 3-kinase (PI3K) signaling pathway plays crucial roles in cell growth, proliferation and survival. Genomic aberrations in the PI3K pathway, such as mutational activation of PI3Kα or loss of function of tumor suppressor PTEN, have been closely linked to the development and progression of a wide range of cancers. Hence, inhibition of the key targets in the pathway, e.g. PI3K, AKT, mTOR, offers great potential for the treatment of cancer. Lead optimization through integration of structure based drug design (SBDD) and physical properties-based optimization (PPBO) led to the discovery of 2-amino-8-[trans-4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxypyridin-3-yl)-4-methylpyrido[2,3-d]pyrimidin-7(8H)-one (PF-04691502, 1) that demonstrated potent in vitro inhibitory activity against both PI3K and mTOR, excellent kinase selectivity, good ADMET, and robust in vivo efficacy in a mouse xenograft tumor growth model. Compound 1 is currently being evaluated in human clinical trials for the treatment of cancer.
The phosphatidylinositol 3-kinase (PI3K) signaling pathway plays crucial roles in cell growth, proliferation and survival.1,2 The PI3K family of enzymes is comprised of 15 lipid kinases with distinct substrate specificities, expression patterns and modes of actions.3 Mutations in PIK3CA, the gene encoding the p110α catalytic subunit of PI3Kα, are very common in human cancers.4–8 Mutational activation of p110α signaling pathways causes cell transformation both in vitro and in vivo.9–12 Phosphatase and tensin homolog (PTEN) deletion, leading to activation of AKT has been observed in many types of malignancies.13–15 mTOR is a class IV PI3K protein kinase.16,17 mTOR regulates growth and proliferation through two functional complexes: mTORC1 and mTORC2. mTORC1 regulates translation via S6 kinase (S6K) and 4E-BP, and is the target of naturally occurring agent rapamycin. mTORC2 regulates AKT S473 phosphorylation. The rapamycin analog CCI-779, which inhibits mTORC1 via its allosteric site, has been approved for the treatment of renal cell carcinoma.18 However, inhibition of mTORC1 alone results in a feedback stimulation of AKT, and thus may be inferior to simultaneous inhibition of both mTORC1 and mTORC2 through blockade of kinase activity.19 Since the kinase domain of mTOR is homologous to the p110α catalytic subunit of the class 1 PI3Ks,20 development of PI3K/mTOR dual kinase inhibitor offers great potential for the treatment of cancer. Several PI3K/mTOR dual inhibitors have progressed to clinical trials.21–23 BEZ-235 and GSK2126458 are the two inhibitors with published structures (Fig. 1). We describe herein the discovery of 2-amino-8-[trans-4-(2-hydroxyethoxy)cyclohexyl]-6-(6-methoxypyridin-3-yl)-4-methylpyrido[2,3-d]pyrimidin-7(8H)-one (Fig. 2, PF-04691502, 1), a highly potent and selective ATP competitive kinase inhibitor of class 1 PI3Ks and mTOR. Compound 1 is being evaluated in phase 1, open label, dose-escalation study in subjects with solid tumors.
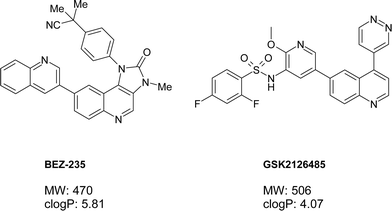 | ||
| Fig. 1 Published clinical PI3K/mTOR dual inhibitors with structures. | ||
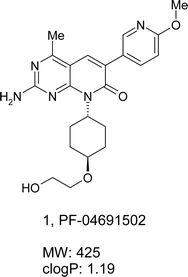 | ||
| Fig. 2 Pfizer clinical candidate PF-04691502. | ||
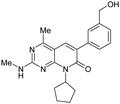
A high throughput biochemical screen using mouse PI3Kα (mPI3Kα) as a surrogate of human PI3Kα was carried out. Screening hits were then tested for activity against mTOR and were further prioritized according to their ligand efficiency (LE)24 and overall kinase selectivity profile. From these hits a series of interest of 4-methylpyridopyrimidinones (MPP) was chosen for follow-up. An early MPP derivative 2 (Table 1) demonstrated potent inhibition of mPI3Kα (1.4 nM), and with a high LE (0.48). When tested in an mTOR kinase domain in vitro biochemical assay, 2 exhibited modest activity with Ki of 840 nM. Compound 2 demonstrated good selectivity over other protein kinases. When tested against 45 protein kinases, compound 2 exhibited less than 20% inhibition at 1 μM against all of the 45 protein kinases. The methyl at the 4-position of the MPP core is necessary to confer excellent overall kinase selectivity to the series. In a BT20 cell assay, measuring inhibition of AKT phosphorylation at S473, 2 exhibited excellent cellular potency with an IC50 of 15 nM.
| Structure | Mouse PI3Kα Ki/nM | mTOR Ki/nM | S473 pAKT IC50/nM | MW | clogP | Human LM ER | Sol/μM | |
|---|---|---|---|---|---|---|---|---|
| a Data is reported in arithmetic mean ± standard deviation. | ||||||||
| 1 |
|
0.57 ± 0.062 | 16 ± 4.9 | 13 ± 6.3 | 425 | 1.19 | <0.26 | 14 ± 1.9 |
| 2 |
|
1.4 ± 0.50 | 840 ± 86 | 15 ± 5.0 | 364 | 3.16 | 0.68 | 65 ± 4.0 |
| 3 |
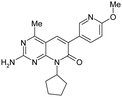
|
1.4 ± 0.3 | 200 ± 30 | 16 ± 3.6 | 351 | 2.7 | <0.26 | 3.9 ± 0.51 |
| 4 |
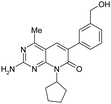
|
1.2 ± 0.12 | 82 ± 24 | — | 350 | 2.33 | — | — |
| 5 |
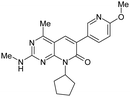
|
1.1 ± 0.028 | 700 ± 139 | — | 365 | 3.52 | — | — |
| 15 |
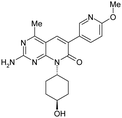
|
0.90 ± 0.017 | 17 ± 0.56 | 12 ± 5.8 | 381 | 1.17 | <0.25 | 16 ± 1.4 |
| 16 |
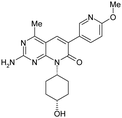
|
3.0 ± 0.092 | 90 ± 9.0 | 22 ± 11 | 381 | 1.17 | <0.26 | 60. ± 19 |
| 17 |
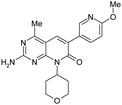
|
3.6 ± 0.23 | 110 ± 32 | — | 367 | 0.86 | <0.26 | 40. ± 1.3 |
| 18 |
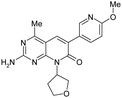
|
6.6 ± 0.43 | 150 ± 26 | — | 353 | 1.05 | <0.26 | 144 ± 7.1 |
A solved co-crystal structure of PI3Kγ, a readily available surrogate of PI3Kα,25–28 with 2 bound into the ATP binding site is illustrated in Fig. 3. The aminopyrimidine forms key hydrogen bonds with the hinge residue, Val 882. Compared to protein kinases excluding mTOR, there is an extra amino acid residue in the hinge region for PI3Ks, which forms a small hydrophobic pocket. The methyl at the 4-position on the MPP core fits tightly into this unique pocket, providing understanding of how this structural feature confers excellent kinase selectivity for the MPP series. The cyclopentyl ring is positioned in the ribose binding pocket, the two methylenes that are one carbon atom away from the MPP core form hydrophobic interactions with the lipophilic side chains of the protein. The benzylic alcohol group is bound in the selectivity pocket, with the alcohol positioned to form hydrogen bonds with the protein Y867 side chain and F965 backbone in the selectivity pocket.
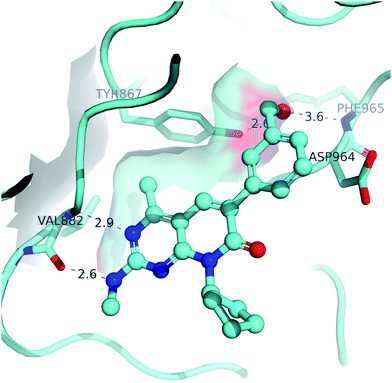 | ||
| Fig. 3 Co-crystal structure of 2 bound to PI3Kγ. | ||
Physical properties-based optimization (PPBO) of multiple drug-like attributes, guided by structure based drug design (SBDD), commenced. Integration of SBDD and PPBO has proven to be an effective strategy to produce cancer clinical candidates.29 Compound 2 had poor pharmacokinetic properties; it was characterized by high in vitro clearance with extraction ratio (ER) of 0.68 in human liver microsomes (HLM). Design focused on strategies to replace metabolic soft spots in the best physical property space (e.g. MW and logP) that will provide the best overlap of desired properties for drug performance in vivo.
The general synthetic route to prepare MPP derivatives is illustrated in Scheme 1.30 The R1 group in compound 6 could be either a free or protected amino group. The protecting group was removed at a stage in the synthesis sequence appropriate for a given target. Compound 6 was reacted with amine 7 to generate 8. Compound 8 was then treated with N-bromosuccinamide to afford 9. Palladium-catalyzed coupling of 9 and 10 yielded compound 11. Upon treatment with DBU at a high temperature, the trans double bond in 11 isomerized to a cis double bond, followed by intramolecular cyclization to yield 12. After 12 was converted to 13 by treatment with N-bromosuccinamide, Suzuki coupling was carried out to afford the desired product 14.
![General synthetic route for the MPP derivatives. Conditions: step 1) R2–NH2, DIEA, K2CO3, DMAC; step 2) NBS, CHCl3; step 3) ethyl acrylate, tri-o-tolylphosphine, palladium(ii) acetate, Et3N; step 4) 1,8-diazabicyclo[5,4,0]undec-7-ene, potassium tert-butoxide, DMA; step 5) NBS, DMF; step 6) boronic acid, K2CO3, Pd(PPh3)2Cl2, DMF–water.](/image/article/2010/MD/c0md00072h/c0md00072h-s1.gif) | ||
| Scheme 1 General synthetic route for the MPP derivatives. Conditions: step 1) R2–NH2, DIEA, K2CO3, DMAC; step 2) NBS, CHCl3; step 3) ethyl acrylate, tri-o-tolylphosphine, palladium(II) acetate, Et3N; step 4) 1,8-diazabicyclo[5,4,0]undec-7-ene, potassium tert-butoxide, DMA; step 5) NBS, DMF; step 6) boronic acid, K2CO3, Pd(PPh3)2Cl2, DMF–water. | ||
In general, metabolic stability correlates with logP/logD; compounds with lower logP/logD tend to exhibit lower clearance.31 In order to address the clearance issue associated with 2, the benzyl alcohol was replaced with more stable isosteric groups that reduced clogP and maintained low MW. The methyl on the 2-amino group is a potential metabolic soft spot for dealkylation, and was removed. Illustrated in Fig. 4 is the plot of HLM ER against clogP for all the MPP derivatives with aryl or heteroaryl groups at the 6-position. Generally, the lower clogP compounds exhibit lower HLM clearance values, as expected, and outliers reveal structural features that are metabolic soft spots on the top, high ER border, or resistant to metabolism on the low ER border. Of these analogues, compound 3 was identified as an early lead that maintained the in vitro potency with mPI3Kα of 1.4 nM, and was stable in the human liver microsome assay with ER less than 0.26. Modification of 2 both by removing the methyl on the 2-amino group and by replacing the benzyl alcohol with methoxypyridine yielded 3, which gained activity against mTOR with Ki of 200 nM. SAR analysis of compounds 2, 3, 4 and 5 suggests that removal of the methyl on the 2-amino group is largely responsible for the increase in mTOR potency. The observed SAR is supported by structural analysis. In a homology model of mTOR, derived from the PI3Kγ crystal structure, the methyl on the 2-amino group of 2 interacted unfavorably with the side chain of TRP-80 that was unique to mTOR.
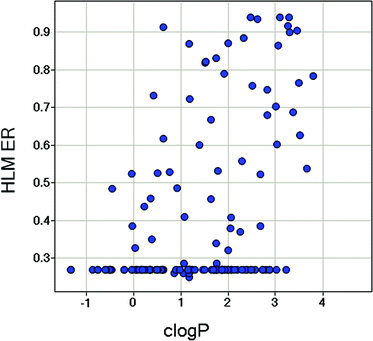 | ||
| Fig. 4 Scatter plot of HLM ER vs. clogP for the MPP derivatives. | ||
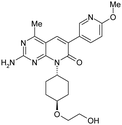
One key issue with 3 is the low solubility (3.9 μM) which resulted in poor absorption (28%) in preliminary in vivo rat PK studies. Since clogP is a critical factor that influences the solubility of an organic molecule,32 the crystal structure of 2 in PI3Kγ (Fig. 3) was examined to determine which regions of the enzyme could accommodate additional polarity. The cyclopentyl ring was bound in the ribose pocket, the methylenes that are two carbon atoms away from the MPP core are lined with polar protein residue side chains. This structural insight supported the cyclopentyl ring as the target location for introduction of polar groups such as an alcohol. Indeed, it appeared that an alcohol could form a hydrogen bond with the polar side chains. Introduction of a cyclohexyl alcohol as an achiral unit was the preferred tactic. Listed in Table 1 are the properties for both the trans-cyclohexanol derivative 15 and the cis-cyclohexanol derivative 16. Both 15 and 16 demonstrated potent inhibition of PI3K and mTOR and improved solubility. For example, 15 was a dual inhibitor with Ki of 0.9 nM against mPI3Kα and Ki of 17 nM against mTOR. 15 exhibited BT20 cell IC50 of 12 nM, low in vitro clearance with ER less than 0.25, and kinetic solubility of 16 μM.
Interestingly, plasma samples from rat and mouse PK studies of 16 were found to contain significant amounts of 15. Similarly a small amount of 16 was detected in animals treated with 15. It is hypothesized that the isomerization occurred via oxidation of the alcohol to the ketone, followed by the reduction of the ketone to give a mixture of 15 and 16, with 15 being the major product.
In order to avoid the potential in vivo isomerization issue associated with the secondary alcohol, the cyclohexanol was modified. Guided by crystal structure analysis, several side chains were investigated,30 and the hydroxylethyl was identified as an optimum group that gave compounds good overall properties. Compound 1 was identified as the lead molecule which demonstrated dual inhibition of PI3Kα and mTOR, with Ki of 0.57 nM against mPI3Kα, and Ki of 16 nM against mTOR. PF-04691502 is an ATP competitive inhibitor of both PI3Kα and mTOR kinases. Compound 1 exhibited BT20 cell IC50 of 13 nM, solubility of 14 μM and low in vitro clearance with ER less than 0.26.
In Fig. 5, the mPI3Kα potency of the compounds described herein is plotted against clogP. The diagonal lines indicate contours of equivalent lipophilic efficiency (LipE = −LogKi − clogP).33 In general, compounds with higher LipE are desired as they tend to have better in vivo properties, through a combination of higher potency and improved PK arising from lower lipophilicity. The original lead 2 had a LipE value of less than 6, and the optimization described above led to analogues with increased LipE values. While the HTS hit 2 is not metabolically stable in the in vitro clearance assay, the clinical candidate 1, with the highest LipE of the analogs presented here, exhibited excellent in vitro metabolic stability.
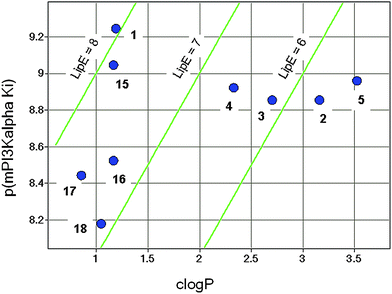 | ||
| Fig. 5 LipE plot. | ||
As illustrated in Fig. 6, the co-crystal structure of 1 with PI3Kγ was determined. Similar to the binding mode of 2 with PI3Kγ, the aminopyrimidine in 1 formed key hydrogen bonds with the hinge residue Val 882. The ring nitrogen on the methoxypyridine formed one key hydrogen bond with a conserved water molecule in the selectivity pocket. In the co-crystal structure of 2 with PI3Kγ protein, this conserved water molecule was replaced by the benzyl alcohol side chain. The terminal alcohol of the hydroxyethyl formed an intramolecular hydrogen bond with the oxygen atom off the cyclohexyl ring, which reduced the effective number of hydrogen bond donors.
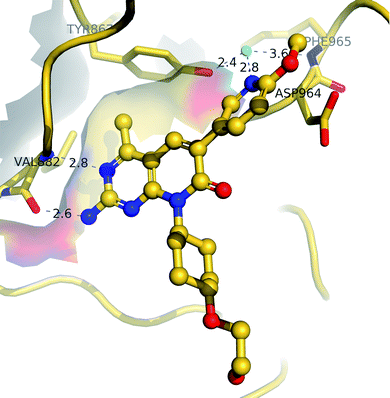 | ||
| Fig. 6 Co-crystal structure of 1 with PI3Kγ. | ||
Compound 1 demonstrated potent cellular IC50 on inhibiting AKT phosphorylation at S473 in the SKOV3 ovarian cancer cell line with IC50 of 7.4 nM. Compound 1 also exhibited robust and dose-dependent inhibition of cell proliferation in vitro and tumor growth inhibition in a mouse subcutaneous model implanted with the SKOV3 tumor cell line.34 Animals were housed in the Pfizer vivarium and all procedures were conducted in accordance with the Pfizer Institutional Animal Care and Use Committee (IACUC).
Compound 1 demonstrated excellent selectivity against protein kinases. It was profiled against 75 protein kinases and the class III PI3K family member hVPS34 utilizing Dundee University and Invitrogen kinase screening services. No significant inhibitory activity was observed for any of the evaluated kinases at concentrations up to 10 μM (>500-fold selectivity).
From in vivo rat PK studies, 1 demonstrated low clearance and good oral bioavailability (Table 2). Compound 1 exhibited moderate plasma protein binding in rat, with unbound fraction (Fu) of 20.2%. Consequently, the unbound clearance (CLu) for 1 is low.
In conclusion, compound 1 is an efficient, low MW and clogP, structurally novel, highly selective and potent dual inhibitor of PI3K and mTOR. These properties have translated to demonstrated robust in vivo performance. Compound 1 has a desirable rat PK profile and in vivo activity in the mouse xenograft models implanted with human, mutated PI3Kα kinase domain SKOV3 tumors.34 Based on the overall profile, 1 was selected as a clinical candidate and has been advanced into a phase 1, open label, dose escalation study in subjects with solid tumors.35
Accession codes: The coordinates for compound 2 and PF-04691502 with PI3Kγ have been deposited, and the PDB ID codes are: 3ML8 for compound 2, 3ML9 for PF-04691502.
Acknowledgements
John Almaden, Tom Carlson, Hieu Lam, Jeff Wang, Peter Wells, Aihua Zou are acknowledged for biochemical screenings. Jeffrey Chen, Jon Engebretsen, John Li, and Xiao-Hong Yu are acknowledged for cellular screening. Hai-Zhi Bu, Christine Del-Carmen, Robert Hunter, Leslie Nguyen, and Wei-Zhu Zhong are acknowledged for their contributions from PDM. Ketan Gajiwala is acknowledged for preparing Fig. 3 and 6, and depositing the X-ray crystal structures.References
- D. A. Fruman, R. E. Myers and L. C. Cantley, Annu. Rev. Biochem., 1998, 67, 481 CrossRef CAS.
- J. A. Engelman, J. Luo and L. C. Cantley, Nat. Rev. Genet., 2006, 7, 606 CrossRef CAS.
- R. Katso, K. Okkenhaug, K. Ahmadi, S. White, J. Timms and M. D. Waterfield, Annu. Rev. Cell Dev. Biol., 2001, 17, 615 CrossRef CAS.
- P. K. Vogt, S. Kang, M. A. Elsliger and M. Gymnopoulos, Trends Biochem. Sci., 2007, 32, 342 CrossRef CAS.
- A. Bardelli and V. E. Velculescu, Curr. Opin. Genet. Dev., 2005, 15, 5 CrossRef CAS.
- Y. Samuels, Z. Wang, A. Bardelli, N. Silliman, J. Ptak, S. Szabo, H. Yan, A. Gazdar, S. M. Powell, G. J. Riggins, J. K. V. Willson, S. Markowitz, K. W. Kinzler, B. Vogelstein and V. E. Velculescu, Science, 2004, 304, 554 CrossRef CAS.
- J. Luo, B. D. Manning and L. C. Cantley, Cancer Cell, 2003, 4, 257 CrossRef CAS.
- B. Karakas, K. E. Bachman and B. H. Park, Br. J. Cancer, 2006, 94, 455 CrossRef CAS.
- S. Kang, A. G. Bader and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 802 CrossRef CAS.
- A. G. Bader, S. Kang and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1475 CrossRef CAS.
- M. Gymnopoulos, M. A. Elsliger and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5569 CrossRef CAS.
- J. J. Zhao, Z. Liu, L. Wang, E. Shin, M. F. Loda and T. M. Roberts, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18443 CrossRef CAS.
- Y. E. Whang, X. Wu, H. Suzuki, R. E. Reiter, C. Tran, R. L. Vessella, J. W. Said, W. B. Isaacs and C. L. Sawyers, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 5246 CrossRef CAS.
- D. L. Dai, M. Martinka and G. Li, J. Clin. Cancer Res., 2005, 23, 1473 Search PubMed.
- Z. Jiang and N. Pore, Cancer Res., 2007, 67, 4467 CrossRef CAS.
- I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489 CrossRef CAS.
- A. S. Strimpakos, E. M. Karapanagiotou, M. Wasif Saif and K. N. Syrigos, Cancer Treat. Rev., 2009, 35, 148 CrossRef CAS.
- B. I. Rini, Clin. Cancer Res., 2008, 14, 1286 CrossRef CAS.
- D. D. Sarbassov, D. A. Guertin, S. M. Ali and D. M. Sabatini, Science, 2005, 307, 1098 CrossRef CAS.
- T. E. Harris and J. C. Lawrence Jr., Science's STKE, 2003, 2003, re15 Search PubMed.
- S.-M. Maira, F. Stauffer, J. Brueggen, P. Furet, C. Schnell, C. Fritsch, S. Brachmann, P. Chene, A. De Pover, K. Schoemaker, D. Fabbro, D. Gabriel, M. Simonen, L. Murphy, P. Finan, W. Sellers and C. Garcia-Echeverria, Mol. Cancer Ther., 2008, 7, 1851 CrossRef CAS.
- S. D. Knight, D. A. Nicholas, J. L. Burgess, A. M. Chaudhari, M. G. Darcy, C. A. Donatelli, J. I. Luengo, K. A. Newlander, C. A. Parrish, L. H. Ridgers, M. A. Sarpong, S. J. Schmidt, G. S. Van Aller, J. D. Carson, M. A. Diamond, P. A. Elkins, C. M. Gardiner, E. Garver, S. A. Gilbert, R. R. Gontarek, J. R. Jackson, K. L. Kershner, L. Luo, K. Raha, C. S. Sherk, C.-M. Sung, D. Sutton, P. J. Tummino, R. J. Wegrzyn, K. R. Auger and D. Dhanak, ACS Med. Chem. Lett., 2010, 1, 39 Search PubMed.
- K. D. Courtney, R. B. Corcoran and J. A. Emgelman, J. Clin. Oncol., 2010, 28, 1075 CrossRef CAS.
- C. H. Reynolds, B. A. Tounge and S. D. Bembenek, J. Med. Chem., 2008, 51, 2432 CrossRef CAS.
- E. H. Walker, O. Perisic, C. Ried, L. Stephens and R. L. Williams, Nature, 1999, 402, 313 CrossRef CAS.
- E. H. Walker, M. E. Pacold, O. Perisic, L. Stephens, P. T. Hawkins, M. P. Wymann and R. L. Williams, Mol. Cell, 2000, 6, 909 CrossRef CAS.
- Z. A. Knight, B. Gonzalez, M. E. Feldman, E. R. Zunder, D. D. Goldenberg, O. Williams, R. Loewith, D. Stokoe, A. Balla, B. Toth, T. Balla, W. A. Weiss, R. L. Williams and K. M. A. Shokat, Cell, 2006, 125, 733 CrossRef CAS.
- C.-H. Huang, D. Mandelker, O. Schmidt-Kittler, Y. Samuels, V. E. Velculescu, K. W. Kinzler, B. Vogelstein, S. B. Gabelli and L. M. Amzel, Science, 2007, 318, 1744 CrossRef CAS.
- M. P. Edwards, Integration of Structure-based Drug Design and Physical Properties-based Optimization to Produce Cancer Clinical Candidates, 2010 AACR invited oral presentation, Washington DC, USA Search PubMed.
- H. Cheng, D. Bhumralkar, K. R. Dress, J. E. Hoffman, C. M. Johnson, R. S. Kania, P. T. Q. Le, M. D. Nambu, M. A. Pairish, M. B. Plewe, K. T. Tran, Pyrido (2,3-D)Pyrimidinone Compounds and Their Use as PI3 Inhibitors, WO 2008/032162 Search PubMed.
- T. W. Johnson, K. R. Dress and M. Edwards, Bioorg. Med. Chem. Lett., 2009, 19, 5560 CrossRef CAS.
- N. Jain and S. H. Yalkowsky, J. Pharm. Sci., 2001, 90, 234 CrossRef CAS.
- P. D. Leeson and B. Springthorpe, Nat. Rev. Drug Discovery, 2007, 6, 881 CrossRef CAS.
- S. Bagrodia, J. Yuan, H. Cheng, S. Sun, J. Chen, K. Luu, E. Zhang, N. V. Lee, J. Engebretsen, K. Rafidi, J. Wang, T. Carlson, J. Almaden, M. Hemkens, A. McHarg, M. A. Marx, J. Kan, A. Pavlicek, L. Ueno, M. Sun, P. Vogt, C. Luo, PF-04691502, A Potent and Selective PI3K/mTOR Dual Inhibitor with Antitumor Activity, 2010 AACR poster abstract # 4479, Washington DC, USA Search PubMed.
- H. Cheng, S. Bagrodia, S. Bailey, D. Bhumalkar, K. Dress, M. Edwards, M. R. Gehring, L. Guo, J. Hoffman, J. Hu, X. Huang, C. Johnson, T. O. Johnson, R. Kania, D. R. Knighton, P. Le, H. Li, S. Li, K. Liu, Z. Liu, M. A. Marx, M. Nambu, S. Ninkovic, D. Nowlin, M. Pairish, A. Pannifer, M. Plewe, C. Rodgers, G. Smith, S. Sun, K. Tran, H. Wang, J. Zbieg, P. Zhu, The Discovery of the Potent and Selective PI3K/mTOR Dual Inhibitor, PF-04691502, through Structure Based Drug Design, 2010 AACR poster abstract # 5779, Washington DC, USA Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/c0md00072h |
| This journal is © The Royal Society of Chemistry 2010 |