Design, synthesis, and structure–activity relationships of indole-3-carboxamides as novel water soluble cannabinoid CB1 receptor agonists†
Julia M.
Adam
*a,
Jim
Cairns
a,
Wilson
Caulfield
a,
Phillip
Cowley
a,
Iain
Cumming
a,
Morag
Easson
a,
Darren
Edwards
a,
Morag
Ferguson
a,
Richard
Goodwin
a,
Fiona
Jeremiah
a,
Takao
Kiyoi
a,
Ashvin
Mistry
a,
Elizabeth
Moir
a,
Richard
Morphy
a,
Jason
Tierney
a,
Mark
York
a,
James
Baker
b,
Jean E.
Cottney
b,
Andrea K.
Houghton
b,
Paul J.
Westwood
b and
Glenn
Walker
b
aDepartment of Chemistry, Schering-Plough Research Institute, Newhouse, Lanarkshire ML1 5SH, UK. E-mail: julia.adam@spcorp.com; Fax: +44 1698 736187; Tel: +44 1698 736000
bDepartment of Pharmacology, Schering-Plough Research Institute, Newhouse, Lanarkshire ML1 5SH, UK
First published on 28th April 2010
Abstract
A novel CB1 receptor agonist lead series was identified using a high-throughput screening approach. The initial screen afforded a single confirmed hit with poor water solubility. Structural variations were explored with the aim of introducing water solubility and improving potency. This led to the discovery of Org 28611, a potent, water soluble CB1 receptor agonist, which was selected for clinical evaluation as a potential intravenous analgesic agent.
Introduction
The CB1 cannabinoid receptor is a G-protein coupled receptor that is widely expressed throughout the central and peripheral nervous system. Activation of CB1 receptors located on presynaptic nerve terminals reduces calcium flux through voltage-dependent calcium channels, resulting in decreased neurotransmitter release.1 Compounds that activate CB1 receptors have been suggested as possible treatments for a wide range of medical disorders including pain, spasticity, inflammation, glaucoma, nausea and emesis, neurodegenerative disorders, anxiety and hypertension.2Limited clinical data indicate that CB1 receptor agonists may have sedative as well as analgesic properties in man.3 A CB1 receptor agonist that could be administered by the intravenous route might be a useful addition to the available therapies for the treatment of peri-operative pain, which are currently limited to opioids and non-steroidal anti-inflammatory drugs (NSAIDs).
The major classes of CB1 receptor agonists known in the literature are exemplified in Fig. 1.4 In general, these molecules are non-selective for CB1 vs. CB2 receptors. WIN 55,212-2, the prototype of indole based cannabinoid receptor agonists, was discovered in the early 1990s by a group at Sterling-Winthrop as a potent CB1 and CB2 receptor agonist with efficacy equivalent to morphine in animal models of acute pain, persistent inflammatory pain and neuropathic pain.5 More recently, indole based compounds with modest to excellent selectivity for CB2 over CB1 receptors have been disclosed.6
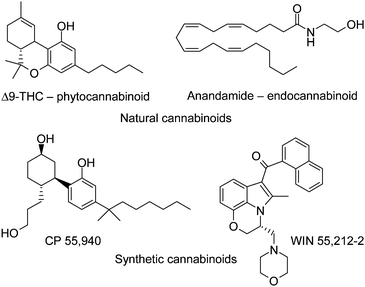 | ||
| Fig. 1 Naturally occurring and synthetic cannabinoid receptor agonists. | ||
In general, the cannabinoid receptor agonists known in the literature are characterized by high lipophilicity, poor water solubility and suboptimal pharmacokinetic profiles, which have hampered their evaluation as therapeutic agents.
We now disclose a new series of intrinsically water soluble, drug-like indole-3-carboxamide CB1 receptor agonists that has been designed and synthesized in our laboratory.
Results and discussion
High throughput screening using CHO cells doubly transfected with human CB1 and a luciferase reporter gene7 resulted in identification of the isatin 1 (Fig. 2). At the time, compound 1 represented a novel scaffold in the cannabinoid receptor agonist field, although recent publications by other research groups have independently disclosed isatin based compounds as cannabinoid receptor ligands.8,9 Compound 1 displayed moderate potency (pEC50 6.2) and good efficacy at the CB1 receptor (119% stimulation at 10−5 M compared to the reference compound CP 55,940) and had the advantages of a low molecular weight and a simple synthetic route. However, in common with the majority of known CB1 receptor ligands, it also exhibited high lipophilicity and extremely poor aqueous solubility.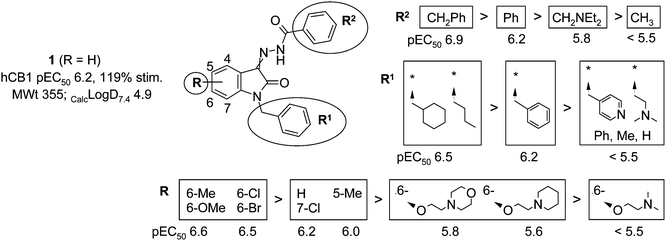 | ||
| Fig. 2 Preliminary SAR around compound 1. | ||
A hit optimisation program was initiated with the aim of improving aqueous solubility and potency, to identify drug candidates suitable for intravenous administration in the peri-operative setting. The potency and solubility criteria for a peri-operative analgesic development candidate were established as a CB1 agonist pEC50 greater than or equal to 7.5, with >50% efficacy compared to CP55,940, together with aqueous solubility greater than 1000 mg L−1 in the pH range 5 to 8. Selectivity over CB2 receptors was not required, as the majority of previously studied CB1 receptor agonists are non-selective vs. CB2 receptors and literature reports suggested that CB2 receptor agonist activity may also produce beneficial analgesic effects.10
Compound 1 and its analogues were synthesised as shown in Scheme 1.8
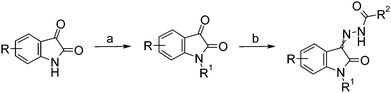 | ||
| Scheme 1 Preparation of isatin analogs. Reagents and conditions: (a) NaH, dimethyl formamide, r.t., 30 min, followed by R1Br, 80 °C, 1 h; (b) H2N–HN–C(O)R2, EtOH, 60 °C, 4 h. | ||
Fig. 2 summarises the results of modifying the substitution around the isatin core. The presence of an R1 substituent was found to be essential for activity at the CB1 receptor. Replacement of the R1 benzyl group with simple alkyl rings or chains was tolerated, with cyclohexylmethyl or nbutyl giving improved potency compared to the benzyl group. A variety of amine containing R1 groups was introduced, but each resulted in complete loss of activity.
The phenyl hydrazide (R2) portion of the molecule was found to be slightly more tolerant to changes. Although replacement of the phenyl ring with a methyl group resulted in loss of activity, replacement with a benzyl group afforded a four-fold improvement in potency. Furthermore, although it was not favorable to introduce an amine substituent in this side chain, in this case it was possible to do so without complete loss of activity.
Introduction of a methyl, methoxy, bromo or chloro substituent in the 6-position of the isatin core was found to increase potency by around 2-fold. 7-Chloro or 5-methyl substituents were tolerated but did not improve potency. These preliminary results encouraged further exploration of the 6-position. Attempts to attach water-solubilizing groups in this position via a phenol ether linker met with some success. Of the polar groups that were introduced, the 2-(morpholin-4-yl)ethoxy substituent was the best tolerated with a pEC50 of 5.8.
The SAR obtained by changing the substitution pattern around the isatin core was found to be approximately additive. The combination of the cyclohexylmethyl R1, benzyl R2 and 6-[2-(morpholin-4-yl)ethoxy] R substituents afforded compound 2 (Fig. 3), which was 10-fold more potent than the original hit. Unfortunately, however, the introduction of the ionizable morpholino group was not sufficient to bestow good aqueous solubility, given the high lipophilicity elsewhere in the molecule. A methanesulfonic acid salt of compound 2 was prepared, but the aqueous solubility of the salt was below 500 mg L−1.
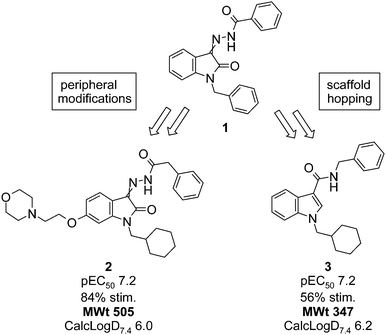 | ||
| Fig. 3 Comparison of peripheral and scaffold hopping approaches for hit optimization of compound 1. | ||
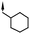
In parallel with the peripheral modifications, replacement of the isatin scaffold with alternative cores was investigated and this scaffold hopping approach soon gave more encouraging results. The simple N-cyclohexylmethyl indole-3-carboxamide 3 was found to have similar potency to compound 2, with a much lower molecular weight (Fig. 3). In addition, the removal of the potentially bio-reactive acyl hydrazine moiety found in 1 and 2 could have advantages in long-term safety. As such, compound 3 was considered a much improved starting point for further optimization and the medicinal chemistry focus shifted to the indole carboxamide scaffold.
Two general synthetic routes to the indole-3-carboxamides were developed, as shown in Scheme 2. Method A was generally employed for single compound synthesis, whereas Method B, in which amide formation was carried out using a novel one pot, three step reaction sequence, was employed for rapid generation of libraries.‡
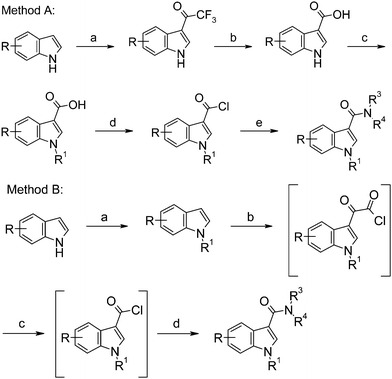 | ||
| Scheme 2 Preparation of indole analogs. Reagents and conditions: Method A: (a) Trifluoroacetic anhydride, DMF, 0 °C to r.t., 1 h; (b) NaOH (4 M, aq), reflux, 1 h; (c) NaH, DMF, r.t., 1 h, followed by R1Br, 60 °C, 1 h; (d) Oxalyl chloride, THF, 18 h, r.t.; (e) NHR3R4, Et3N, CH2Cl2, r.t., 18 h. Method B: (a) NaH, DMF, r.t., 1 h min, followed by R1Br, 60 °C, 1 h; (b) Oxalyl chloride, 1,1,2,2-tetrachloroethane, 0 °C to r.t., 1 h; (c) 120 °C, 1.5 h; (d) NHR3R4, Et3N, r.t., 15 h. | ||
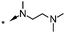
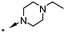
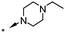
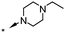
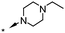
A focused indole-3-carboxamide library was synthesized to investigate a range of diamines as potential water solubilizing replacements of the benzylamine group. This speculative library led to the surprising identification of N-methyl and N-ethyl piperazines as benzylamine replacements (Table 1, compounds 6 and 7). These piperazine amides were less potent than compound 3, but had the advantage that stable salts with excellent aqueous solubility (>1000 mg L−1 in citrate buffer, pH 5) could be easily prepared. To our knowledge, these indole-3-piperazine carboxamides represent the first known CB1 receptor agonists with high intrinsic water solubility.
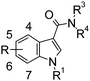
| Compound | R | R1 | NR3R4 | pEC50/Ma | % stim @ 10−5Ma,b |
|---|---|---|---|---|---|
| a Values are means of three experiments performed in triplicate. b Mean % stimulation at 10−5 M compared to CP 55,940. c Data for trifluoroacetic acid salt. d Data for hydrochloric acid salt. e Data for maleic acid salt. | |||||
| CP 55,940 | 7.7 | 100 | |||
| WIN 55,212-2 | 7.3 | 61 | |||
| 3 | H |
|

|
7.2 | 56 |
| 4 | H |

|
|
<5.5 | — |
| 5 | H |

|
|
<5.5 | — |
| 6 | H |

|
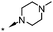
|
6.3 | 39 |
| 7 | H |
|
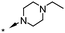
|
6.5c | 45c |
| 8 | 4-F |
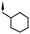
|
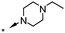
|
6.6d | 36d |
| 9 | 4-Me |

|

|
<5.5 | — |
| 10 | 5-F |
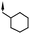
|

|
6.3d | 28d |
| 11 | 5-Cl |
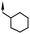
|
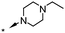
|
5.9d | 31d |
| 12 | 5-Me |
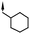
|
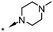
|
5.7 | 28 |
| 13 | 5-OMe |
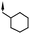
|
|
<5.5d | — |
| 14 | 6-F |
|
|
6.6d | 30d |
| 15 | 6-Cl |
|
|
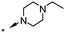
5.8d | 52d |
| 16 | 6-Br |
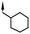
|
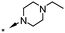
|
6.0d | 50d |
| 17 | 6-Me |
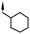
|
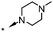
|
6.5 | 48 |
| 18 | 6-OMe |
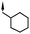
|

|
6.8e | 46e |
| 19 | 7-F |
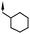
|
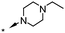
|
7.0d | 41d |
| 20 | 7-Cl |
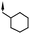
|
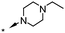
|
6.3d | 80d |
| 21 | 7-Br |
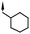
|
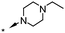
|
6.1d | 111d |
| 22 | 7-Me |
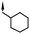
|

|
6.7 | 83 |
| 23 | 7-OMe |
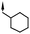
|
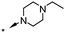
|
6.8e | 71e |
| 24 | 7-OMe |
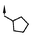
|
|
6.5d | 56d |
| 25 | 7-OMe |
|
|
6.1d | 61d |
| 26 | 7-OMe |
|

|
<5.5d | — |
| 27 | 7-OMe |

|
|
<5.5 | — |
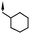
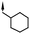
Substitution of the indole ring was revisited in the indole-3-carboxamide series in an attempt to increase potency and efficacy while retaining water solubility (compounds 8–23). All of these aims could be advanced by substitution in the 7-position. The effect of the 7-substituent on CB1 receptor agonist efficacy appeared to be driven by the size of the 7-substituent, rather than by electron withdrawing or electron donating effects. It is possible that this group helps to direct the cyclohexylmethyl group into a favorable agonist binding orientation.
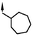
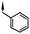
The 7-methoxy group gave the best balance of potency, efficacy and physico-chemical properties and so with this group in place, variations of the cyclohexylmethyl moiety at R1 were explored (compounds 24–27). As before, however, this region of the molecule was found to be intolerant to changes. Reducing the size to cyclopentylmethyl or increasing to cycloheptylmethyl gave reduced potency and efficacy. Also, the benzyl group of the original hit and the ethylmorpholino group of the prototypical aminoalkylindole CB1 receptor agonist WIN 55,212-25 were not tolerated.
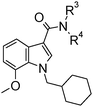
Compound 23, 1-{[1-(cyclohexyl)methyl-7-methoxy-1H-indol-3-yl]carbonyl}-4-ethylpiperazine maleic acid salt, was selected as a lead compound for further optimization. This compound had moderate potency (pEC50 6.8) and efficacy (71% stimulation at 10−5 M compared to the reference compound CP 55,940), combined with good aqueous solubility [>3000 mg L−1 at pH 5 and >200 mg L−1 at pH 6.7 (Table 2)].
| Compound | NR3R4 | pEC50/Ma | % stim at 10−5 Ma,b | MSF solubility/mg L−1 | |
|---|---|---|---|---|---|
| PBS pH 7.4 (final pH) | Citrate pH 5 (final pH) | ||||
| a Values are means of three experiments performed in triplicate. b Mean % stimulation at 10−5M compared to CP 55,940. c Data for hydrochloric acid salt. d Data for maleic acid salt. | |||||
| 23 |
|
6.8d | 71d | 216 (6.7) | 3398 (5.0) |
| 28 |

|
6.2c | 63c | 74 (7.1) | >4000 (5.0) |
| 29 |
|
7.4c | 75c | 37 (7.1) | >4000 (5.0) |
| 30 |
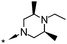
|
8.0c | 60c | 9 (7.0) | >4000 (5.0) |
| 31 |

|
6.9c | 88c | 181 (7.1) | 3831 (5.0) |
| 32 |
|
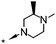
6.9c | 70c | 88 (7.1) | 2859 (5.0) |
| 33 Org 28611 |

|
7.6c | 77c | 129 (6.9) | 2809 (4.9) |
| 34 Org 28312 |
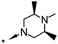
|
7.6c | 73c | 12 (7.0) | >4000 (5.0) |
| 35 |
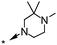
|
6.8c | 78c | 129 (7.0) | 2158 (5.0) |
A methyl survey of compound 23 revealed substitution in the 3- and 5-positions of the piperazine ring to be the key to achieving the desired improvements in potency. Introduction of a single methyl substituent in the 3-position afforded a pair of enantiomers, 28 and 29, of which the (S)-enantiomer 29 was the most potent. cis-3,5-Di-substitution afforded achiral compound 30, which showed a greater than 10-fold improvement in potency over compound 23 but reduced aqueous solubility in the neutral pH range (pEC50 8.0; solubility 9 mg L−1 at pH 7). The achiral compound arising from 3,3-dimethyl substitution showed better aqueous solubility at neutral pH, but was less potent (compound 31, pEC50 6.9; solubility 181 mg L−1 at pH 7.1). The 4-ethyl group could be shortened to a methyl group with minimal effects on potency, efficacy or aqueous solubility (compounds 32 to 35).
Compounds 33 (Org 28611, pEC50 7.6; solubility 2809 mg L−1 at pH 4.9 and 129 mg L−1 at pH 6.9) and 34 (Org 28312, pEC50 7.6; solubility >4000 mg L−1 at pH 5.0 and 12 mg L−1 at pH 7.0) were selected for further profiling based on their overall solubility, potency and efficacy profiles (Fig. 4).
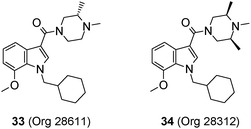 | ||
| Fig. 4 Structures of compounds 33 (Org 28611) and 34 (Org 28312). | ||
Org 28611 and Org 28312 exhibited high affinity for both CB1 and CB2 cannabinoid receptors, as determined by radioligand competition binding assays using [3H]CP 55,940 binding to either hCB1 or hCB2 receptors expressed in insect Sf9 membranes (Table 3). Both compounds exhibited good general selectivity, with greater than 100-fold selectivity against a panel of 42 unrelated molecular targets, including the hERG channel (data not shown).
| CB1 pKi | CB2 pKi | |
|---|---|---|
| CP 55,940 | 9.5 | 9.5 |
| WIN 55,212-2 | 7.9 | 8.6 |
| Org 28611 | 8.9 | 8.8 |
| Org 28312 | 8.9 | 9.1 |
The in vitro and in vivo DMPK profiles of Org 28611 and Org 28312 are summarized in Table 4. Org 28611 and Org 28312 were rapidly metabolized by mouse and human hepatic microsomes. Mouse brain and plasma levels were determined following intravenous administration of a 3 μmol kg−1 dose (terminal sampling using CO2). Both compounds were rapidly cleared in vivo, as predicted from the rapid microsomal metabolism. Both Org 28611 and Org 28312 showed higher total levels in brain compared to plasma (B![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :P ratio of 4.0 and 2.7 for Org 28611 and Org 28312, respectively). Brain concentration vs. time profiles for both compounds mirrored the profiles seen in plasma indicating rapid equilibration across the blood–brain barrier.
:P ratio of 4.0 and 2.7 for Org 28611 and Org 28312, respectively). Brain concentration vs. time profiles for both compounds mirrored the profiles seen in plasma indicating rapid equilibration across the blood–brain barrier.
| Org 28611 | Org 28312 | |
|---|---|---|
| Microsomal stability | ||
| Human CLint/μl min−1 mg−1 | >270 | >270 |
| Mouse CLint/μl min−1 mg−1 | >270 | >270 |
| PK (ICR Mouse, 3 μmol kg−1, i.v.) | ||
| Vehicle | Water | Water |
| Plasma Cmax (ng ml−1; t = 0.05 h) | 620 | 593 |
| AUCplasma, i.v. (h ng ml−1) | 248 | 253 |
| Clearance/ml min−1 kg−1 | 77.3 | 77.4 |
| T1/2 elimination/h | 0.41 | 0.32 |
| Vss/L kg−1 | 2.1 | 2.0 |
| Brain Penetration (ICR mouse) same studies as above | ||
| Brain Cmax/ng g−1 | 2457 | 1619 |
| Brain tmax/h | 0.17 | 0.05 |
| Brain AUC0–1 h/h ng g−1 | 804 | 616 |
| Brain : Plasma Cmax ratio | 4.0 | 2.7 |
Org 28611 and Org 28312 demonstrated potent antinociceptive activity in the mouse tail flick test, a preclinical threshold model of nociception in which a mouse reacts to a noxious thermal stimulus by flicking its tail away from a focused beam of radiant heat. Intravenous administration of either Org 28611 or Org 28312 increased tail flick latency in a dose-dependent manner (Table 5). Both compounds showed a rapid onset of action. Org 28611 exhibited a longer duration of action in this model compared to Org 28312; the time taken for the tail flick latency to return to within the mean plus two standard deviations of the baseline value following a twice ED50 dose being 90 min for Org 28611 and 50 min for Org 28312.
| Test compound | Dose/μmol kg−1 | %MPE |
|---|---|---|
| a Denotes that the effect was significantly greater than in vehicle treated mice (P < 0.01, Dunn's post-test). | ||
| Vehicle (10% Tween 80) | 10 ml kg−1 | 4.3 ± 1.3 |
| Org 28611 | 0.2 | 18.3 ± 1.8 |
| Org 28611 | 0.4 | 50.1 ± 5.3a |
| Org 28611 | 1.0 | 100 ± 0a |
| Vehicle (10% Tween 80) | 10 ml kg−1 | 2.9 ± 2.8 |
| Org 28312 | 0.1 | 14.5 ± 3.7 |
| Org 28312 | 0.3 | 72.3 ± 9.7a |
| Org 28312 | 1.0 | 100 ± 0a |
Org 28611 and Org 28312 also demonstrated potent antinociceptive activity in the mouse formalin paw test (FPT), a model that assesses behavioral responses to continuous, noxious stimulation generated by injection of a dilute solution of formalin into one hindpaw. The injection of formalin induces an inflammatory response in the paw that elicits licking behavior. There are two distinct phases of nociceptive behavior, the first period begins immediately after formalin injection and lasts 4–5 min. This early phase is followed by a quiescent phase of 5–10 min duration, after which a second phase of nociceptive behavior occurs; this phase continues for a further 20–30 min. Both Org 28611 and Org 28312, administered intravenously (0.01–0.3 μmol kg−1), dose-dependently reduced the amount of time spent licking the injected paw during both phases of licking (Table 6).
| Test compound | Dose/μmol kg−1 | % inhibition of counts compared to vehiclea | |
|---|---|---|---|
| Phase 1 | Phase 2 | ||
| a * (P < 0.05) and ** (P < 0.01) denotes that the effect was significantly greater than in vehicle treated mice (Dunn’s post-test). | |||
| 10% Tween 80 in saline | 10 mL kg−1 | — | — |
| Org 28611 | 0.01 | 25.7 ± 8.2 | 49.2 ± 22.0 |
| Org 28611 | 0.03 | 66.0 ± 6.0 | 79.7 ± 10.4 |
| Org 28611 | 0.1 | 99.0 ± 0.6** | 93.4 ± 5.0* |
| Org 28611 | 0.3 | 100 ± 0** | 100 ± 0** |
| 10% Tween 80 in saline | 10 mL kg−1 | — | — |
| Org 28312 | 0.01 | 16.4 ± 7.9 | 7.8 ± 12.2 |
| Org 28312 | 0.03 | 49.5 ± 7.3 | 86.9 ± 4.0 |
| Org 28312 | 0.1 | 84.0 ± 3.3* | 93.3 ± 2.5* |
| Org 28312 | 0.3 | 97.8 ± 1.8** | 99.8 ± 0.2** |
Antinociception produced by either compound in this test was antagonized by the selective CB1 receptor antagonist SR141716A (rimonabant), indicating that their activity in this model was mediated by activation of CB1 receptors (data not shown).
Following further preclinical evaluation, Org 28611 was selected as a development candidate for clinical evaluation as a potential intravenous peri-operative analgesic agent.11
Conclusion
A new lead-like series of potent, water soluble CB1 receptor agonists was identified through high-throughput screening followed by parallel optimization of potency and physico-chemical properties. Attempts to improve the poor physico-chemical properties of the HTS hit 1 by peripheral modifications were unsuccessful, resulting in high molecular weight, lipophilic molecules. The successful approach was to first apply scaffold hopping together with exploratory library synthesis to fix the physico-chemical properties of the HTS hit, followed by small, systematic peripheral modifications to improve the potency. This resulted in identification of compound 33 (Org 28611) which was taken forward for clinical evaluation.Org 28611 and its analogues are suitable for administration in aqueous vehicles by the intravenous route and as such provide useful tools for further preclinical and clinical exploration of the potential of CB1 receptor agonists as therapeutic agents.
Notes and references
- A. C. Howlett, F. Barth, T. I. Bonner, G. Cabral, P. Casellas, W. A. Devane, C. C. Felder, M. Herkenham, K. Mackie, B. R. Martin, R. Mechoulam and R. G. Pertwee, International Union of Pharmacology. XXVII. Classification of Cannabinoid Receptors, Pharmacol. Rev., 2002, 54, 161–202 CrossRef CAS.
- Additional reviews on cannabinoid receptor agonists: (a) Handbook of Experimental Pharmacology, ed. R. G. Pertwee, Springer, 2005, Vol. 168 Search PubMed; (b) L. Iversen and V. Chapman, Curr. Opin. Pharmacol., 2002, 2, 50–55 CrossRef CAS.
- A. K. Jain, J. R. Ryan, F. G. McMahon and G. Smith, J. Clin. Pharmacol., 1981, 21, 320S–326S CAS.
- (a) J. Adam, P. M. Cowley, T. Kiyoi, A. J. Morrison and C. J. W. Mort, in Progress in Medicinal Chemistry, ed. F. D. King and G. Lawton, Elsevier, 2006, Vol. 44, pp 207–329 Search PubMed; (b) G. A. Thakur, R. Tichkule, S. Bajaj and A. Makriyannis, Expert Opin. Ther. Pat., 2009, 19, 1647–1673 Search PubMed.
- (a) T. E. D'Ambra, K. G. Estep, M. R. Bell, M. A. Eissenstat, K. A. Josef, S. J. Ward, D. A. Haycock, E. R. Baizman, F. M. Casiano, N. C. Beglin, S. M. Chippari, J. D. Grego, R. K. Kullnig and G. T. Daley, J. Med. Chem., 1992, 35, 124–135 CrossRef CAS; (b) M. A. Eissenstat, M. R. Bell, T. E. D'Ambra, E. J. Alexander, S. J. Daum, J. H. Ackerman, M. D. Gruett, V. Kumar, K. G. Estep, E. M. Olefirowicz, J. R. Wetzel, M. D. Alexander, J. D. Weaver, D. A. Haycock, D. A. Luttinger, F. M. Casiano, S. M. Chippari, J. E. Kuster, J. I. Stevenson and S. J. Ward, J. Med. Chem., 1995, 38, 3094–3105 CrossRef CAS.
- (a) M. Gallant, C. Dufresne, Y. Gareau, D. Guay, Y. Leblanc, P. Prasit, C. Rochette, N. Sawyer, D. M. Slipetz, N. Tremblay, K. M. Metters and M. Labelle, Bioorg. Med. Chem. Lett., 1996, 6, 2263–2268 CrossRef CAS; (b) J. W. Huffman, G. Zengin, M.-J. Wu, J. Lu, G. Hynd, K. Bushell, A. L. S. Thompson, S. Bushell, C. Tartal, D. P. Hurst, P. H. Reggio, D. E. Selley, M. P. Cassidy, J. L. Wiley and B. R. Martin, Bioorg. Med. Chem., 2005, 13, 89–112 CrossRef CAS; (c) J. Hynes, Jr., K. Leftheris, H. Wu, C. Pandit, P. Chen, D. J. Norris, B.-C. Chen, R. Zhao, P. A. Kiener, X. Chen, L. A. Turk, V. Patil-Koota, K. M. Gillooly, D. J. Shuster and K. W. McIntyre, Bioorg. Med. Chem. Lett., 2002, 12, 2399–2402 CrossRef; (d) G. M. P. Giblin, A. Billinton, M. Briggs, A. J. Brown, I. P. Chessell, N. M. Clayton, A. J. Eatherton, P. Goldsmith, C. Haslam, M. R. Johnson, W. L. Mitchell, A. Naylor, A. Perboni, B. P. Slingsby and A. W. Wilson, J. Med. Chem., 2009, 52, 5785–5788 CrossRef CAS.
- M. R. Price, G. L. Baillie, A. Thomas, L. A. Stevenson, M. Easson, R. Goodwin, A. McLean, L. McIntosh, G. Goodwin, G. Walker, P. Westwood, J. Marrs, F. Thomson, P. Cowley, A. Christopoulos, R. G. Pertwee and R. A. Ross, Mol. Pharmacol., 2005, 68, 1484–1495 CrossRef CAS.
- (a) P. Diaz, J. Xu, F. Astruc-Diaz, H.-M. Pan, D. L. Brown and M. Naguib, J. Med. Chem., 2008, 51, 4932–4947 CrossRef CAS; (b) P. Diaz, S. S. Phatak, J. Xu, F. Astruc-Diaz, C. N. Cavasotto and M. Naguib, J. Med. Chem., 2009, 52, 433–444 CrossRef CAS.
- R. Bhogal; J. Chugh; H. Meldrum (Unilever PLC) PCT Int. Appl. 2006, WO 2006097193 A1.
- P. Anand, G. Whiteside, C. J. Fowler and A. G. Hohmann, Brain Res. Rev., 2009, 60, 255–266 CrossRef CAS.
- L. Zuurman, P. C. C. M. Passier, M. L. de Kam, H. J. Kleijn, A. F. Cohen and J. M. A. van Gerven, J. Psychopharmacol., 2009, 23, 633–644 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data for compounds 1 to 35 and materials and methods for in vitro and in vivo assays. See DOI: 10.1039/c0md00022a |
| ‡ Example: synthesis of compound 10 according to Method B: 1-{[1-(Cyclohexyl)methyl-5-fluoro-1H-indol-3-yl]carbonyl}-4-ethylpiperazine, hydrochloride salt To a solution of 5-fluoroindole (1.0 g, 7.4 mmol) in dimethyl formamide (20 ml) was added sodium hydride (60% dispersion in mineral oil; 327 mg, 8.14 mmol). The mixture was stirred at room temperature for 10 min before the addition of bromomethylcyclohexane (1.3 ml, 9.3 mmol). The resulting mixture was stirred at room temperature for 15 h. A further addition of sodium hydride (170 mg, 4.23 mmol) then bromomethylcyclohexane (0.65 ml, 4.65 mmol) was made and the reaction stirred for a further 15 h. The reaction was quenched with 2-propanol (10 ml) and then concentrated. The resulting brown gum was partitioned between ethyl acetate (50 ml) and 5% sodium hydrogen carbonate solution (50 ml). The organic layer was washed with water (50 ml), dried over sodium sulfate and concentrated. The crude intermediate was then purified by flash chromatography using 95% dichloromethane, 5% methanol as eluent, to afford 1-(cyclohexylmethyl)-5-fluoroindole (1.26 g, 5.45 mmol). To a solution of 1-(cyclohexylmethyl)-5-fluoroindole (208 mg, 0.9 mmol) in 1,1,2,2-tetrachloroethane (15 ml) at 0 °C was added oxalyl chloride (0.122 ml, 0.945 mmol) with stirring under a stream of nitrogen. The mixture was allowed to warm to room temperature over 1 h, then heated to 120 °C for a further 1.5 h. The mixture was cooled to room temperature and triethylamine (0.138 ml, 0.99 mmol) was added. Stirring was continued for a further 10 min before the addition of N-ethylpiperazine (0.125 ml, 0.99 mmol). The mixture was stirred at room temperature for 15 h and then partitioned between 0.4 M sodium hydroxide solution (10 ml) and dichloromethane (10 ml). The organic layer was washed with water (10 ml), dried over Na2SO4 and concentrated. The resulting brown oil was purified by flash chromatography using 95% dichloromethane, 5% methanol as eluent to yield the title compound as the free base. Hydrochloride salt formation was achieved by the addition of hydrogen chloride 2 M solution in diethyl ether (3 ml) to a solution of the free base in diethyl ether (5 ml). The precipitate was filtered and dried. The solid was crystallised from diethyl ether and methanol to afford 10 (1 |
| This journal is © The Royal Society of Chemistry 2010 |