Chemical space as a source for new drugs
Jean-Louis
Reymond
*,
Ruud
van Deursen
,
Lorenz C.
Blum
and
Lars
Ruddigkeit
Department of Chemistry and Biochemistry, University of Berne, Freiestrasse 3, CH-3012, Berne, Switzerland. E-mail: jean-louis.reymond@ioc.unibe.ch; Fax: +41 31 631 80 57
First published on 28th April 2010
Abstract
The chemical space is the ensemble of all possible molecules, which is believed to contain at least 1060 organic molecules below 500 Da of possible interest for drug discovery. This review summarizes the development of the chemical space concept from enumerating acyclic hydrocarbons in the 1800's to the recent assembly of the chemical universe database GDB. Chemical space travel algorithms can be used to explore defined regions of chemical space by generating focused virtual libraries. Maps of the chemical space are produced from property spaces visualized by principal component analysis or by self-organizing maps, and from structural analyses such as the scaffold-tree or the MQN-system. Virtual screening of virtual chemical space followed by synthesis and testing of the best hits leads to the discovery of new drug molecules.
 Jean-Louis Reymond | Jean-Louis Reymond is Professor of Chemistry and Chemical Biology at the University of Berne, Switzerland. He studied chemistry and biochemistry at the ETH Zürich and obtained his PhD in 1989 at the University of Lausanne in the area of natural products synthesis. He then joined the Scripps Research Institute in La Jolla, CA, and became an assistant Professor there in 1992. In 1997 he joined the Department of Chemistry and Biochemistry at the University of Berne as an Associate and in 1998 Full Professor. His research interests focus on exploring molecular diversity using combinatorial chemistry, computer-aided drug design and cheminformatics. |
 Ruud van Deursen | Ruud van Deursen was born in 1979 in Helmond (Netherlands). He received his MSc in Chemical Engineering and Chemistry from the Eindhoven University of Technology in 2004. After master courses in Biochemistry and Molecular Biology at Ecole Normale Supérieure de Lyon (France), he wrote his master thesis on using alcohol dehydrogenases for biotransformations in the group of Professors Kurt Faber and Wolfgang Kroutil at Karl-Franzens-University in Graz (Austria). In December 2005 he joined the group of Prof. Jean-Louis Reymond at the University of Berne. Current research is focused on development of chemoinformatic tools for the understanding of chemical space and screening for bioactive molecules. |
 Lorenz Blum | Lorenz Christian Blum, was born in Berne (Switzerland) in 1983. He studied chemistry at the University of Berne and received his MSc degree in physical chemistry in 2006. Thereupon he started his PhD studies in chemoinformatics under the supervision of Prof. Jean-Louis Reymond. His current research interests are the assembly, analysis and applications of large virtual molecular databases. |
 Lars Ruddigkeit | Lars Ruddigkeit, born in Hamm (Germany) in 1982, studied chemical biology at the Technical University of Dortmund, and wrote his MSc on molecular probes for EGFR with Prof. Dr Herbert Waldmann. In June 2009, he started his PhD under the supervision of professor Jean-Louis Reymond at the University of Berne (Switzerland). His current research interests include the exploration of chemical space and in silico generated molecule databases. |
1. Introduction
Drug discovery was historically based on serendipity, more precisely on the chance discovery of activities in certain classes of compounds as they came under investigation. As the molecular understanding of disease and drug action has progressed, a very broad knowledge base has accumulated that can be exploited to perform rationally guided searches for active compounds in silico using virtual screening.1–6 Methods include the application of QSAR models,7 similarity measures to known reference drugs for molecular topology8 and three-dimensional structure (shape alignment),9–12 and modeling binding interactions to protein active sites (docking).13–17 Scoring functions are first developed by reproducing existing sets of bioactivity data, and then applied to rank compounds available from commercial or in-house collections. The highest scoring compounds are collected to form a focused library which is subjected to actual testing in vitro.One can also use scoring functions to rank compounds from virtual libraries prior to their synthesis, with the aim of exploring yet unknown chemical space and accessing new compound classes. This review focuses on this strategy and summarizes approaches to generate virtual libraries, to visualize the chemical space by producing maps, and to perform de novo drug discovery by virtual screening of virtual libraries followed by synthesis and testing of the best hits. Such exploration of yet unknown chemical space might help to solve the problem of the high attrition rates in drug development by giving more compounds to choose from at the hit prioritization level, which should increase the chances of success at later stages.18,19 Exploring a broader range of structures by virtual screening might also allow to address the problem of target promiscuity that is apparent in many drugs and allow the design of safer drugs.20,21
2. From molecule enumeration to chemical space
Synthetic chemistry is about making covalent bonds between atoms. The combinatorial possibilities of this simple concept have fascinated chemists from the early days of organic chemistry.22 Initial inquiries focused on calculating the total number of possible molecules of a given type. For instance Cayley and Schiff both independently considered in 1875 the problem of calculating the number of possible acyclic hydrocarbon isomers.23,24 The question was correctly solved in 1931 by Henze and Blair,25 predicting for example that there are 366![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 319 isomers with formula C20H42, a result which is easily confirmed using the GENG program26 for generating the corresponding graphs.
319 isomers with formula C20H42, a result which is easily confirmed using the GENG program26 for generating the corresponding graphs.
While these early considerations focused on counting only, the idea of actually enumerating and representing molecular structures in a computer was addressed in the 1960's by Lederberg and Djerassi, who invented DENDRAL, a program designed to help structure elucidation by mass spectrometry.27–29 DENDRAL produced all possible organic molecules with a given elemental formula. It was possible to exclude undesirable functional groups from a “badlist” and enforce functional groups specified in a “goodlist” to restrict the output. Provided enough such constraints, the list of structures would automatically be reduced to a handful of possibilities. This project gave rise to the topic of computer-assisted structure elucidation (CASE), which addresses automatic structure assignment from analytical data such as MS and NMR spectra and uses various structure generators30,31 as a key component.32–36
Enumeration by synthesis replaced virtual enumeration with the advent of combinatorial chemistry in the early 1990's. The key triggers were the inventions of (1) solid-supported split-and-mix synthesis,37–39 and (2) surface synthesis of two-dimensional arrays on glass or paper support.40,41 These methods allowed the simultaneous synthesis of thousands to millions of compounds as physically segregated and identifiable products. Solid-supported combinatorial chemistry was pursued first for iterative syntheses of oligomers such as peptides,37–39 peptoids42 and oligonucleotides,43 and later extended to include a broad arsenal of synthetic reactions leading to compounds of ever increasing complexity, in particular in the elegant diversity-oriented syntheses of Schreiber and coworkers.44,45 Latest advances in combinatorial chemistry include improvements in library decoding46 and screening methods,47 and the preparation of libraries of billions of compounds using DNA-encoded chemistry.48 The concept of combinatorial chemistry also led to automated parallel synthesis, which is used to systematically enlarge compound collections in pharmaceutical companies and at commercial providers.49 Databases of many of these compounds are publicly available in which the structures are written as SMILES,50–52 or related formats such as InChI.53 Examples include catalogs from commercial providers and public databases such as ZINC,54a BindingDB,54b Chembl54c or PubChem.55
The availability of collections of millions of compounds for drug discovery has suggested the concept of chemical space for describing the ensemble of all the molecules.56–58 The chemical space metaphor offers a more inspiring imagery than the older “needle in a haystack” paradigm in the context activity screening, and has been broadly embraced by the medicinal chemistry community to talk about drug discovery. All the known molecules form the “available chemical space”. There also exists a much larger space containing all the chemically possible molecules, which we call the chemical universe. Although chemical space is not uniquely defined, one generally considers that structurally related molecules form close groups, and that drug discovery can be guided geographically in chemical space. Areas of interest mark the biologically relevant chemical space, which includes natural products that have co-evolved with protein and nucleic acid binding sites in the course of the evolution of life, and all the drugs so far crafted by homo sapiens sapiens in his own fight for survival.
Is chemical space finite? Yes, if boundaries are defined. For small molecule drug discovery the natural limit is the molecular weight, which must be capped at 300–500 Da to ensure reasonable bioavailability.59 This chemical space of drug-like molecules has been estimated to be in excess of 1060 molecules.56,60 Our group has pushed the concept one step further and produced actual lists of all molecules that are possible up to a certain size following simple constraints of chemical stability and synthetic feasibility, forming the GDB database.61–63 The database is constructed from an exhaustive list of graphs produced by the program GENG,26 which are transformed into molecules by replacing graph nodes by atoms (C, N, O, F, Cl, S) and graph edges by single, double or triple bonds following simple valency rules, and retaining only chemically meaningful ring systems and functional groups (Fig. 1). It should be noted that exotic yet sometimes known molecules such as a molecule corresponding to a non-planar graph,64 or those containing strained fused ring systems such as cubane or prismane, are not considered in such enumerations.
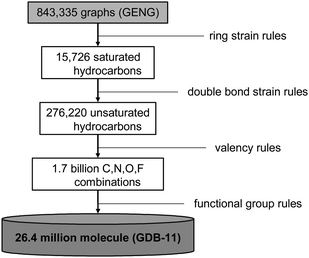 | ||
| Fig. 1 Process for generating the chemical universe database GDB-11. | ||
GDB has been published for the enumeration up to 11 atoms (GDB-11, with C, N, O, F, 26.4 million cpds with 152.9 ± 7.3 Da)63 and 13 atoms (GDB-13, with C, N, O, Cl and S, 980 million cpds 179.9 ± 8.3 Da),62 and completed in-house for 15 atoms (GDB-15, 28.8 billion cpds 206.8 ± 5.4 Da). GDB consists in large part of relatively rigid molecules, with bicyclic and tricyclic topologies being the most abundant. Most GDB-molecules are generated at intermediate ratios of polar atoms to carbon at clogP values between −2 and 2. These molecules fulfill Lipinski's criteria for oral bioavailability59 as well as lead-likeness65 and fragment-likeness66 criteria, mostly because these criteria primarily restrain molecular size. The GDB approach is limited to relatively small molecules due to the combinatorial explosion. An analysis of chemical space for larger molecules has been recently proposed by focusing on scaffold topologies.67 This description does not explicitly enumerate molecules but allows understanding of structural types in broad terms and was used to show that only a small subset of the possible scaffold topologies occur in known molecules.68
3. Chemical space travel
The complete enumeration of all possible molecules up to 500 Da, if summing up to at least 1060, is practically out of range. In most cases, however, one needs only to enumerate focused libraries featuring a small yet relevant subset of chemical space. Generating a focused library corresponds to traveling within a limited region of chemical space. A large part of the initial efforts to use cheminformatics for drug discovery consisted in the enumeration of virtual libraries to assist the design of synthetic combinatorial libraries, either towards predetermined targets or for optimal diversity.69 Several programs enumerate virtual libraries on the basis of known synthetic reactions and building blocks, and explore a subset of readily synthesizable structures for virtual screening.70,71 This approach is limited in its potential for structural innovation, but offers a very practical framework for transition from virtual screening to wet chemistry.One can also travel in chemical space with genetic algorithms that combine molecule generation with a fitness function in iterative cycles.72–74 One of the first examples was the SPROUT algorithm of Johnson and coworkers, which grows molecules into a targeted protein binding site by coupling building blocks following retrosynthesis rules.75–77 SPROUT selects synthetically feasible products that have a maximum fitness as estimated by docking to the target protein. The same strategy is followed in SYNOPSIS,78 which restricts itself to directly realizable reactions, and in EVOLUATOR,79 which allows interactive molecule selection as the molecule population evolves to its highest fitness. Other genetic algorithms include Skelgen,80 TOPAS,81 Flux,82,83 ADAPT,84 and the more recent multi-objective optimization algorithms GANDI85 and MEGA.86
Chemical space travel has also been realized using formal molecular evolution rules that are independent of synthetic schemes, resulting in a much deeper and structurally more innovative exploration of chemical space. In one case, Gasteiger and coworkers reported a molecular breeding algorithm based on the recombination of molecular fragments that was used to generate median molecules maximizing common features of two different starting molecules.87 The fitness function in this algorithm optimized the Pareto rank relative to the Tanimoto similarity coefficients of structural fingerprints to both starting molecules. Genetic algorithms breeding random fragments were similarly reported that assemble any target molecule by iterative cycles,88 evolve a molecular population to maximum fitness as defined by QSAR,89 and generate new inhibitors by cross-breeding known ones.90
The approach is exemplified by our own version of chemical space travel, which uses a SPACESHIP to travel between a starting molecule A and a target molecule B by iterative cycles of mutation and selection (Fig. 2).91 In the SPACESHIP, the mutation generator is the engine, which is driven by exhausting mutants containing elementary structural changes in bond and atom types. Motion is directed by a compass, which points towards the target B by selecting mutants with the highest Tanimoto similarity coefficient to the target for the next step.
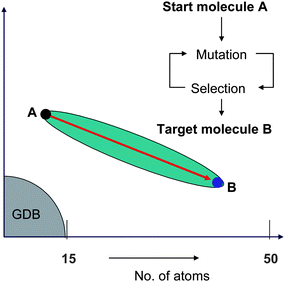 | ||
| Fig. 2 The SPACESHIP algorithm travels from A to B in the chemical space of molecules up to 50 heavy atoms not accessible to GDB. | ||
SPACESHIP explores chemical space for molecules up to 50 heavy atoms which is not accessible to exhaustive enumeration by GDB. The algorithm can join any pair of molecules in a few tens of mutations and selection cycles and generates “trajectory libraries”, which are filtered for chemical consistency by eliminating strained rings and impossible functional groups. Trajectory libraries contain up to several million intermediate molecules between A and B that may later be used for virtual screening. In a model study, a trajectory library of 500000 compounds linking AMPA, an agonist of the corresponding glutamate receptor, with CNQX,92 was ranked by high-throughput docking. A strong enrichment of high-scoring hits such as the β-amino acid 1 formed at intermediate distances between AMPA and CNQX was observed in this library compared to docking with non-selected libraries, suggesting that the trajectory libraries explore privileged regions of chemical space (Fig. 3).
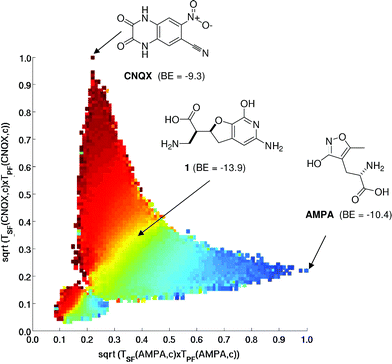 | ||
| Fig. 3 Chemical space travel trajectories between AMPA and CNQX represented in the 2-dimensional Tanimoto similarity space. The trajectory library is colored according to the distance from CNQX to AMPA in number of mutation steps. Binding energies as estimated by docking with Autodock 3.0.5 to the AMPA-receptor 1FTK.pdb are indicated for start and target and a strong-docking intermediate. | ||
4. Maps of the chemical space
The concept of chemical space implies the existence of dimensions and of a map, which in their most simple implementation should define distances between compounds.93–95 In the perspective of drug discovery, the most important dimension is the fitness value during virtual screening, which defines a one-dimensional chemical space. The fitness value is derived from a scoring function, which may be the Tanimoto similarity coefficient for structural or pharmacophore fingerprints or the shape similarity to a reference bioactive molecule, or the score of a docking pose in a given protein binding pocket. The concept can be extended to two or more dimensions if one considers fitness to several targets simultaneously, as proposed by Gasteiger et al. and their concept of median molecules as discussed above.87 For example, the trajectory libraries produced by the SPACESHIP are shown above in a two dimensional space of Tanimoto similarity to the starting molecule A and the target B, in which the iterative cycles of mutation and selection gradually move molecules from one to the other (Fig. 3).91While fitness values produce a different chemical space for every application, it is also possible to define generally valid dimensions using descriptors, which represent structural and physico-chemical properties of the molecules. Thousands of descriptors have been reported in the literature, allowing practically limitless possibilities to construct chemical spaces.58,96 Maps to represent these spaces can be produced by principal component analysis (PCA) and representation of the plane of the first two PCs or the space of the first three PCs. In such property space maps, compounds with related structural, physicochemical and sometimes biological activities are generally grouped together. Notable examples include the ChemGPS system97,98 and related approaches to classify drugs and natural products.99,100 The multidimensional property spaces defined by descriptors can also be visualized using self-organizing maps, which are grids of neurons to which similar compounds are assigned.101 SOM-maps have been used successfully to differentiate various bioactivity classes.102,103 A simple structure-based classification of the chemical universe database GDB-11 can be obtained using a SOM trained with autocorrelation vectors of atomic properties101 as descriptors. In this representation, molecules are organized by their structural types.63 SOM are limited to classifying, at most, a few million molecules due to the computational time needed to train the map.
The periodic system, which is arguably the oldest and best known map of a chemical space, came out of a historical breakthrough when classification of the elements was attempted based on the atomic weights and later the atomic number rather than on the properties of their compounds.104 Similarly, a unified and generally useful classification of organic molecules might arise by using a system based purely on structural features rather than on properties as in the examples above. Two recent approaches have proposed structure-based classification concepts for organic molecules that lead to a mapping of the chemical space.
In the first case, Schuffenhauer et al. reported a so-called scaffold-tree classification by gradually deconstructing molecules in successive steps of functional groups and cycle removals following a simple set of priority rules.105 The analysis defines linkages called brachiation between related molecules. Most remarkably, the scaffold-tree reveals natural families of bioactive scaffolds when annotated with known bioactivities, suggesting new activities for known scaffolds and new scaffolds for known activities.106 For example, analysis of the brachiating structure for inhibitors of the pyruvate kinase led to the identification of three activators (AC50 ≤ 10 μM) and six inhibitors (IC50 ≤ 10 μM) from databases of known compounds.107
In the second case, we have reported a classification of organic molecules based on molecular quantum numbers (MQNs).108 A set of 42 MQNs are defined as counts for elementary constituents of molecules such as atoms, bonds, polar groups, and topological features. MQNs reflect purely structural elements rather than calculated properties as described earlier. The analysis produces a very straightforward map of chemical space when the 42 MQN-dimensions are projected in the PC1/PC2 plane using a non-normalized PCA. For example the MQN-map of the GDB-11 database groups molecules in islands containing molecules with increasing numbers of rings and decreasing number of rotatable bonds. In each island, the north end contains polar molecules and the south end apolar molecules (Fig. 4a–c). Molecules are also well separated into different categories in such maps (Fig. 4d), as was previously observed in a SOM-classification of the database.63 Distances between molecules in MQN-space can be calculated by using a city-block distance, which is the sum of the absolute differences between MQN values of each molecule. MQN-space groups structurally related molecules, as illustrated for the closest MQN-neighbors of diazepam 2–4 found in ZINC, while compounds with high structural similarity as measured by structural fingerprints such as 5 and 6 are more distant (Fig. 5A). MQN-distance classification provides a simple and efficient enrichment scheme for virtual screening of ZINC (Fig. 5B).
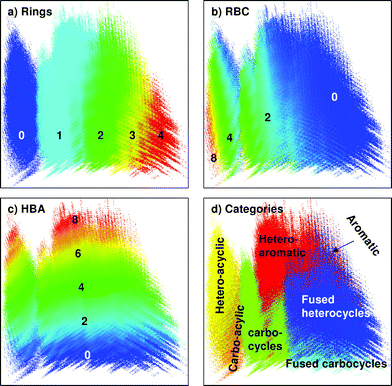 | ||
| Fig. 4 MQN-map of GDB-11 colored by (a) number of cycles, (b) number or rotatable bonds, (c) number of hydrogen-bond acceptor atoms, and (d) molecule categories. In (d) the category of molecules was assigned using the following priority rule: 1. Heteroaromatic (red) > 2. Aromatic (magenta) > 3. Fused heterocyclic (blue) > 4. Fused carbocyclic (cyan) > 5. Heterocyclic (green) > 6. Carbocyclic (bright green) > 7. Heteroacyclic (yellow) > 8. Carboacyclic (orange). Each point in the map is colored according to the majority category for the compounds grouped at that point, with grey shading (saturation in HSL scale) indicating category purity. | ||
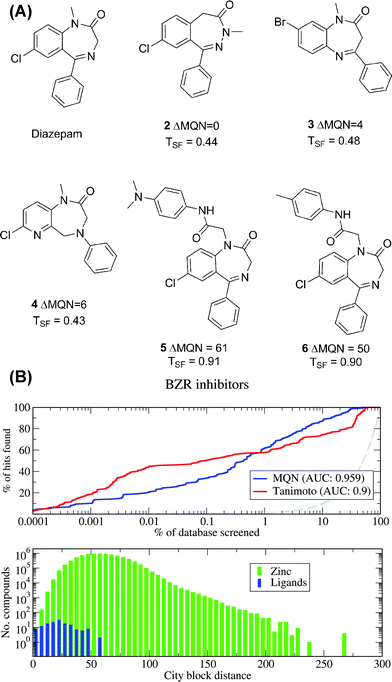 | ||
| Fig. 5 MQN-city block distances for virtual screening. A. Analogs of Diazepam by MQN-distance (2–4) and by structural fingerprint measure (5–6). B. Enrichment curves of recovering known bioactive ligand analogs of diazepam from ZINC using MQN-distances or Tanimoto similarity coefficients of structural fingerprints. | ||
5. Drug discovery from virtual libraries
Over the last few years, many reports have shown that virtual screening actually works, which means that the focused libraries assembled on the basis of scoring functions display a significant percentage of active compounds (up to 50% hit rate) and thus allow the discovery of initial lead compounds much faster and at much lower cost than by blind high-throughput screening (0–0.1% hit rate). This strategy includes the bulk of structure-based drug discovery programs ongoing in medicinal chemistry laboratories worldwide, in particular all prioritization programs applied to in-house and commercial databases to guide retrieval and purchase. In the spirit of this review we focus on cases in which large libraries of yet unknown virtual molecules were subjected to virtual screening to identify potentially active compounds prior to their synthesis.The chemical space travel algorithms discussed above have successfully been implemented in a number of case studies.103 SYNOPSIS was validated by successfully guiding a focused library of 200 possible HIV inhibitors featuring mostly heteroaromatic amides, of which 18 were successfully synthesized and led to 10 non-toxic inhibitors that show significant activity, such as compound 7 (IC50 = 80 μM) (Fig. 6).78 EVOLUATOR has been used to identify compound 8 as an inhibitor that is active on both the α1- and α1-adrenergic receptors and shows a displacement of >50% at a concentration of 10 μM in the radioligand binding assay.79 Skelgen has been used to discover estrogen inhibitors. From the 17 synthesized structures, 5 show inhibition in μM range, such as 9 (IC50 = 0.34 μM).80 Flux was applied for the identification of inhibitors for the disruption of the interaction between the Tat-Peptide and TAR RNA, which is part of the human immunodeficiency virus (HIV-1), such as 10 (IC50 = 500 μM).109
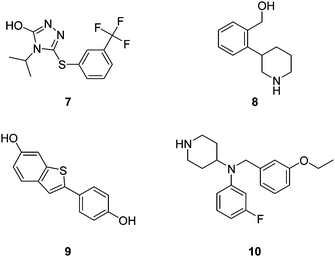 | ||
| Fig. 6 Examples of bioactive molecules identified from virtual libraries prior to synthesis. | ||
In the above examples, molecule generation is coupled to fitness selection, and the database of generated structures is never discussed or explicitly exposed. This strategy eludes the questions of completeness, i.e. have all the possibilities been examined? and of intellectual property protection, i.e. are the generated molecules lost to the public domain? In the case of the chemical universe databases GDB, completeness is addressed because the database is exhaustive, implying that the best possible molecules should be found in the database for any given target provided that a perfect virtual screen is available. Interestingly, the molecules exposed in GDB are not lost to the public domain. Indeed, although GDB-molecules are in principle possible because they contain chemically stable structural elements such as functional groups and ring systems, they are by no means trivial to synthesize. A claim to a structure from GDB will therefore only be possible and valid once the compound has actually been made in the laboratory. Note that this may not necessarily apply if extremely focused GDB-subsets containing molecules that are entirely trivial to make were exposed.
As proof of concept for the use of GDB in drug discovery, we have investigated the case of the glycine binding site of the NMDA-receptor, an important neurotransmitter receptor implicated in various neurological diseases.110 Docking GDB-molecules to the binding site defined in the crystal structure of its glycine complex showed that known ligands such as D-alanine, D-serine, or glycine itself, are indeed among the best (top 1.03%) docking compounds. In one implementation,110 we selected a GDB-subset of 15061 structures using a Bayesian classifier trained with known NMDA-receptor ligands, and carried out high-throughput docking of the corresponding 69367 stereoisomers generated using CORINA.111 Synthesis and testing of a selection of 23 compounds among the 712 compounds docking better than glycine led to the identification of simple dipeptides such as 11–12 as a new class of NMDA-glycine site inhibitors, as well as the D-alanine analog 13 (Fig. 7). Lead optimization was performed by attaching hydrophobic alkyl groups to the terminal amino group, providing the N-ethyl β-alanine dipeptide 14 as optimal ligand. The preference of the NMDA-glycine site for amino acids was confirmed when we docked a random selection of 8000 (31121 stereoisomers) molecules from GDB, which featured non-cyclic amino acids similar to the previously identified ligands in the best docking hits.112 This non-directed screening campaign pointed to the yet unknown diketopiperazines 15 and 16 as possible new types of ligands for the receptor. Indeed synthesis and testing showed that compound 15 was a weak inhibitor of the glycine site, while 16 was inactive. Further discovery programs ongoing in our laboratory have largely confirmed that high-throughput docking of GDB-derived molecules followed by synthesis and testing provides a reliable entry into new ligands.
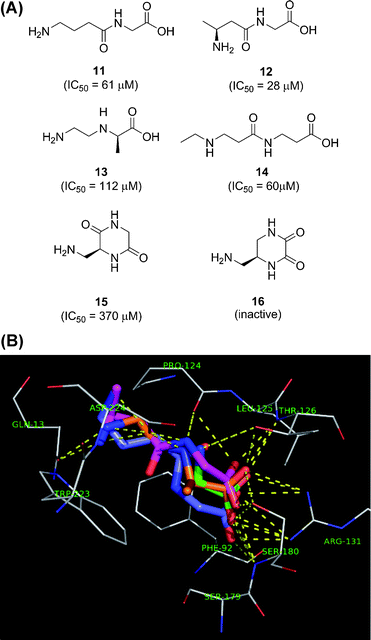 | ||
| Fig. 7 A. Structural formulae of virtual hits 11–16 identified from GDB-11. B. Binding modes within the NMDA-glycine site (1PB7.pdb) for glycine (green), virtual hit 11 (blue), virtual hit 12 (magenta) and virtual hit 13 (orange). | ||
6. Conclusion and outlook
When considering the immensity of chemical space as revealed by exhaustive analyses such as GDB, one must conclude that organic chemistry has not even begun. The unexplored molecular diversity is so large that it is tempting to declare it useless or irrelevant.113 However reassuring, this view is probably mistaken. On the contrary, chemistry should be driven into the unknown chemical space by the pressing need for innovation in small molecule drug discovery.Acknowledgements
This work was supported financially by the University of Berne, the Swiss National Science Foundation, the Office Fédéral de l'Education et de la Science, and the COST program Angiokem.References
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Docking and scoring in virtual screening for drug discovery: methods and applications, Nat. Rev. Drug Discovery, 2004, 3, 935–49 CrossRef CAS.
- T. I. Oprea and H. Matter, Integrating virtual screening in lead discovery, Curr. Opin. Chem. Biol., 2004, 8, 349–58 CrossRef CAS.
- W. L. Chen, Chemoinformatics: past, present, and future, J. Chem. Inf. Model., 2006, 46, 2230–55 CrossRef CAS.
- G. Klebe, Virtual ligand screening: strategies, perspectives and limitations, Drug Discovery Today, 2006, 11, 580–94 CrossRef CAS.
- H. Koeppen, Virtual screening - what does it give us?, Curr. Opin. Drug Discov. Devel., 2009, 12, 397–407 Search PubMed.
- G. Schneider and H. J. Bohm, Virtual screening and fast automated docking methods, Drug Discov. Today, 2002, 7, 64–70 CrossRef CAS.
- A. Nicholls, N. E. MacCuish and J. D. MacCuish, Variable selection and model validation of 2D and 3D molecular descriptors, J. Comput.-Aided Mol. Des., 2004, 18, 451–74 CrossRef CAS.
- P. Willett, Similarity-based virtual screening using 2D fingerprints, Drug Discovery Today, 2006, 11, 1046–53 CrossRef CAS.
- G. Wolber, T. Seidel, F. Bendix and T. Langer, Molecule-pharmacophore superpositioning and pattern matching in computational drug design, Drug Discovery Today, 2008, 13, 23–9 CrossRef CAS.
- J. Kirchmair, S. Ristic, K. Eder, P. Markt, G. Wolber, C. Laggner and T. Langer, Fast and efficient in silico 3D screening: toward maximum computational efficiency of pharmacophore-based and shape-based approaches, J. Chem. Inf. Model., 2007, 47, 2182–96 CrossRef CAS.
- T. S. Rush, 3rd, J. A. Grant, L. Mosyak and A. Nicholls, A shape-based 3-D scaffold hopping method and its application to a bacterial protein-protein interaction, J. Med. Chem., 2005, 48, 1489–95 CrossRef.
- S. Kortagere, M. D. Krasowski and S. Ekins, The importance of discerning shape in molecular pharmacology, Trends Pharmacol. Sci., 2009, 30, 138–47 CrossRef CAS.
- N. Moitessier, P. Englebienne, D. Lee, J. Lawandi and C. R. Corbeil, Towards the development of universal, fast and highly accurate docking/scoring methods: a long way to go, Br. J. Pharmacol., 2008, 153(Suppl. 1), S7–26 CrossRef CAS.
- P. Kolb, R. S. Ferreira, J. J. Irwin and B. K. Shoichet, Docking and chemoinformatic screens for new ligands and targets, Curr. Opin. Biotechnol., 2009, 20, 429–36 CrossRef CAS.
- B. K. Shoichet, S. L. McGovern, B. Wei and J. J. Irwin, Lead discovery using molecular docking, Curr. Opin. Chem. Biol., 2002, 6, 439–46 CrossRef CAS.
- P. C. D. Hawkins, A. G. Skillman and A. Nicholls, Comparison of shape-matching and docking as virtual screening tools, J. Med. Chem., 2007, 50, 74–82 CrossRef CAS.
- G. L. Warren, C. W. Andrews, A. M. Capelli, B. Clarke, J. LaLonde, M. H. Lambert, M. Lindvall, N. Nevins, S. F. Semus, S. Senger, G. Tedesco, I. D. Wall, J. M. Woolven, C. E. Peishoff and M. S. Head, A critical assessment of docking programs and scoring functions, J. Med. Chem., 2006, 49, 5912–5931 CrossRef CAS.
- I. Kola and J. Landis, Can the pharmaceutical industry reduce attrition rates?, Nat. Rev. Drug Discovery, 2004, 3, 711–5 CrossRef CAS.
- T. Wunberg, M. Hendrix, A. Hillisch, M. Lobell, H. Meier, C. Schmeck, H. Wild and B. Hinzen, Improving the hit-to-lead process: data-driven assessment of drug-like and lead-like screening hits, Drug Discovery Today, 2006, 11, 175–80 CrossRef CAS.
- A. L. Hopkins, Network pharmacology: the next paradigm in drug discovery, Nat. Chem. Biol., 2008, 4, 682–90 CrossRef CAS.
- J. Mestres, E. Gregori-Puigjane, S. Valverde and R. V. Sole, The topology of drug-target interaction networks: implicit dependence on drug properties and target families, Mol. BioSyst., 2009, 5, 1051–7 RSC.
- P. J. Hansen and P. C. Jurs, Chemical applications of graph theory. Part II. Isomer enumeration, J. Chem. Educ., 1988, 65, 661 CrossRef CAS.
- E. Cayley, Ueber die analytischen Figuren, welche in der Mathematik Bäume genannt werden und ihre Anwendung auf die Theorie chemischer Verbindungen, Chem. Ber., 1875, 8, 1056–1059 CrossRef.
- H. Schiff, Zur Statistik Chemischer Verbindungen, Chem. Ber., 1875, 8, 1542–1547 CrossRef.
- H. R. Henze and C. M. Blair, The Number Of Isomeric Hydrocarbons Of The Methane Series, J. Am. Chem. Soc., 1931, 53, 3077–3085 CrossRef CAS.
- B. D. McKay, Practical Graph Isomorphism, Congressus Numerantium, 1981, 30, 45–87 Search PubMed.
- A. M. Duffield, A. V. Robertson, C. Djerassi, B. G. Buchanan, G. L. Sutherland, E. A. Feigenbaum and J. Lederberg, Applications of artificial intelligence for chemical inference. II. Interpretation of low-resolution mass spectra of ketones, J. Am. Chem. Soc., 1969, 91, 2977–2981 CrossRef CAS.
- J. Lederberg, G. L. Sutherland, B. G. Buchanan, E. A. Feigenbaum, A. V. Robertson, A. M. Duffield and C. Djerassi, Applications of artificial intelligence for chemical inference. I. Number of possible organic compounds. Acyclic structures containing carbon, hydrogen, oxygen, and nitrogen, J. Am. Chem. Soc., 1969, 91, 2973–2976 CrossRef CAS.
- B. G. Buchanan, D. H. Smith, W. C. White, R. J. Gritter, E. A. Feigenbaum, J. Lederberg and C. Djerassi, Applications of artificial intelligence for chemical inference. 22. Automatic rule formation in mass spectrometry by means of the meta-DENDRAL program, J. Am. Chem. Soc., 1976, 98, 6168–6178 CrossRef CAS.
- S. Bohanec and J. Zupan, Structure generation of constitutional isomers from structural fragments, J. Chem. Inform. Comp. Sci., 1991, 31, 531–540 CrossRef CAS.
- M. S. Molchanova and N. S. Zefirov, Irredundant Generation of Isomeric Molecular Structures with Some Known Fragments, J. Chem. Inform. Comp. Sci., 1998, 38, 8–22 CrossRef CAS.
- W. A. Warr, Computer-assisted structure elucidation. Part II: Indirect database approaches and established systems, Anal. Chem., 1993, 65, 1087A–1095A CAS.
- M. Elyashberg, K. Blinov, S. Molodtsov, Y. Smurnyy, A. J. Williams and T. Churanova, Computer-assisted methods for molecular structure elucidation: realizing a spectroscopist's dream, Journal of Cheminformatics, 2009, 1, 3 Search PubMed.
- C. Steinbeck, SENECA: A Platform-Independent, Distributed, and Parallel System for Computer-Assisted Structure Elucidation in Organic Chemistry, J. Chem. Inform. Comp. Sci., 2001, 41, 1500–1507 CrossRef CAS.
- M. Will, W. Fachinger and J. R. Richert, Fully Automated Structure ElucidationA Spectroscopist's Dream Comes Trueâ€, J. Chem. Inform. Comp. Sci., 1996, 36, 221–227 CrossRef CAS.
- C. Steinbeck, Recent developments in automated structure elucidation of natural products, Nat. Prod. Rep., 2004, 21, 512–8 RSC.
- A. Furka, F. Sebestyen, M. Asgedom and G. Dibo, General method for rapid synthesis of multicomponent peptide mixtures, Int. J. Pept. Protein Res., 1991, 37, 487–93 CAS.
- R. A. Houghten, C. Pinilla, S. E. Blondelle, J. R. Appel, C. T. Dooley and J. H. Cuervo, Generation and use of synthetic peptide combinatorial libraries for basic research and drug discovery, Nature, 1991, 354, 84–6 CrossRef CAS.
- K. S. Lam, S. E. Salmon, E. M. Hersh, V. J. Hruby, W. M. Kazmierski and R. J. Knapp, A new type of synthetic peptide library for identifying ligand-binding activity, Nature, 1991, 354, 82–4 CrossRef CAS.
- S. P. Fodor, J. L. Read, M. C. Pirrung, L. Stryer, A. T. Lu and D. Solas, Light-directed, spatially addressable parallel chemical synthesis, Science, 1991, 251, 767–73 CrossRef CAS.
- R. Frank, SPOT-Synthesis - An Easy Technique for the Positionally Adressable, Parallel Chemical Synthesis on a Membrane Support, Tetrahedron, 1992, 48, 9217–9232 CrossRef CAS.
- R. N. Zuckermann and T. Kodadek, Peptoids as potential therapeutics, Curr. Opin. Mol. Ther., 2009, 11, 299–307 Search PubMed.
- S. L. Beaucage and R. P. Iyer, Advances in the Synthesis of Oligonucleotides by the Phosphoramidite Approach, Tetrahedron, 1992, 48, 2223–2311 CrossRef CAS.
- S. L. Schreiber, Target-oriented and diversity-oriented organic synthesis in drug discovery, Science, 2000, 287, 1964–9 CrossRef CAS.
- T. E. Nielsen and S. L. Schreiber, Towards the optimal screening collection: a synthesis strategy, Angew. Chem., Int. Ed., 2008, 47, 48–56 CrossRef CAS.
- J. Kofoed and J. L. Reymond, A general method for designing combinatorial peptide libraries decodable by amino acid analysis, J. Comb. Chem., 2007, 9, 1046–52 CrossRef CAS.
- N.l. Maillard, T. Darbre and J.-L. Reymond, Identification of Catalytic Peptide Dendrimers by Off-Bead in Silica High-Throughput Screening of Combinatorial Libraries, J. Comb. Chem., 2009, 11, 667–675 CrossRef CAS.
- S. Melkko, C. E. Dumelin, J. Scheuermann and D. Neri, Lead discovery by DNA-encoded chemical libraries, Drug Discovery Today, 2007, 12, 465–71 CrossRef CAS.
- P. J. Edwards, Current parallel chemistry principles and practice: application to the discovery of biologically active molecules, Curr. Opin. Drug Discov. Devel., 2009, 12, 899–914 Search PubMed.
- J. Braun, R. Gugisch, A. Kerber, R. Laue, M. Meringer and C. Rucker, MOLGEN-CID–A canonizer for molecules and graphs accessible through the Internet, J. Chem. Inform. Comp. Sci., 2004, 44, 542–8 CrossRef CAS.
- D. Weininger, Smiles, a Chemical Language and Information-System.1. Introduction to Methodology and Encoding Rules, J. Chem. Inform. Comp. Sci., 1988, 28, 31–36 CrossRef CAS.
- D. Weininger, A. Weininger and J. L. Weininger, Smiles.2. Algorithm for Generation of Unique Smiles Notation, J. Chem. Inform. Comp. Sci., 1989, 29, 97–101 CrossRef CAS.
- S. R. Heller, S. E. Stein and D. V. Tchekhovskoi, InChI: Open access/open source and the IUPAC international chemical identifier, Abstracts of Papers of the American Chemical Society, 2005, 230, 60–CINF Search PubMed.
- (a) J. J. Irwin and B. K. Shoichet, ZINC - A free database of commercially available compounds for virtual screening, J. Chem. Inf. Model., 2005, 45, 177–182 CrossRef CAS; (b) T. Liu, Y. Lin, X. Wen, R. N. Jorrisen and M. K. Gilson, BindingDB: a web-accessible database of experimentally determined protein-ligand binding affinities, Nucleic Acids Res., 2007, 35, D198–D201 CrossRef CAS; (c) J. Overington, J. Comput. Aided Mol. Des., 2009, 23, 195–198 CrossRef.
- E. E. Bolton, PubChem: Integrated Platform of Small Molecules and Biological Activities, Annu. Rep. Comput. Chem., 2008, 4, 217–241 Search PubMed.
- R. S. Bohacek, C. McMartin and W. C. Guida, The art and practice of structure-based drug design: a molecular modeling perspective, Med. Res. Rev., 1996, 16, 3–50 CrossRef CAS.
- C. M. Dobson, Chemical space and biology, Nature, 2004, 432, 824–8 CrossRef CAS.
- I. Vogt and J. Bajorath, Design and exploration of target-selective chemical space representations, J. Chem. Inf. Model., 2008, 48, 1389–95 CrossRef CAS.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef.
- P. Ertl, Cheminformatics analysis of organic substituents: identification of the most common substituents, calculation of substituent properties, and automatic identification of drug-like bioisosteric groups, Journal of Chemical Information and Computer Sciences, 2003, 43, 374–380 Search PubMed.
- T. Fink, H. Bruggesser and J. L. Reymond, Virtual exploration of the small-molecule chemical universe below 160 Daltons, Angew. Chem., Int. Ed., 2005, 44, 1504–8 CrossRef CAS.
- L. C. Blum and J. L. Reymond, 970 million druglike small molecules for virtual screening in the chemical universe database GDB-13, J. Am. Chem. Soc., 2009, 131, 8732–3 CrossRef CAS.
- T. Fink and J. L. Reymond, Virtual exploration of the chemical universe up to 11 atoms of C, N, O, F: assembly of 26.4 million structures (110.9 million stereoisomers) and analysis for new ring systems, stereochemistry, physicochemical properties, compound classes, and drug discovery, J. Chem. Inf. Model., 2007, 47, 342–53 CrossRef CAS.
- S. A. Benner, J. E. Maggio and H. E. Simmons, Rearrangement of a geometrically restricted triepoxide to the first topologically nonplanar molecule: a reaction path elucidated by using oxygen isotope effects on carbon-13 chemical shifts, J. Am. Chem. Soc., 1981, 103, 1581–1582 CrossRef CAS.
- S. J. Teague, A. M. Davis, P. D. Leeson and T. Oprea, The Design of Leadlike Combinatorial Libraries, Angew. Chem., Int. Ed., 1999, 38, 3743–3748 CrossRef CAS.
- M. Congreve, R. Carr, C. Murray and H. Jhoti, A rule of three for fragment-based lead discovery?, Drug Discovery Today, 2003, 8, 876–877 CrossRef.
- S. N. Pollock, E. A. Coutsias, M. J. Wester and T. I. Oprea, Scaffold topologies. 1. Exhaustive enumeration up to eight rings, J. Chem. Inf. Model., 2008, 48, 1304–10 CrossRef CAS.
- M. J. Wester, S. N. Pollock, E. A. Coutsias, T. K. Allu, S. Muresan and T. I. Oprea, Scaffold topologies. 2. Analysis of chemical databases, J. Chem. Inf. Model., 2008, 48, 1311–24 CrossRef CAS.
- A. R. Leach and M. M. Hann, The in silico world of virtual libraries, Drug Discovery Today, 2000, 5, 326–336 CrossRef CAS.
- X. Q. Lewell, D. B. Judd, S. P. Watson and M. M. Hann, RECAP–retrosynthetic combinatorial analysis procedure: a powerful new technique for identifying privileged molecular fragments with useful applications in combinatorial chemistry, J. Chem. Inform. Comp. Sci., 1998, 38, 511–22 CrossRef CAS.
- H. Patel, M. J. Bodkin, B. Chen and V. J. Gillet, Knowledge-based approach to de novo design using reaction vectors, J. Chem. Inf. Model., 2009, 49, 1163–84 CrossRef CAS.
- P. Willett, Genetic algorithms in molecular recognition and design, Trends Biotechnol., 1995, 13, 516–21 CrossRef CAS.
- G. Schneider and U. Fechner, Computer-based de novo design of drug-like molecules, Nat. Rev. Drug Discovery, 2005, 4, 649–63 CrossRef CAS.
- J. Gasteiger, De novo design and synthetic accessibility, J. Comput.-Aided Mol. Des., 2007, 21, 307–309 CrossRef CAS.
- V. J. Gillet, W. Newell, P. Mata, G. Myatt, S. Sike, Z. Zsoldos and A. P. Johnson, Sprout - Recent Developments in the De-Novo Design of Molecules, J. Chem. Inform. Comp. Sci., 1994, 34, 207–217 CrossRef CAS.
- V. J. Gillet, G. Myatt, Z. Zsoldos and A. P. Johnson, SPROUT, HIPPO and CAESA: Tools for de novo structure generation and estimation of synthetic accessibility, Perspect. Drug Discovery Des., 1995, 3, 34–50 Search PubMed.
- P. Mata, V. J. Gillet, A. P. Johnson, J. Lampreia, G. J. Myatt, S. Sike and A. L. Stebbings, Sprout - 3d Structure Generation Using Templates, J. Chem. Inform. Comp. Sci., 1995, 35, 479–493 CrossRef CAS.
- H. M. Vinkers, M. R. de Jonge, F. F. Daeyaert, J. Heeres, L. M. Koymans, J. H. van Lenthe, P. J. Lewi, H. Timmerman, K. van Aken and P. A. Janssen, SYNOPSIS: SYNthesize and OPtimize System in Silico, J. Med. Chem., 2003, 46, 2765–73 CrossRef CAS.
- E. W. Lameijer, J. N. Kok, T. Back and A. P. Ijzerman, The molecule evoluator. An interactive evolutionary algorithm for the design of drug-like molecules, J. Chem. Inf. Model., 2006, 46, 545–52 CrossRef CAS.
- S. Firth-Clark, H. M. Willems, A. Williams and W. Harris, Generation and selection of novel estrogen receptor ligands using the de novo structure-based design tool, SkelGen, J. Chem. Inf. Model., 2006, 46, 642–7 CrossRef CAS.
- G. Schneider, M. L. Lee, M. Stahl and P. Schneider, De novo design of molecular architectures by evolutionary assembly of drug-derived building blocks, J. Comput.-Aided Mol. Des., 2000, 14, 487–94 CrossRef CAS.
- U. Fechner and G. Schneider, Flux (1): a virtual synthesis scheme for fragment-based de novo design, J. Chem. Inf. Model., 2006, 46, 699–707 CrossRef CAS.
- U. Fechner and G. Schneider, Flux (2): comparison of molecular mutation and crossover operators for ligand-based de novo design, J. Chem. Inf. Model., 2007, 47, 656–67 CrossRef CAS.
- S. C. Pegg, J. J. Haresco and I. D. Kuntz, A genetic algorithm for structure-based de novo design, J. Comput.-Aided Mol. Des., 2001, 15, 911–33 CrossRef CAS.
- F. Dey and A. Caflisch, Fragment-Based de Novo Ligand Design by Multiobjective Evolutionary Optimization, J. Chem. Inf. Model., 2008, 48, 679–690 CrossRef CAS.
- C. A. Nicolaou, J. Apostolakis and C. S. Pattichis, De novo drug design using multiobjective evolutionary graphs, J. Chem. Inf. Model., 2009, 49, 295–307 CrossRef CAS.
- N. Brown, B. McKay, F. Gilardoni and J. Gasteiger, A graph-based genetic algorithm and its application to the multiobjective evolution of median molecules, J. Chem. Inform. Comp. Sci., 2004, 44, 1079–1087 CrossRef CAS.
- A. Globus, J. Lawton and T. Wipke, Automatic molecular design using evolutionary techniques, Nanotechnology, 1999, 10, 290–299 CrossRef.
- D. Douguet, E. Thoreau and G. Grassy, A genetic algorithm for the automated generation of small organic molecules: Drug design using an evolutionary algorithm, J. Comput.-Aided Mol. Des., 2000, 14, 449–466 CrossRef CAS.
- A. C. Pierce, G. Rao and G. W. Bemis, BREED: Generating novel inhibitors through hybridization of known ligands. Application to CDK2, P38, and HIV protease, J. Med. Chem., 2004, 47, 2768–2775 CrossRef CAS.
- R. van Deursen and J. L. Reymond, Chemical Space Travel, ChemMedChem, 2007, 2, 636–640 CrossRef CAS.
- H. Brauner-Osborne, J. Egebjerg, E. O. Nielsen, U. Madsen and P. Krogsgaard-Larsen, Ligands for glutamate receptors: design and therapeutic prospects, J. Med. Chem., 2000, 43, 2609–45 CrossRef.
- J. Bajorath, Selected concepts and investigations in compound classification, molecular descriptor analysis, and virtual screening, J. Chem. Inform. Comp. Sci., 2001, 41, 233–45 CrossRef CAS.
- J. W. Godden and J. Bajorath, A distance function for retrieval of active molecules from complex chemical space representations, J. Chem. Inf. Model., 2006, 46, 1094–7 CrossRef CAS.
- Y. A. Ivanenkov, N. P. Savchuk, S. Ekins and K. V. Balakin, Computational mapping tools for drug discovery, Drug Discovery Today, 2009, 14, 767–75 CrossRef CAS.
- P. Kolb and A. Caflisch, Automatic and efficient decomposition of two-dimensional structures of small molecules for fragment-based high-throughput docking, J. Med. Chem., 2006, 49, 7384–92 CrossRef CAS.
- T. I. Oprea and J. Gottfries, Chemography: The art of navigating in chemical space, J. Comb. Chem., 2001, 3, 157–166 CrossRef CAS.
- J. Rosen, J. Gottfries, S. Muresan, A. Backlund and T. I. Oprea, Novel chemical space exploration via natural products, J. Med. Chem., 2009, 52, 1953–62 CrossRef CAS.
- J. L. Medina-Franco, K. Martinez-Mayorga, A. Bender, R. M. Marin, M. A. Giulianotti, C. Pinilla and R. A. Houghten, Characterization of activity landscapes using 2D and 3D similarity methods: consensus activity cliffs, J. Chem. Inf. Model., 2009, 49, 477–91 CrossRef CAS.
- N. Singh, R. Guha, M. A. Giulianotti, C. Pinilla, R. A. Houghten and J. L. Medina-Franco, Chemoinformatic analysis of combinatorial libraries, drugs, natural products, and molecular libraries small molecule repository, J. Chem. Inf. Model., 2009, 49, 1010–24 CrossRef CAS.
- H. Bauknecht, A. Zell, H. Bayer, P. Levi, M. Wagener, J. Sadowski and J. Gasteiger, Locating biologically active compounds in medium-sized heterogeneous datasets by topological autocorrelation vectors: Dopamine and benzodiazepine agonists, J. Chem. Inform. Comp. Sci., 1996, 36, 1205–1213 CrossRef CAS.
- M. Schmuker and G. Schneider, Processing and classification of chemical data inspired by insect olfaction, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20285–9 CrossRef CAS.
- G. Schneider, M. Hartenfeller, M. Reutlinger, Y. Tanrikulu, E. Proschak and P. Schneider, Voyages to the (un)known: adaptive design of bioactive compounds, Trends Biotechnol., 2009, 27, 18–26 CrossRef CAS.
- S. G. Wang and W. H. Schwarz, Icon of chemistry: the periodic system of chemical elements in the new century, Angew. Chem., Int. Ed. Engl., 2009, 48, 3404–3415 CrossRef CAS.
- A. Schuffenhauer, P. Ertl, S. Roggo, S. Wetzel, M. A. Kock and H. Waldmann, The scaffold tree - Visualization of the scaffold universe by hierarchical scaffold classification, J. Chem. Inf. Model., 2007, 47, 47–58 CrossRef CAS.
- S. Wetzel, K. Klein, S. Renner, D. Rauh, T. I. Oprea, P. Mutzel and H. Waldmann, Interactive exploration of chemical space with Scaffold Hunter, Nat. Chem. Biol., 2009, 5, 581–3 CrossRef CAS.
- S. Renner, W. A. van Otterlo, M. Dominguez Seoane, S. Mocklinghoff, B. Hofmann, S. Wetzel, A. Schuffenhauer, P. Ertl, T. I. Oprea, D. Steinhilber, L. Brunsveld, D. Rauh and H. Waldmann, Bioactivity-guided mapping and navigation of chemical space, Nat. Chem. Biol., 2009, 5, 585–92 CrossRef CAS.
- K. T. Nguyen, L. C. Blum, R. van Deursen and J. L. Reymond, Classification of Organic Molecules by Molecular Quantum Numbers, ChemMedChem, 2009, 4, 1803–1805 CrossRef CAS.
- A. Schuller, M. Suhartono, U. Fechner, Y. Tanrikulu, S. Breitung, U. Scheffer, M. W. Gobel and G. Schneider, The concept of template-based de novo design from drug-derived molecular fragments and its application to TAR RNA, J. Comput.-Aided Mol. Des., 2008, 22, 59–68 CrossRef.
- K. T. Nguyen, S. Syed, S. Urwyler, S. Bertrand and J. L. Reymond, Discovery of NMDA glycine site inhibitors from the chemical universe database GDB, ChemMedChem, 2008, 3, 1520–4 CrossRef CAS.
- J. Sadowski and J. Gasteiger, From Atoms and Bonds to 3-Dimensional Atomic Coordinates - Automatic Model Builders, Chem. Rev., 1993, 93, 2567–2581 CrossRef CAS.
- K. T. Nguyen, E. Luethi, S. Syed, S. Urwyler, S. Bertrand, D. Bertrand and J. L. Reymond, 3-(Aminomethyl)piperazine-2,5-dione as a novel NMDA glycine site inhibitor from the chemical universe database GDB, Bioorg. Med. Chem. Lett., 2009, 19, 3832–5 CrossRef CAS.
- J. Hert, J. J. Irwin, C. Laggner, M. J. Keiser and B. K. Shoichet, Quantifying biogenic bias in screening libraries, Nat. Chem. Biol., 2009, 5, 479–83 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |