Measurement of the cellular deacetylase activity of SIRT1 on p53 via LanthaScreen® technology†‡
Matthew B.
Robers
a,
Christine
Loh
b,
Coby B.
Carlson
a,
Hongying
Yang
b,
Elizabeth A.
Frey
a,
Spencer B.
Hermanson
a and
Kun
Bi
*a
aDiscovery Assays and Services, Life Technologies Corporation, Madison, WI 53719, USA. E-mail: kun.bi@lifetech.com
bSirtris, A GSK Company, 200 Technology Square, Cambridge, MA 02139, USA
First published on 8th October 2010
Abstract
Upon genomic insult, the tumor suppressor p53 is phosphorylated and acetylated at specific serine and lysine residues, increasing its stability and transactivation function. Deacetylases, including the type III histone deacetylase SIRT1, remove acetyl groups from p53 and counterbalance acetyltransferase activity during a DNA damage response. This report describes a series of high-throughput LanthaScreen® time-resolved Förster resonance energy transfer (TR-FRET) immunoassays for detection of intracellular p53 phosphorylation of Ser15 and acetylation of Lys382 upon treatment with DNA damage agents, such as etoposide. These assays were used to measure the deacetylase activity of SIRT1 and/or Type I/II Histone deacetylases (HDACs). First, BacMam-mediated overexpression of SIRT1 resulted in dose-dependent deacetylation of GFP-p53 following etoposide treatment of U-2 OS cells, confirming that GFP-p53 serves as a SIRT1 substrate in this assay format. Further, overexpression of the acetyltransferase p300 via BacMam increased the acetylation of GFP-p53 at Lys382. Next, siRNA-mediated knockdown of SIRT1 resulted in increased GFP-p53 acetylation, indicating that endogenous SIRT1 activity can also be measured in U-2 OS cells. Consistent with these results, GFP-p53 acetylation was also increased upon treatment of cells with a small-molecule inhibitor of SIRT1, EX-527. The effect of this compound was dramatically increased when used in combination with chemotherapeutic drug and/or the HDAC inhibitor Trichostatin A, confirming a proposed synergistic mechanism of p53 deacetylation by SIRT1 and Type I/II HDACs. Taken together, the cellular assays described here can be used as high-throughput alternatives to traditional immunoassays such as western blotting for identifying pharmacological modulators of specific p53-modifying enzymes.
Introduction
In response to DNA damage, the tumor suppressor protein p53 is capable of regulating the expression of nearly 100 genes responsible for determining cell fate, division arrest, repair of the damaged DNA, or apoptosis.1,2 In unstressed cells, p53 is degraded via an MDM2–ubiquitin–proteasome pathway. Upon genotoxic stress, such as chemical DNA damage agents or UV treatment, p53 is decoupled from the degradation pathway and phosphorylated at numerous residues (Fig. 1). One of the early events at the sites of DNA damage involves recruitment of ataxia telangiectasia-mutated kinase (ATM) and its phosphorylation of p53 at residues including Ser15.3 Besides phosphorylation, p53 is also acetylated by the acetyltransferases Tip60, p300, and PCAF at numerous residues, including Lys382.4,5 While the precise role of each of these modifications in modulating p53 function remains to be defined, it has been proposed that both phosphorylation and acetylation of these residues enhance p53 stability and/or transcriptional activity,4,5 and impact the functional outcomes of senescence, cell cycle arrest, and apoptosis.6,7 Therefore, enzymes responsible for removing these modifications play an equally important role in determining the p53-mediated downstream functional responses.2,4,5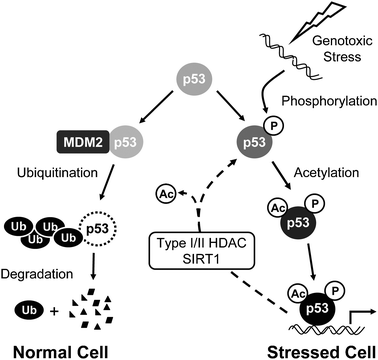 | ||
| Fig. 1 Schematic description of the p53 pathway induced by genotoxic stress. In unstressed cells, p53 is constitutively degraded via the ubiquitin–proteasome pathway. In response to genotoxic stress such as chemical DNA damage, p53 is phosphorylated at Ser15 and acetylated at Lys382, decoupling it from the degradation pathway, resulting in increased transcription from p53-dependent genes. Deacetylases, such as SIRT1 and Type I/II HDACs, remove acetyl groups from p53 and counterbalance acetyltransferase activity during a DNA damage response. | ||
Histone deacetylases (HDACs) have received considerable attention recently as regulators of gene expression and cell cycle progression. At specific genetic loci or at the global genomic level, HDACs induce histone deacetylation and impart a condensed nucleosomal conformation in a cell-cycle dependent manner. In addition to regulating the function and structure of chromatin, HDACs also catalyze the removal of acetyl groups from non-histone proteins such as p53,8–10 counterbalancing acetyltransferase activity following genotoxic stress. Mammalian HDACs are separated into three categories based on homology. Type I (HDACs 1, 2, 3, and 8) and the larger molecular weight Type II HDACs (HDACs 4, 5, 6, 7, 9, and 10) are catalytically dependent on a Zn2+ cofactor and are inhibited by a broad class of small-molecule inhibitors, including hydroxamate-based compounds such as Trichostatin A (TSA). The observation that TSA treatment is capable of potentiating acetylation of p53 (Lys382) implicates Type I/II HDACs in regulation of p53 function.8 Type III HDACs, commonly referred to as Sirtuins (SIRTs 1–7), are mechanistically distinct from Type I/II HDACs, requiring nicotinamide adenine dinucleotide (NAD+) as a cofactor, and are proposed to play a role in metabolism and age-related diseases.11 At the molecular level, SIRT enzymes are virtually unaffected by Type I/II HDAC inhibitors such as TSA, but can be inhibited by a diverse set of molecules, including their own reaction product, nicotinamide. p53 was one of the earliest defined substrates for SIRT1, and among the various acetylated lysine residues in p53, Lys382 was determined to be specifically deacetylated by SIRT1.9 Using combinations of TSA to inhibit Type I/II HDACs and the highly-selective small-molecule inhibitor of SIRT1, EX-527,12 one report suggests a synergistic mechanism between Type I/II HDACs and SIRT1 in deacetylating p53 (Lys382) in various cell lines, including osteosarcoma.8 The role of SIRT1 in oncogenic transformation is still being elucidated, and evidence exists that both small-molecule SIRT1 inhibitors or activators may be valuable for induction of apoptosis in cancer cells.13–17
Current cell-based methods for the measurement of p53 post-translational modifications are limited in terms of sensitivity and throughput, providing a challenge for high-throughput screening (HTS) of small-molecule modulators of enzymes such as Type I/II HDACs and SIRT1. This report describes HTS-compatible cellular assays for measuring phosphorylation and acetylation of p53 proteins using a time-resolved Förster resonance energy transfer (TR-FRET, LanthaScreen®) technology, which has been described previously.18 These assays utilize terbium (Tb)-labeled antibodies in combination with cells expressing a genetic fusion of green fluorescent protein (GFP) with p53 via a baculovirus-mediated gene delivery system known as BacMam (Fig. 2).18–21 First, the GFP-p53 fusion protein was validated as a physiologically-relevant substrate in this assay format, using chemical DNA damage agents to induce the phosphorylation (Ser15) and acetylation (Lys382) of the GFP-p53 proteins, consistent with anticipated effects on endogenous p53 proteins. Next, increased acetylation at Lys382 following ectopic overexpression of the p300 histone acetyltransferase (HAT) was detected. Conversely, BacMam-mediated expression of the deacetylase SIRT1 attenuated acetyl-p53 (Lys382) back to basal levels. Inhibition of SIRT1 at the genetic level viasiRNA resulted in potentiated acetyl-p53 (Lys382), confirming the role of endogenous SIRT1 in modulating p53 Lys382 deacetylation. Finally, the small-molecule inhibitors, EX-527 and TSA, were used to characterize a proposed synergistic mechanism of p53 deacetylation in osteosarcoma cells involving Type I/II HDACs and SIRT1.
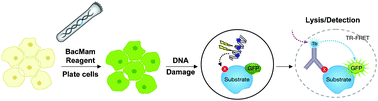 | ||
| Fig. 2 Schematic description of the use of TR-FRET to monitor post-translational modifications of GFP-p53 . Live cells are engineered to express a fusion of GFP with p53 using BacMam as a gene delivery tool. Next, cells are treated with a DNA damaging agent to promote p53 post-translational modifications such as phosphorylation at Ser15 or acetylation at Lys382. Following stimulation, cells are lysed in the presence of a modification-specific antibody chemically labeled with a terbium (Tb) chelate. The association between the antibody and the GFP-p53 analyte allows energy transfer to occur between the excited-state donor fluorophore (Tb) and acceptor fluorophore (GFP), leading to an increase in TR-FRET signal. | ||
The cellular TR-FRET assay described within enables measurement of the tuning of p53 acetylation (Lys382) in response to modulation of these enzymes at the genetic or small molecule level. These results indicate that p53 acetylation occurs in a delicate balance between HATs, HDACs, and SIRT1. Importantly, the parallel measurement of p53 phosphorylation (Ser15) serves as an elegant negative control to confirm the specificity of small-molecule SIRT1/HDAC inhibitors. The method employed here could serve as a template for the HTS-compatible measurement of these activities in a variety of cell types, shedding light on the roles of acetyltransferases and deacetylases in modulating the function of p53.
Results
TR-FRET enables measurement of DNA damage-induced GFP-p53 phosphorylation (Ser15) and acetylation (Lys382) in U-2 OS cells
We first confirmed that the GFP-p53 fusion proteins retain the physiological properties of the endogenous p53 with respect to DNA-damage induced post-translational modifications. Etoposide was selected as a suitable chemical DNA damage agent, as this compound is known to induce double-strand breaks by inhibiting topoisomerase II during cell division. These lesions induce a cellular response resulting in p53 phosphorylation, acetylation, and stabilization.22 Therefore, we first attempted to measure the inducible Ser15 phosphorylation, and Lys382 acetylation of GFP-p53 expressed in U-2 OS cells using two distinct Tb-labeled antibodies. When U-2 OS cells were transduced with BacMam GFP-p53 and treated for 18 hours with serially-diluted etoposide, a dose-dependent increase in TR-FRET signal was observed for GFP-p53 phosphorylation (Ser15) and acetylation (Lys382) using Tb-labeled antibodies specific to these modification events (Fig. 3A). The EC50 for induced phosphorylation and acetylation were 2.7 μM and 5.5 μM, respectively (Fig. 3A). In addition to etoposide, doxorubicin and mitomycin C also induce a DNA damage response that can be quantified in this TR-FRET assay format (data not shown). Etoposide-induced GFP-p53 phosphorylation and acetylation were also confirmed via western blot analysis (Fig. 3B). In the absence of etoposide treatment, little phosphorylation or acetylation of p53 was detected; however, phosphorylation at Ser15 and acetylation at Lys382 were clearly induced with 40 μM etoposide. Taken together, these results support that the GFP-p53 protein serves as a surrogate for endogenous p53 with respect to the DNA damage pathway, and that these assays enable specific detection of p53 modifications.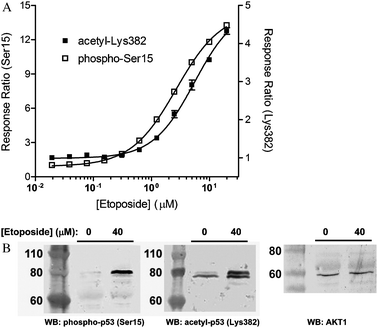 | ||
| Fig. 3 Chemical DNA damage induces GFP-p53 phosphorylation ( Ser15 ) and p53 acetylation ( Lys382 ), as measured using TR-FRET. (A) For measurement of p53 phosphorylation at Ser15 and acetylation at Lys382 using TR-FRET, U-2 OS cells were transduced with 1% BacMam GFP-p53. Treatment with serially-diluted etoposide for 18 hours results in a dose-dependent increase in TR-FRET signal for both phosphorylated p53 at Ser15 (□, left axis) and acetylated p53 at Lys382 (■, right axis). Error bars represent the average of at least 4 data points ± S.E. The Response Ratio (y-axis) is generated by dividing the TR-FRET emission ratio of each stimulated sample by the average value of unstimulated samples. Response ratio of the unstimulated sample is therefore 1. This term simply defines the fold change induced by the treatment. (B) Western blot analysis showing etoposide induced GFP-p53 acetylation at Lys382 and phosphorylation at Ser15. Anti-AKT1 blot was used as a loading control. | ||
Ectopic overexpression of SIRT1 and p300 enzymes regulate the acetylation status of GFP-p53
In order to determine if the cellular TR-FRET assay for p53 acetylation at Lys382 described above can be used to measure SIRT1 deacetylase activity, we examined the effect of ectopic overexpression of SIRT1 on DNA-damage induced p53 acetylation. U-2 OS cells were treated with varying concentrations of BacMam SIRT1 in the presence of 40 μM etoposide and BacMam GFP-p53, followed by the TR-FRET readouts using Tb-anti-p53 AcLys382 or pSer15 antibodies (Fig. 4A). Increasing concentrations of BacMam SIRT1 attenuated etoposide-induced p53 (Lys382) acetylation and resulted in complete depletion of acetyl-p53 TR-FRET signal at 2.5% BacMam SIRT1. The p53 (Ser15) TR-FRET readout served as a negative control, as dosing of BacMam SIRT1 did not affect the phosphorylation status of p53 (similarly, non-SIRT1 encoded BacMam virus did not affect acetyl-p53 levels, data not shown). Western blot analysis was performed to confirm the SIRT1 protein expression (Fig. S1, ESI‡). A dose-dependent SIRT1 expression was observed in U-2 OS cells, indicated by bands of a reported molecular weight 110 kDa for SIRT1 on SDS-PAGE.23 Taken together, these results suggest that SIRT1 overexpression alone is sufficient to suppress DNA-damage induced p53 acetylation at Lys382, and that dose-dependent SIRT1 deacetylase activity can be measured in this TR-FRET assay format.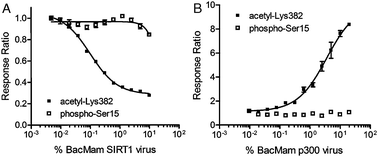 | ||
| Fig. 4 Ectopic overexpression of SIRT1 and p300 enzymes via BacMam results in dose-dependent regulation of GFP-p53 acetylation . (A) U-2 OS cells transduced with BacMam GFP-p53 were treated with serially-diluted BacMam SIRT1 virus in the presence of a fixed concentration of etoposide (40 μM), resulting in a dose-dependent decrease in acetyl-GFP-p53 (Lys382) TR-FRET levels. The signal for phosphorylated GFP-p53 (Ser15) was unaffected upon treatment with BacMam SIRT1; (B) U-2 OS cells were transduced with BacMam GFP-p53 as above, and then treated with serially-diluted BacMam p300 virus. The result was a dose-dependent increase in the levels of acetyl-GFP-p53 (Lys382) and no change in TR-FRET signal for phospho-GFP-p53 (Ser15). | ||
Next, to test whether increased levels of the p300 acetyltransferase are sufficient to induce accumulation of acetylated p53 (Lys382) in the absence of DNA damage, BacMam-mediated overexpression of p300 was tested on U-2 OS cells in the p53 TR-FRET assay (Fig. 4B). Consistent with the observed SIRT1-mediated deacetylation of p53 (Lys382), a dose-dependent increase in the TR-FRET signal for acetylation was observed with increasing amounts of BacMam p300 reagent used, whereas no effect on the phosphorylation was detected.
siRNA knockdown of endogenous SIRT1 increases the level of p53 acetylation at Lys382
Since ectopic overexpression of SIRT1 resulted in dose-dependent deacetylation of p53, knockdown of endogenous SIRT1 was anticipated to induce increased levels of acetylated p53. We next measured the acetylation of GFP-p53 (Lys382) following knockdown of endogenous SIRT1viasiRNA. Lipid transfection of SIRT1-specific RNAi duplex resulted in depletion of SIRT1 protein to undetectable levels, as monitored by Western blot analysis (Fig. 5A). The same RNAi duplex was then used in the TR-FRET assay. As shown in Fig. 5B, SIRT1 knockdown potentiated acetyl-p53 TR-FRET signal in U-2 OS cells compared to negative control oligos, even in the absence of DNA damage induction, confirming the notion that in unstressed cells, the levels of acetylated p53 depend on the equilibrium between constitutive HAT and HDAC activities. Furthermore, SIRT1 knockdown enhanced the acetylated p53 levels induced by etoposide treatment. Taken together, these data show that etoposide treatment did not maximally induce acetylation of Lys382, and that the assay is able to detect a range of p53 deacetylation activities. Furthermore, the data indicate endogenous SIRT1 activity is present in U-2 OS cells and that this assay enables measurement of SIRT1-mediated deacetylation of p53 under conditions of genotoxic stress or under unstressed conditions.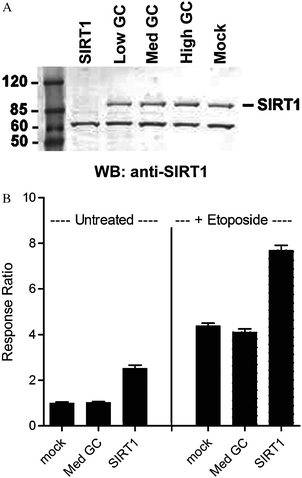 | ||
| Fig. 5 Knockdown of SIRT1 using siRNA results in increased acetyl GFP-p53 levels. (A) Anti-SIRT1 Western blot analysis following siRNA-mediated knockdown of SIRT1 in U-2 OS cells results in complete depletion of SIRT1 to undetectable levels compared to various control oligos (Low, Med, and High GC). A non-specific band of lower molecular weight is observed, indicating that total protein levels are equivalent in each lane; (B) transfection of SIRT1-specific RNAi duplexes in U-2 OS cells results in potentiated acetyl-GFP-p53 TR-FRET signal compared to untreated cells or upon etoposide treatment. Values represent the average of 3 data points, ±S.E. | ||
Small-molecule inhibitors of Type I/II HDAC and SIRT1 activity modulate the acetylation status of p53 (Lys382) in U-2 OS cells
We next determined whether small-molecule inhibitors of HDACs could mediate a change in acetyl-p53 (Lys382). Consistent with the data above showing that SIRT1 knockdown increased acetyl-p53, treatment of cells with 10 μM EX-527, a potent and selective inhibitor of SIRT1, induced an increase (1.6 fold) in acetyl-p53 TR-FRET signal (Fig. 6), indicating that constitutive SIRT1 activity is present in U-2 OS cells, and inhibition of SIRT1 enzyme at the molecular level shifts the equilibrium, resulting in higher levels of acetylated p53. Interestingly, treatment with etoposide in the presence of EX-527 led to a cumulative increase in acetyl-p53 when compared to etoposide alone (to an extent similar to that enhanced by SIRT1 knockdown). TSA enhanced the etoposide-induced acetyl-p53 signal to a lesser extent (Fig. 6A). Notably, neither EX-527 nor TSA induced phosphorylation of Ser 15 on GFP-p53, again suggesting that these compounds do not induce p53 acetylation through a stress response that mimics DNA damage.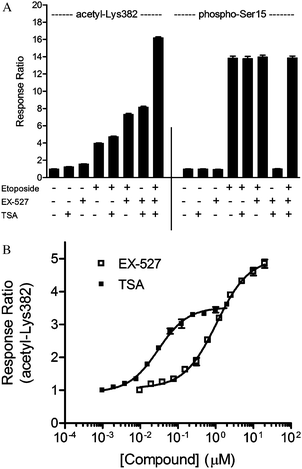 | ||
| Fig. 6 Small-molecule inhibitor confirms the role of Type I/II HDACs and SIRT1 in deacetylation of GFP-p53 ( Lys382 ). (A) As described in Experimental section, treatment of U-2 OS cells with the SIRT1 inhibitor EX-527 (10 μM) or a Type I/II HDAC inhibitor TSA (1 μM) results in increases in acetyl-p53 (Lys382) TR-FRET signal, but does not affect phospho-p53 (Ser15) TR-FRET levels. Compound effects were cumulative with respect to acetyl-p53 TR-FRET signal, indicating synergism between SIRT1 and HDACs in the deacetylation of p53; (B) treatment of U-2 OS cells with serially-diluted EX-527 (□, in the presence of 1 μM TSA) or serially-diluted TSA (■, in the presence of 10 μM EX-527) resulted in dose-dependent increases in acetyl-p53 (Lys382) TR-FRET signal. Values represent the average of at least 4 data points, ±S.E. | ||
Previous reports suggested a synergistic role of Type I/II HDACs and SIRT1 in deacetylation of p53 at Lys382.8 As shown in Fig. 6A, concurrent treatment with EX-527 (10 μM) and TSA (1 μM) resulted in a dramatic increase in acetyl-p53 (Lys382) TR-FRET signal (compared to cells treated with etoposide, EX-527, or TSA alone, concurrent treatment resulted in 2-fold, 5-fold, and 7-fold increase, respectively, Fig. 6A). Such treatment did not affect the phosphorylation of p53 at Ser15. Increases in acetyl-p53 (Lys382) TR-FRET signal upon treatment with serially-diluted EX-527 (in the presence of 1 μM TSA) or serially-diluted TSA (in the presence of 10 μM EX-527) were dose-dependent, generating EC50 values of 1.2 μM and 14 nM, respectively (Fig. 6B). Finally, simultaneous treatment with EX-527, TSA, and etoposide led to a cumulative effect on p53 acetylation, resulting in a 4-fold increase in TR-FRET signal compared to etoposide treatment alone and a 16-fold increase in TR-FRET signal compared to unstimulated cells. As measured in this assay format, the acetylation status of p53 is regulated at the molecular level by SIRT1, Type I/II HDAC, and HAT enzymes. The observation that the effects of these small-molecule inhibitors are cumulative with respect to acetylation of Lys382 on p53 provides additional evidence that pharmacological intervention at multiple steps in the p53 pathway may be necessary to maximize the acetylation status of p53.
Discussion
In response to DNA double strand breaks, p53 undergoes a host of post-translational modifications, including phosphorylation and acetylation.1,2,4,5,24 Evidence suggests that these modifications play a role in p53 stabilization, transcriptional activation, and the role of p53 in determining cell fate.6 Despite an increasing level of understanding of the role of phosphorylation in regulating p53 signaling, the roles of acetylation of p53 by acetyltransferases such as p300 and Tip60, and deacetylation by Type I/II HDACs and Sirtuins (SIRTs) have been only recently described.8,9,24 The NAD+-dependent deacetylase SIRT1 was the first sirtuin characterized to deacetylate p53, specifically on Lys382.9 Several studies have shown that overexpression of SIRT1 reduces the levels of acetyl-p53 and has a significant impact on p53 function, suggesting a role for SIRT1 in suppressing p53-mediated responses including cell cycle arrest and apoptosis.7 Our results showing that SIRT1 overexpression markedly attenuates DNA-damage induced acetyl-p53 in the TR-FRET assay mirror the previous studies using the endogenous p53. We have extended those findings to show that SIRT1 siRNA knockdown and treatment with the SIRT1 inhibitor, EX-527, result in increased acetyl-p53, and further enhance the levels of acetyl-p53 induced by etoposide, indicating that endogenous SIRT1 activity regulates p53 acetylation.Originally linked to aging and longevity in yeast, SIRT1 has been shown to play a central role in metabolism, mitochondrial function, nutrient sensing, inflammation, and circadian oscillations in higher organisms.11,16 Therefore, it is not surprising that substrates for SIRT1 are diverse, and include transcription factors other than p53, such as FOXO, PGC1α, and NFκB for example.16 Due to the diverse roles and substrate specificities of SIRT1, both inhibitors and activators may be of therapeutic value in a tissue or cell context-dependent manner. Small molecules capable of potentiating SIRT1-mediated substrate deacetylation have been shown to promote cellular responses mimicking calorie restriction.25 Among this class of compounds, resveratrol has been shown to also inhibit proliferation of certain cancer cell types, further substantiating the potential value of such compounds.13 Conversely, small-molecule SIRT1 inhibitors, such as EX-527 and sirtinol, have been shown to prevent cell growth and promote senescence in other transformed cell lines.26,27 Therefore, SIRT1 has been the subject of screening campaigns to identify a diverse set of chemical modulators.
A commonly used method for interrogation of SIRT1 activity utilizes purified SIRT1 enzyme and fluorescently-conjugated peptide substrates derived from the p53 amino acid sequence. While convenient and scalable for HTS, such methods utilizing substrate fragments may not faithfully represent the relevant physiological substrates well.28 Recently a biochemical TR-FRET assay using GFP-p53 (full-length p53) was described overcoming the limitation with the fragmented substrates.29 Furthermore, methods for measuring the activity of SIRT1 in a cellular context more accurately represent the natural environment in which the enzymes and regulatory proteins function.30 Current cell-based methods, such as Western blot and ELISA, for measurement of p53 post-translational modifications are not scalable for HTS, limiting the ability to screen for small-molecule SIRT1 modulators in a physiologically-relevant context. The assays described in this report require minimal hands-on time, are compatible for 384-well format, and are statistically robust for use in HTS (consistently generating Z′ values >0.6).18 Analysis of endogenous p53 proteins can also be hindered due to the fact that p53 levels are often undetectable in some cell types.31 When measurement of SIRT1 activity is desired in such cell lines, alternate substrate analytes are a requirement (and antibodies specific to alternate acetylated SIRT1 substrates can be limiting). To address this limitation, BacMam can be used as a gene delivery tool for the transient expression of GFP-p53. Notably, using BacMam-mediated delivery of GFP-p53 in combination with the TR-FRET assay described here, we have successfully measured p53 phosphorylation and acetylation in a diverse set of cell types, including HCT-116 and PC-3, the latter of which has been shown to lack p53 at detectable levels31 (Fig. S2, ESI‡). Our data also support that dose-dependent BacMam GFP-p53 expression can be achieved, enabling the fine-tuning of intracellular levels of GFP-p53 analyte so that DNA-damage induced p53 stabilization can be monitored (data not shown). BacMam-mediated expression has many benefits over other transient expression systems,20,32 providing an attractive alternative to analysis of endogenous p53 proteins.
The roles that post-translational modifications (PTMs) of p53 have on its function continue to expand and become increasingly complex.6 Further adding to the complexity are the findings that these PTMs are coordinately linked. For example, phosphorylation of p53 at serine residues 15, 33, and 37 appear to potentiate subsequent recruitment of p300/CBP and acetylation of p53.33 More recently, HIPK2 has been shown to mediate not only phosphorylation of Ser46, but also acetylation of Lys382.34 In this context, the BacMam GFP-p53 system described here also lends itself to investigation of other protein modifiers of p53. One can study both HAT and Type I/II HDAC activities, as highlighted by our observations that the acetylation state of p53 can be modulated by p300 overexpression and Type I/II HDAC inhibitor TSA. As etoposide induced a dramatic 14-fold increase in phospho-p53 TR-FRET signal, this assay may also serve as a tool for the interrogation of ATM (or related kinases) on the p53 substrate. Therefore, this assay principle should serve as an expandable template for the development of assays measuring alternate p53 post-translational modifications, as well as enable measurement of alternate SIRT enzymes or substrates. The cell-based acetyl-p53 assay described here may be an ideal complement for methods that measure the phenotypic effects of small-molecule acetyltransferase and deacetylase modulators on the DNA damage pathway. This technology may therefore serve as a tool to better define the role of SIRT1 in regulation of p53 function. Given the enormous interest in the potential therapeutic value of many regulators of p53, including SIRT1, this approach should enable investigations into the diverse roles of p53 post-translational modifications in cells and subsequent drug discovery.
Experimental
Molecular biology and production of BacMam particles
To generate a GFP-p53 BacMam expression vector, the cDNA for full-length human p53 (Life Technologies Ultimate™ ORF IOH4961 matching nucleotide reference sequence BC003596.1) was subcloned into a pDEST8-CMV-n-GFP vector as described previously.20 To generate a SIRT1 BacMam expression vector, the cDNA for full-length SIRT1 (encoding a protein matching reference sequence NP_036370.2) was subcloned into an analogous vector but lacking the GFP open reading frame. The resulting expression constructs were then transformed into DH10BAC bacterial strain for processing into bacmid DNA. PCR-qualified bacmid DNA was then transfected into SF9 insect cells for BacMam virus production according to Invitrogen Bac-to-Bac® Baculovirus Expression Systems manual32,35 (Life Technologies).Cell culture and transduction of mammalian cells with BacMam virus
All cell culture media and supplements were from Life Technologies. U-2 OS cells were purchased from ATCC. For transduction of U-2 OS cells with BacMam GFP-p53, cells were suspended in growth medium (consisting of McCoy's 5A medium supplemented with 10% fetal bovine serum, non-essential-amino acids, and penicillin/streptomycin) at a density of approximately 2 × 105cells mL−1. Prior to seeding, cells were mixed with BacMam GFP-p53 to a final concentration indicated (for measurement of p53 phosphorylation and acetylation, 1% BacMam GFP-p53 (v/v) was consistently used). For BacMam-mediated expression of SIRT1 or p300, cells were transduced as described above but in the presence of serially-diluted BacMam SIRT1 or p300 enzymes, with 20% (v/v) the highest concentration tested. 32 μL per well of the cell/virus mixture were then seeded into white, 384-well plates (Corning), corresponding to approximately 8000 cells per well.Compound treatment and TR-FRET-based assay for GFP-p53 phosphorylation at Ser15 and acetylation at Lys382
For measurement of p53 phosphorylation and acetylation, cells were stimulated with compound immediately at the time of seeding in 384-well plates in the presence of BacMam GFP-p53 virus (1% v/v) as indicated above. 4 μL additions of diluted compound or vehicle were used such that the final volume was 40 μL per well. Cells were incubated under normal growth conditions in a humidified incubator set to 37 °C and 5% CO2. Unless indicated otherwise, the final concentration of etoposide (Sigma) was 40 μM, EX-527 (6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide, Tocris) was 10 μM, and TSA (Trichostatin A, Sigma) was 1 μM. Stimulation occurred for 18 h with etoposide or EX-527 and 4 h with TSA. Immediately following stimulation, cell medium was removed by inverting the plate and blotting the contents onto a dry paper towel. Tb-labeled antibodies specific to acetylated p53 (Lys382) and phosphorylated p53 (Ser15) were obtained from Life Technologies.29 Following media removal, 20 μL of cell lysis buffer (Life Technologies), supplemented as described previously,18,20 but including 2 nM Tb-labeled antibody specific to phosphorylated p53 (Ser15) or acetylated p53 (Lys382) was immediately added to each well using a multichannel pipette. Lysates were allowed to equilibrate for 2 h at room temperature before TR-FRET values were measured on a plate reader (BMG Labtech PHERAstar) set to standard TR-FRET settings as described previously.18 Emission ratios were calculated using the time-gated fluorescence intensity at 520 nm/490 nm (340 nm excitation). To generate relative Response Ratios (which is an equivalent term for fold increase, fold change, or assay window), the emission ratio values of the etoposide or compound-treated sample were divided by the average of the values of the vehicle treated samples.SIRT1 siRNA analysis
Lipofectamine® RNAi Max transfection reagent, validated Stealth RNAi™ SIRT1 siRNA and negative control (Medium, Low, and High GC Control) duplexes were from Life Technologies. Cells were transfected by first preparing cell suspensions (as indicated above for the TR-FRET assays), and adding a 1/5 volume of lipid:RNA complexes such that the final concentration of each duplex was 40 nM. For TR-FRET assays, immediately after mixing cells with RNAi:lipid complexes, 32 μL per well of cell suspension (including RNAi oligos) were added to white 384-well plates (Corning). Cells were then incubated 24 h prior to addition of 4 μL per well of undiluted BacMam GFP-p53 and 4 μL per well of etoposide to a final concentration of 40 μM. Cells were then incubated an additional 24 h prior to cell lysis and TR-FRET measurement as described above.Western blot analysis
For measurement of SIRT1 expression via Western blot, 4 × 105U-2 OS cells were seeded into each well of a 6-well plate. Cells were treated with siRNA duplexes or BacMam particles at the time of seeding. Following 48 h of incubation with siRNA duplexes, 24 h incubation with BacMam SIRT1, or 18 h of 40 μM etoposide treatment, cell media were removed and cells were lysed using 0.2 mL per well of M-PER protein extraction reagent (Pierce) supplemented with protease inhibitor cocktail (Sigma). 10 μL of each sample were then subjected to SDS-PAGE and transferred to nitrocellulose as described previously.18 Blots were probed overnight with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ∶400 dilution of rabbit anti-SIRT1 antibody (Abcam) (while SIRT1 consists of 750 amino acids with a predicted molecular weight of ∼90 kDa, both the endogenous and overexpressed SIRT1 protein were detected at ∼110 kDa, consistent with what has been reported in the literature23 and is presumably due to post-translational modifications of SIRT1 in cells), 1∶1000 anti-p53 antibody (Cell Signaling Technologies), 1 μM Tb-anti acetyl-p53 (Lys382) antibody, or 1 μM Tb-anti-phospho-p53 (Ser15), followed by chromogenic detection with AP-conjugated anti-species antibodies (Life Technologies).
∶400 dilution of rabbit anti-SIRT1 antibody (Abcam) (while SIRT1 consists of 750 amino acids with a predicted molecular weight of ∼90 kDa, both the endogenous and overexpressed SIRT1 protein were detected at ∼110 kDa, consistent with what has been reported in the literature23 and is presumably due to post-translational modifications of SIRT1 in cells), 1∶1000 anti-p53 antibody (Cell Signaling Technologies), 1 μM Tb-anti acetyl-p53 (Lys382) antibody, or 1 μM Tb-anti-phospho-p53 (Ser15), followed by chromogenic detection with AP-conjugated anti-species antibodies (Life Technologies).
Conclusion
We have developed a robust cellular assay for detecting the deacetylase activities of SIRT1 and type I/II HDAC on p53, which can be used for screening for SIRT1/HDAC modulators in a high-throughput compatible format. The combination of BacMam-mediated gene delivery and the TR-FRET cellular assay technology can be expanded to the interrogation of acetyltransferase activity and DNA-damage activated kinases that phosphorylate p53.References
- M. F. Lavin and N. Gueven, The complexity of p53 stabilization and activation, Cell Death Differ., 2006, 3(6), 941–950 CrossRef.
- A. Olsson, C. Manzl, A. Strasser and A. Villunger, How important are post-translational modifications in p53 for selectivity in target-gene transcription and tumour suppression?, Cell Death Differ., 2007, 14(9), 1561–1575 CrossRef CAS.
- N. Tomimatsu, C. G. Tahimic, A. Otsuki, S. Burma, A. Fukuhara, K. Sato, G. Shiota, M. Oshimura, D. J. Chen and A. Kurimasa, Ku70/80 modulates ATM and ATR signaling pathways in response to DNA double strand breaks, J. Biol. Chem., 2007, 282(14), 10138–10145 CrossRef CAS.
- L. Feng, T. Lin, H. Uranishi, W. Gu and Y. Xu, Functional analysis of the roles of posttranslational modifications at the p53 C terminus in regulating p53 stability and activity, Mol. Cell. Biol., 2005, 25(13), 5389–5395 CrossRef CAS.
- Y. Tang, W. Zhao, Y. Chen, Y. Zhao and W. Gu, Acetylation is indispensable for p53 activation, Cell, 2008, 133(4), 612–626 CrossRef CAS.
- J. P. Kruse and W. Gu, Modes of p53 regulation, Cell, 2009, 137(4), 609–622 CrossRef CAS.
- J. Yi and J. Luo, SIRT1 and p53, effect on cancer, senescence and beyond, Biochim. Biophys. Acta, 2010, 1804(8), 1684–1689 CAS.
- J. M. Solomon, R. Pasupuleti, L. Xu, T. McDonagh, R. Curtis, P. S. DiStefano and L. J. Huber, Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage, Mol. Cell. Biol., 2006, 26(1), 28–38 CrossRef CAS.
- H. Vaziri, S. K. Dessain, E. Ng Eaton, S. I. Imai, R. A. Frye, T. K. Pandita, L. Guarente and R. A. Weinberg, hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase, Cell, 2001, 107(2), 149–159 CrossRef CAS.
- J. Luo, A. Y. Nikolaev, S. Imai, D. Chen, F. Su, A. Shiloh, L. Guarente and W. Gu, Negative control of p53 by Sir2alpha promotes cell survival under stress, Cell, 2001, 107(2), 137–148 CrossRef CAS.
- D. M. Taylor, M. M. Maxwell, R. Luthi-Carter and A. G. Kazantsev, Biological and potential therapeutic roles of sirtuin deacetylases, Cell. Mol. Life Sci., 2008, 65(24), 4000–4018 CrossRef CAS.
- A. D. Napper, J. Hixon, T. McDonagh, K. Keavey, J. F. Pons, J. Barker, W. T. Yau, P. Amouzegh, A. Flegg, E. Hamelin, R. J. Thomas, M. Kates, S. Jones, M. A. Navia, J. O. Saunders, P. S. DiStefano and R. Curtis, Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1, J. Med. Chem., 2005, 48(25), 8045–8054 CrossRef CAS.
- M. H. Aziz, M. Nihal, V. X. Fu, D. F. Jarrard and N. Ahmad, Resveratrol-caused apoptosis of human prostate carcinoma LNCaP cells is mediated via modulation of phosphatidylinositol 3′-kinase/Akt pathway and Bcl-2 family proteins, Mol. Cancer Ther., 2006, 5(5), 1335–1341 CrossRef CAS.
- C. X. Deng, SIRT1, is it a tumor promoter or tumor suppressor?, Int. J. Biol. Sci., 2009, 5(2), 147–152 Search PubMed.
- N. Kabra, Z. Li, L. Chen, B. Li, X. Zhang, C. Wang, T. Yeatman, D. Coppola and J. Chen, SirT1 is an inhibitor of proliferation and tumor formation in colon cancer, J. Biol. Chem., 2009, 284(27), 18210–18217 CrossRef CAS.
- S. Lavu, O. Boss, P. J. Elliott and P. D. Lambert, Sirtuins—novel therapeutic targets to treat age-associated diseases, Nat. Rev. Drug Discovery, 2008, 7(10), 841–853 CrossRef CAS.
- T. Liu, P. Y. Liu and G. M. Marshall, The critical role of the class III histone deacetylase SIRT1 in cancer, Cancer Res., 2009, 69(5), 1702–1705 CrossRef CAS.
- M. B. Robers, R. A. Horton, M. R. Bercher, K. W. Vogel and T. Machleidt, High-throughput cellular assays for regulated posttranslational modifications, Anal. Biochem., 2008, 372(2), 189–197 CrossRef CAS.
- C. B. Carlson, M. B. Robers, K. W. Vogel and T. Machleidt, Development of LanthaScreen™ Cellular Assays for Key Components within the PI3K/AKT/mTOR Pathway, J. Biomol. Screening, 2009, 14(2), 121–132 CrossRef CAS.
- K. G. Huwiler, T. Machleidt, L. Chase, B. Hanson and M. B. Robers, Characterization of serotonin 5-hydroxytryptamine-1A receptor activation using a phospho-extracellular-signal regulated kinase 2 sensor, Anal. Biochem., 2009, 393(1), 95–104 CrossRef CAS.
- M. B. Robers, T. Machleidt, C. B. Carlson and K. Bi, Cellular LanthaScreen and beta-lactamase reporter assays for high-throughput screening of JAK2 inhibitors, Assay Drug Dev. Technol., 2008, 6(4), 519–529 CrossRef CAS.
- A. Vazquez, E. E. Bond, A. J. Levine and G. L. Bond, The genetics of the p53 pathway, apoptosis and cancer therapy, Nat. Rev. Drug Discovery, 2008, 7(12), 979–987 CrossRef CAS.
- H. L. Cheng, R. Mostoslavsky, S. Saito, J. P. Manis, Y. Gu, P. Patel, R. Bronson, E. Appella, F. W. Alt and K. F. Chua, Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice, Proc. Natl. Acad. Sci. U. S. A., 2003, 100(19), 10794–10799 CrossRef CAS.
- M. Kruszewski and I. Szumiel, Sirtuins (histone deacetylases III) in the cellular response to DNA damage—facts and hypotheses, DNA Repair, 2005, 4(11), 1306–1313 CrossRef CAS.
- K. T. Howitz, K. J. Bitterman, H. Y. Cohen, D. W. Lamming, S. Lavu, J. G. Wood, R. E. Zipkin, P. Chung, A. Kisielewski, L. L. Zhang, B. Scherer and D. A. Sinclair, Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan, Nature, 2003, 425(6954), 191–196 CrossRef CAS.
- B. Jung-Hynes, M. Nihal, W. Zhong and N. Ahmad, Role of sirtuin histone deacetylase SIRT1 in prostate cancer. A target for prostate cancer management via its inhibition?, J. Biol. Chem., 2009, 284(6), 3823–3832 CAS.
- B. Peck, C. Y. Chen, K. K. Ho, P. Di Fruscia, S. S. Myatt, R. C. Coombes, M. J. Fuchter, C. D. Hsiao and E. W. Lam, SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2, Mol. Cancer Ther., 2010, 9(4), 844–855 CrossRef CAS.
- M. T. Borra, B. C. Smith and J. M. Denu, Mechanism of human SIRT1 activation by resveratrol, J. Biol. Chem., 2005, 280(17), 17187–17195 CrossRef CAS.
- J. M. Dudek and R. A. Horton, TR-FRET biochemical assays for detecting posttranslational modifications of p53, J. Biomol. Screening, 2010, 15(5), 569–575 CrossRef CAS.
- K. Vogel, Z. Zhong, K. Bi and B. Pollok, Developing assays for drug discovery—where have the advances come from, Expert Opin. Drug Discovery, 2008, 3, 115–125 Search PubMed.
- B. Jung-Hynes and N. Ahmad, Role of p53 in the anti-proliferative effects of Sirt1 inhibition in prostate cancer cells, Cell Cycle, 2009, 8(10), 1478–1483 CrossRef CAS.
- T. A. Kost, J. P. Condreay, R. S. Ames, S. Rees and M. A. Romanos, Implementation of BacMam virus gene delivery technology in a drug discovery setting, Drug Discovery Today, 2007, 12(9–10), 396–403 CrossRef CAS.
- K. Sakaguchi, J. E. Herrera, S. Saito, T. Miki, M. Bustin, A. Vassilev, C. W. Anderson and E. Appella, DNA damage activates p53 through a phosphorylation-acetylation cascade, Genes Dev., 1998, 12(18), 2831–2841 CrossRef CAS.
- R. Puca, L. Nardinocchi, A. Sacchi, G. Rechavi, D. Givol and G. D'Orazi, HIPK2 modulates p53 activity towards pro-apoptotic transcription, Mol. Cancer, 2009, 8, 85 CrossRef.
- R. Ames, J. Fornwald, P. Nuthulaganti, J. Trill, J. Foley, P. Buckley, T. Kost, Z. Wu and M. Romanos, BacMam recombinant baculoviruses in G protein-coupled receptor drug discovery, Recept. Channels, 2004, 10(3–4), 99–107 CrossRef CAS.
Footnotes |
| † This article is part of a themed issue of Molecular BioSystems on Post-translational modifications. |
| ‡ Electronic supplementary information (ESI) available: 2 supplementary figures. See DOI: 10.1039/c0mb00026d |
| This journal is © The Royal Society of Chemistry 2011 |