Visualization of PKA activity in plasma membrane microdomains†
Charlene
Depry
a,
Michael D.
Allen
ab and
Jin
Zhang
*ac
aDepartment of Pharmacology and Molecular Sciences, The Johns Hopkins University School of Medicine, Baltimore, Maryland, USA
bCurrently at the Clinical Endocrinology Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, Maryland, USA
cThe Solomon H. Snyder Department of Neuroscience and Department of Oncology, The Johns Hopkins University School of Medicine, Maryland, USA
First published on 14th September 2010
Abstract
Membrane rafts are sphingolipid- and cholesterol-rich microdomains that contain dynamic arrangements of signaling proteins. Notably, various components of the ubiquitous cAMP/Protein Kinase A (PKA) pathway, including β-adrenergic receptors (β-ARs), G proteins, and adenylyl cyclases (ACs), have been shown to localize differentially between membrane rafts and non-raft regions of the plasma membrane. As PKA participates in regulating diverse fundamental cellular functions, a number of which require membrane rafts, it is important to understand how PKA activity is specifically regulated in these membrane microdomains. To this end, we developed an improved FRET-based PKA activity biosensor, and targeted it to both membrane raft and non-raft regions of the plasma membrane to examine PKA activity dynamics in different plasma membrane microdomains. Disruption of membrane raftsviacholesterol depletion was shown to enhance β-AR stimulated PKA activity at the plasma membrane, suggesting that membrane rafts play a negative role in β-AR stimulated PKA activation. Furthermore, we found that membrane rafts possess higher basal PKA activity in the resting state compared to non-raft regions, which depends on the integrity of membrane rafts and proper localization of PKA. This study shows that membrane rafts play an important role in regulating the activity of PKA at the plasma membrane, and demonstrates the ability of live-cell FRET-based assays to reveal dynamic differences amongst plasma membrane microdomains, laying a foundation for further dissection of membrane regulated signal transduction.
Introduction
Membrane rafts are small, heterogeneous microdomains enriched in cholesterol and glycosphingolipids, which are more ordered and rigid in comparison to the rest of the plasma membrane.1 As “organizing centers”, membrane rafts have been shown to function in basic cellular processes, such as signal transduction, trafficking, endocytosis, and exocytosis.2–6 The roles that membrane rafts play in signal transduction are not well understood, yet one current theory is that they enhance the specificity of signaling,1 by segregating signaling components, such as cell surface receptors and downstream effector proteins.1,4,6The cAMP/Protein Kinase A (PKA) pathway is ubiquitously expressed and regulates a plethora of fundamental cellular functions, including metabolism, gene expression, growth and proliferation.7,8 In order to attain proper cell responses, PKA phosphorylation of its many substrates requires meticulous organization and regulation. This is in part achieved by compartmentalization of PKA with its substrates. A Kinase Anchoring Proteins (AKAPs) localize PKA, along with its substrates and regulators (e.g., phosphodiesterases, phosphatases, and adenylyl cyclases), to discrete subcellular compartments, promoting precise phosphorylation and regulation of PKA substrates.9,10 For example, it has been shown that type I PKA, which is localized to membrane raftsvia the AKAP Ezrin, mediates inhibition of T cell activation by phosphorylating Csk. Knockdown of Ezrin or disruption of PKA binding to Ezrin was shown to eliminate type I PKA from membrane rafts, as well as the ability of this kinase to inhibit T cell activation.11
It has been shown that various signaling components upstream of PKA, including G protein-coupled receptors (GPCRs), G proteins, and adenylyl cyclases (ACs), are differentially localized within plasma membrane microdomains. For example, the β2-AR, a canonical GPCR, is sequestered outside of membrane rafts, whereas Gα stimulatory (Gαs) proteins and ACs are dispersed throughout the plasma membrane. Moreover, upon cholesterol depletion with methyl-β-cyclodextrin (MβCD), cAMP accumulation is enhanced, as a result of increased β2-AR-Gαs-AC associations.12 These findings shed light onto the roles that membrane rafts play in regulating the cAMP pathway. However, the effects at the PKA activity level have yet to be examined and the membrane compartmentalization of PKA activity is not well understood.
Fluorescence resonance energy transfer (FRET)-based biosensors are powerful tools that can be used to visualize activity dynamics of signaling molecules with high spatiotemporal resolution in live cells. We have successfully engineered FRET-based A kinase Activity Reporters (AKAR) to examine real-time PKA activity dynamics, in single cells, using live-cell fluorescence microscopy. These reporters were designed by sandwiching a molecular switch between two green fluorescent protein (GFP) variants. The molecular switch consists of a surrogate substrate for PKA and a phosphoamino acid-binding domain (PAABD). Following phosphorylation of the surrogate substrate by PKA, the PAABD binds the phosphorylated substrate, inducing a conformational change, which results in an increase in FRET. To monitor compartmentalized PKA activity, previously, we targeted AKAR to varied subcellular locations and visualized differences in localized PKA activity.13 In this study, to visualize PKA activity in plasma membrane microdomains, we targeted AKAR to membrane rafts or non-raft regions of the plasma membrane and found that PKA activity dynamics are differentially regulated in these compartments.
Results
Characterization of new AKARs
In order to facilitate detection of subtle changes in PKA activity we set out to generate a new version of AKAR with an enhanced dynamic range. Taking advantage of brighter fluorescent protein variants available from protein engineering efforts,14 we replaced the enhanced cyan fluorescent protein (eCFP) of AKAR313 with cerulean, a brighter variant of CFP15 (Fig. 1A). The cerulean containing reporter was transfected in HEK293 cells and found to maintain the same uniform distribution throughout the cell as AKAR3 (Fig. 1B), but exhibit brighter cyan fluorescence (data not shown). When HEK293 cells expressing this reporter were stimulated with isoproterenol (Iso), a general β-AR agonist, with illumination of the cyan fluorophore, yellow fluorescence increased at the expense of cyan fluorescence, yielding a change in yellow-to-cyan emission ratio of 58 ± 1.7% (n = 7) [average ± SEM, n = number of cells]. This new probe, designated AKAR4, has an improved dynamic range, showing 67% improvement over AKAR3 (Fig. 1C).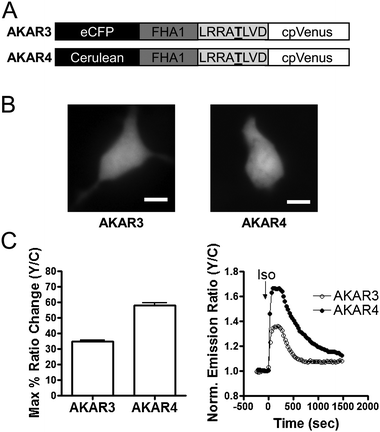 | ||
| Fig. 1 Development and Characterization of AKAR4 in HEK293 cells. (A) Domain structures of AKAR3 and AKAR4 biosensors. The underlined T is phosphorylated by PKA. (B) CFP images of AKAR3 and AKAR4 showing their distribution within the entire cell. Scale bar = 10 μm. (C) Summary (left) and representative curves (right) of AKAR3 (○) and AKAR4 (●) in response to 1 μM Iso. | ||
In order to visualize compartmentalized PKA activity in plasma membrane microdomains, we targeted AKAR4 to the plasma membrane using different lipid modification domains. A motif derived from the N-terminus of Lyn kinase is known to direct reporters to membrane raftsviamyristoylation and palmitoylation, while a CaaX (C = cysteine, a = aliphatic amino acid, and X = any amino acid) sequence along with a poly lysine motif, adapted from K-Ras, anchors reporters to non-raft regions of the plasma membrane16 (Fig. 2A). Both reporters were found to localize to the plasma membrane of HEK293 cells. We further validated the specific localization of Lyn-AKAR4 and AKAR4-Kras in HEK293 cells using sucrose density gradient fractionation followed by western analysis (Fig. 2B). Among the fractions separated via the sucrose density gradient method, membrane rafts are known to accumulate in the more buoyant fractions, while non-raft regions accumulate in higher density fractions. Cholera toxin B subunit (CtxB) was used to mark membrane rafts as it binds to a known raft-associated ganglioside, GM1. Probing all fractions with a GFP specific antibody revealed that Lyn-AKAR4 partitions into membrane rafts, whereas AKAR4-Kras is excluded from membrane rafts. We see imperfect colocalization between the raft marker and the membrane raft targeted biosensor. Since CtxB binds gangliosides and Lyn-AKAR4 is tethered viamyristoylation and palmitoylation lipid anchors, CtxB and Lyn-AKAR4 may not localize to the same microdomains within heterogeneous membrane rafts.
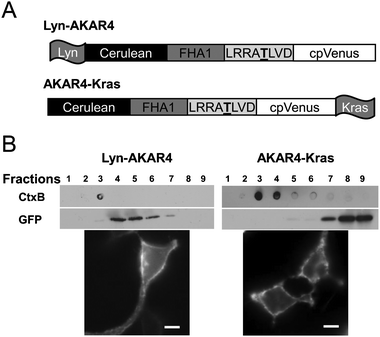 | ||
| Fig. 2 Design and Localization of PM Targeted AKARs in HEK293 cells. (A) Schematic of Lyn-AKAR4 and AKAR4-Kras biosensors. (B) The microdomain localization of Lyn-AKAR4 and AKAR4-Kras was confirmed with sucrose density gradient fractionation. Total cell lysates from HEK293 cells overexpressing Lyn-AKAR4 or AKAR4-Kras were subjected to sucrose density gradient fractionation followed by western analysis using an anti-GFP antibody. Cholera toxin B subunit was used as a membrane raft marker. CFP images show their distribution at the plasma membrane. Scale bar = 10 μm. | ||
HEK293 cells expressing either plasma membrane targeted biosensor were stimulated with activators of the cAMP/PKA pathway to assess their responses. Upon β-AR receptor stimulation using Iso, Lyn-AKAR4 and AKAR4-Kras show a ratio change of 9.4 ± 0.8% (n = 45) and 9.4 ± 1.4% (n = 23), respectively (Fig. 3A and B). Similarly, activation of ACs using the pharmacological agonist forskolin (Fsk) generated an emission ratio change of 8.8 ± 0.8% (n = 28) and 8.2 ± 0.9% (n = 25) from Lyn-AKAR4 and AKAR4-Kras, respectively (Fig. 3C and D). Thus the plasma membrane microdomain targeted versions can detect PKA activity within their individual compartments.
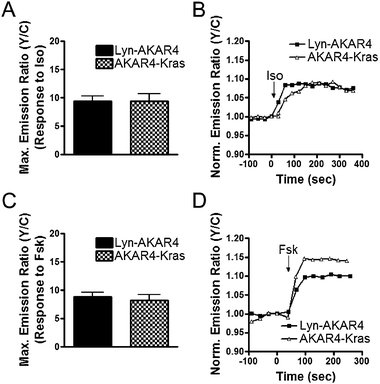 | ||
| Fig. 3 Characterization of Plasma Membrane Targeted AKARs. Summary of Lyn-AKAR4 and AKAR4-Kras responses to Iso (A) and Fsk (C). Representative curves of Lyn-AKAR4 (■) and AKAR4-Kras (△) responses to Iso (B) and Fsk (D). All data in bar graphs are presented as average ± SEM. | ||
Cholesterol depletion enhances stimulated PKA activity throughout the plasma membrane
Using our plasma membrane targeted AKARs, we investigated the role of membrane microdomains in regulating PKA activity upon receptor stimulation. Stimulation of HEK293 cells expressing Lyn-AKAR4 with Iso, to activate β2-ARs that couple to the classic Gs-AC-cAMP-PKA signaling pathway,17 produced a 9.8 ± 0.9% (n = 25) increase in emission ratio. Subsequent addition of the PKA inhibitor, H89, generated a 23 ± 1.9% (n = 25) decrease in yellow-to-cyan emission ratio (Fig. 4A and B), as a result of dephosphorylation of the reporter by endogenous phosphatases. We hypothesize that a substantial amount of Lyn-AKAR4 is phosphorylated prior to β-AR activation due to basal PKA activity within membrane rafts, given that the decrease generated by H89 treatment exceeds the Iso stimulated response.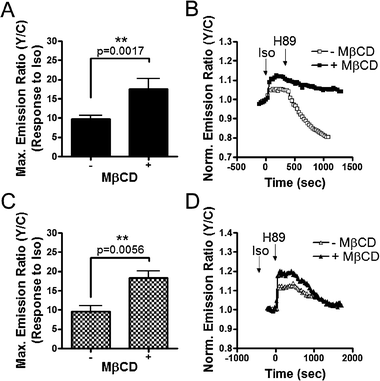 | ||
| Fig. 4 β2-Adrenergic receptor stimulated PKA activity is enhanced at the plasma membrane upon raft disruption. Response summary (A) and representative curves (B) of Lyn-AKAR4 in the presence (■) or absence (□) of 4 mM MβCD. Response summary (C) and representative curves (D) of AKAR4-Kras in the presence (▲) or absence (△) of 4 mM MβCD. All data in bar graphs are presented as average ± SEM. | ||
To investigate the effect of raft disruption on β2-AR stimulated PKA activity, we incubated HEK293 cells with MβCD, which depletes cholesterol thereby disrupting rafts. Cells were pretreated with 4 mM MβCD for 1 h, followed by stimulation with Iso and then PKA inhibition with H89. In the presence of MβCD, the Iso stimulated Lyn-AKAR4 response was significantly heightened. The 17.5 ± 2.8% (n = 7) emission ratio change represents a 78% increase over control cells. Following the plateau of the Iso-stimulated response, addition of H89 generated an emission ratio decrease of 13.7 ± 2.5% (n = 7) (Fig. 4A and B).
We then examined the effect of raft disruption on PKA activity in non-raft regions of the plasma membrane. In HEK293 cells expressing AKAR4-Kras, Iso stimulation yields a response of 9.7 ± 1.4% (n = 9). Subsequent addition of H89 led to a decrease in the emission ratio of 6.5 ± 1.1% (n = 9). Similarly, pretreatment with MβCD enhanced the Iso response by 88% over control cells, with an Iso-induced response of 18.3 ± 1.8% (n = 5), which was fully reversed by subsequent H89 treatment (Fig. 4C and D).
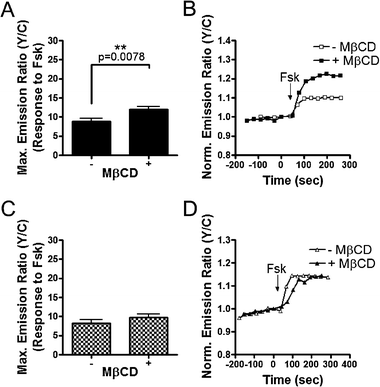
Next we tested if the enhancement of PKA activity by raft disruption is dependent on G-protein coupled receptors by directly stimulating ACs, thus bypassing the receptor. In HEK293 cells, the response of AKAR4-Kras to 50 μM Fsk was not sensitive to raft disruption with an emission ratio increase of 9.8 ± 0.9% (n = 24) observed in MβCD-treated cells compared to a response of 8.2 ± 0.9% (n = 25) in control cells (Fig. 5C and D). These results suggest that enhancement of PKA activity upon raft disruption is dependent on the involvement of the receptors, consistent with previous studies on β2-AR stimulated cAMP in membrane microdomains.18
 | ||
| Fig. 5 Effects of Raft Disruption on PKA Activity Stimulated by Adenylyl Cyclase Activation. Response summary (A) and representative curves (B) of Lyn-AKAR4 in the presence (■) or absence (□) of 4 mM MβCD. Response summary (C) and representative curves (D) of AKAR4-Kras in the presence (▲) or absence (△) of 4 mM MβCD. All data in bar graphs are presented as average ± SEM. | ||
Contrarily, when preincubated with MβCD, HEK293 cells expressing Lyn-AKAR4 displayed an increased response to Fsk, generating an emission ratio change of 12.0 ± 0.8% (n = 38) compared to an 8.8 ± 0.8% (n = 28) change in emission ratio in control cells (Fig. 5A and B). We speculate that the observed enhanced response of Lyn-AKAR4 upon raft disruption is due to differences in levels of phosphorylated probe prior to Fsk addition. The reporter responses likely reached saturation upon stimulation. In theory, if raft disruption diminished basal PKA activity and thereby decreased the amount of pre-phosphorylated Lyn-AKAR4 in the basal state, we would observe a larger response upon stimulation.
In summary, plasma membrane PKA activity stimulated by β-AR activation is enhanced upon raft disruption, suggesting that membrane rafts play a negative role in β-AR stimulation of PKA activity. This finding is consistent with previous studies investigating the effect of membrane rafts on cAMP accumulation.12,18 Furthermore, multiple lines of evidence suggest the existence of basal PKA activity in membrane rafts.
Membrane rafts have high levels of basal PKA activity
As the aforementioned data suggests that membrane rafts possess basal PKA activity, we set out to investigate PKA activity dynamics within plasma membrane microdomains of unstimulated cells by directly inhibiting PKA without stimulation. If membrane rafts indeed contain elevated levels of basal PKA activity, inhibition of PKA in unstimulated cells should lead to dephosphorylation of Lyn-AKAR4, seen as a decrease in the emission ratio. To test this, H89 was added to resting cells, allowing endogenous phosphatases to dephosphorylate AKAR. In HEK293 cells expressing Lyn-AKAR4, H89 addition generated a 12.0 ± 0.8% (n = 59) decrease in emission ratio (Fig. 6A and B). The large decrease below starting emission ratio levels indicates that an ample amount of Lyn-AKAR4 is phosphorylated in resting cells. In contrast, AKAR4-Kras only displayed a minimal change of 2.8 ± 0.5% (n = 26) in yellow-to-cyan emission ratio upon H89 addition (Fig. 6C and D). These results provide evidence for differential regulation of basal PKA activity in plasma membrane microdomains with high basal PKA activity specifically found in membrane rafts.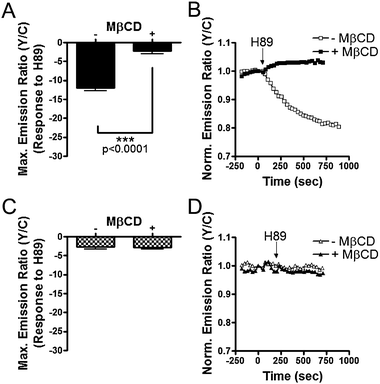 | ||
| Fig. 6 Varied Basal PKA Activity at the Plasma Membrane in HEK293 cells. Response summary (A) and representative curves (B) of Lyn-AKAR4 in the presence (■) or absence (□) of 4 mM MβCD. Response summary (C) and representative curves (D) of AKAR4-Kras in the presence (▲) or absence (△) of 4 mM MβCD. All data in bar graphs are presented as average ± SEM. | ||
To further test the importance of membrane rafts in regulating basal PKA activity, we used MβCD to deplete cholesterol and disrupt membrane rafts. Upon MβCD treatment, the H89 induced Lyn-AKAR4 response is significantly lessened, yielding a small 2.3 ± 0.7% (n = 31) decrease below baseline (Fig. 6A and B). As a control, we tested the response of AKAR4-Kras to H89 addition in MβCD treated cells. A 2.9 ± 0.4% (n = 31) decrease in emission ratio was observed and no statistically significant difference was found between untreated and MβCD treated cells (Fig. 6C and D). These data indicate that basal PKA activity observed here depends on raft integrity.
Proper localization of PKA contributes to high levels of basal kinase activity in membrane rafts
Having ascertained that membrane rafts specifically possess high levels of basal PKA activity, we examined the contribution made by localized PKA. We used mitochondrially-targeted AKAP derived peptides specific for type I and type II PKA, mitoAKB-RI and mitoAKB-RII, respectively, which can disrupt PKA localization to endogenous sites by relocalizing PKA to mitochondria.19 Previous studies utilizing these mitoAKB constructs show that they can be used to relocalize PKA that is present at the leading edge of migrating cells, demonstrating the usefulness of mitoAKBs to investigate the role of localized PKA.20We conducted initial experiments using individual mitoAKBs, in an attempt to decipher PKA isoform specific effects on raft-localized basal PKA activity. However, for both Lyn-AKAR4 and AKAR4-Kras, we observed no difference in the presence or absence of individual mitoAKB constructs indicative of redundant or compensatory mechanisms between type I and type II PKA. Consequently, we transfected both mitoAKB constructs to gather information on the combined effect caused by relocalization of type I and type II PKA. In the presence of both mitoAKB-RI and mitoAKB-RII, the Lyn-AKAR4 response to H89 treatment is reduced, showing an emission ratio decrease of 6.9 ± 0.5% (n = 13). As a control, mitoAKBs were expressed in AKAR4-Kras-containing cells and found to have no effect on the H89-induced response (Fig. 7). These results suggest that basal PKA activity in membrane rafts of resting cells depends on proper localization of PKA at the plasma membrane.
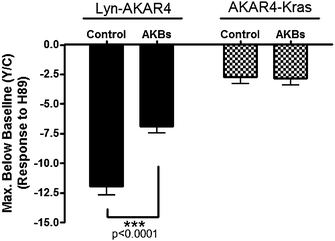 | ||
| Fig. 7 Relocalization of Endogenous PKA Affects Basal PKA Activity at the Plasma Membrane. Summary of Lyn-AKAR4 and AKAR4-Kras responses to H89 in the presence or absence of both mitoAKB-RI and mitoAKB-RII (AKBs). All data in bar graphs are presented as average ± SEM. | ||
Discussion
Our results suggest that membrane rafts play an important role in regulating PKA activity. On one hand, we observed enhanced PKA activity in response to β2-AR stimulation upon raft disruption. This finding adds to previous studies12,18 and suggests that the raft-disruption induced enhancement in cAMP accumulation can be translated into an increase in PKA activity. On the other hand, we show that membrane rafts contain basal PKA activity, providing another example of membrane compartmentalized kinase activities. This conclusion is based on multiple lines of evidence. First, H89 treatment led to a decrease in emission ratio in HEK293 cells expressing Lyn-AKAR4 but not those expressing Kras-AKAR4, suggesting basal PKA activity in membrane rafts resulted in phosphorylation of Lyn-AKAR4 prior to any stimulation. Secondly, upon raft disruption, Lyn-AKAR4 showed a heightened response to AC activation, whereas the response of AKAR4-Kras is unchanged. Consistent with the previous observation that enhanced activity upon raft disruption is specific to receptor stimulation, the variations in AC stimulated PKA activity are most likely due to differences in basal levels of biosensor phosphorylation resulting from raft-integrity dependent basal PKA activity. In fact, this reduced level of basal biosensor phosphorylation should also contribute to the enhanced PKA activity induced by Iso upon raft disruption (Fig. 4), in addition to the enhanced β2-AR-Gαs-AC associations.17 Thirdly, when HEK293 cells expressing Lyn-AKAR4 were stimulated with Iso and then treated with H89, the decrease generated by H89 treatment exceeded the stimulated Iso response, again pointing to the existence of basal PKA activity. Further experiments revealed that the basal PKA activity in membrane rafts depends on raft integrity and proper PKA localization. Additionally, it is possible that phosphatase activities are also differentially regulated in membrane microdomains. For instance, although cells expressing Lyn-AKAR4 or AKAR4-Kras responded similarly to Iso stimulation, the kinetics of H89-induced responses appeared to be faster in membrane rafts when compared to non-raft regions (Fig. 4). Future studies will examine the roles of membrane rafts in regulating phosphatases.Our study invokes an interesting question as to what role basally activated PKA plays in membrane rafts. Membrane raft-localized PKA is known to play an important role in tuning T cell responses,11 however it is not clear whether this functional effect involves basally activated PKA. Another intriguing possibility is that this active pool of PKA in rafts may play a role in exocytosis. Interestingly, exocytosis is known to depend on membrane rafts and PKA.2,4,5,21 It has been previously suggested that tonic PKA activity near the plasma membrane aids in the regulation of neurotransmitter release.22,23 Moreover, raft disruption decreases neurotransmitter releases in the model neurosecretory cell line, PC12.24 Future studies will investigate the importance of raft-localized basal PKA activity in the exocytotic processes of various cell types.
This study highlights the need to further understand how plasma membrane microdomains, such as membrane rafts, regulate signaling. However, these microdomains can be challenging to study as they are, by nature, small and highly dynamic. Commonly used methods, such as isolation of raft fractions viasucrose density gradients only provide static glimpses of the signaling components within different fractions. These biochemical approaches therefore need to be combined with live-cell based assays that are suitable for capturing activity dynamics of signaling molecules. Towards this goal, we and others have successfully created membrane microdomain targeted activity biosensors,16,18,25 which allowed us to track kinase activity dynamics in specific plasma membrane microdomains in living cells in order to achieve a better understanding of membrane compartmentalized signaling.
Experimental
Development and subcellular localization of AKAR4
AKAR4 was generated by removing truncated eCFP1-227 from AKAR3 in pcDNA3 vector and exchanged with the brighter CFP variant, Cerulean, via BamHI and SphI restriction sites. AKAR4 was targeted to membrane rafts by a 5′ myristoylation and palmitoylation lipid modification sequence (GCIKSKRKDK) derived from Lyn kinase and to non-rafts by a 3′ farnesylation lipid modification sequence (KKKKKKSKTKCVIM) derived from K-Ras kinase.Cell culture
HEK293 cells were cultured in DMEM supplemented with 10% FBS and 1% Pen-Strep and maintained at 37 °C with 5% CO2. For imaging, cells were plated onto sterilized glass cover slips in 35 mm dishes, transfected with calcium phosphate at 50–60% confluency, and then grown for approximately 24 h before imaging.Imaging
Cells were washed twice with Hanks’ Balanced Salt Solution (HBSS) and imaged in the dark at room temperature. When indicated, cells were treated with isoproterenol (Iso; Sigma), forskolin (Fsk; Calbiochem), and H89 (Sigma). Cholesterol depletion experiments were carried out by treating cells with 4 mM methyl-β-cyclodextrin (MβCD; Sigma) for 30 min at 37 °C before imaging. Images were acquired on an Axiovert 200M microscope (Carl Zeiss, Thornwood, NY) with a cooled charge-coupled device camera (Roper Scientific, Trenton, NJ) controlled by Metafluor 6.2 software (Molecular Devices, Downingtown, PA). Dual emission ratio imaging used a 420DF20 excitation filter, a 450DRLP dichroic mirror, and two emission filters (475DF40 for CFP and 535DF25 for YFP). Exposure time was 50–500 ms and images were acquired every 30 s. Background correction of the fluorescence images was performed by subtracting autofluorescence intensities of untransfected cells or regions of the imaging dish with no cells. Graph curves were normalized by setting the emission ratio before drug addition equal to one.Sucrose density gradient fractionation and western blot analysis
Membrane rafts and non-rafts were isolated from HEK293 cellsviasucrose density gradient fractionation using an adapted version of a previously described protocol.12Cells were lysed in 1 mL of TNE buffer (10 mM Tris, pH 7.4, 150 mM NaCl, 5 mM EDTA) containing 2 mM PMSF, 2 mM NaVO4, 2 mM NaF, 10 nM Calyculin A, and a protease inhibitor cocktail. Cell lysates were incubated on ice for 30 min, and then spun down for 5 min at 5000 RPM. The supernatant was then spun for 50 min at 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 RPM. The pelleted plasma membrane was then resuspended in 1 mL TNE buffer with 1% Triton-X 100 and incubated on ice for 1 h with periodic mixing. The membrane mixture was then diluted 1:1 with 85% sucrose and layered with 4 ml of 35% sucrose, then 1 mL of 5% sucrose, and finally 4.5 mL of TNE buffer. Ultracentrifugation was performed at 39000 x g for 18 h in a Beckman SW41-Ti rotor (Beckman Coulter, Fullerton, MA). All experimental steps were performed at 4 °C and using ice cold buffer. After ultracentrifugation, the top 3.5 mL was discarded and nine 825 μL fractions were collected, starting from the top of the gradient.
000 RPM. The pelleted plasma membrane was then resuspended in 1 mL TNE buffer with 1% Triton-X 100 and incubated on ice for 1 h with periodic mixing. The membrane mixture was then diluted 1:1 with 85% sucrose and layered with 4 ml of 35% sucrose, then 1 mL of 5% sucrose, and finally 4.5 mL of TNE buffer. Ultracentrifugation was performed at 39000 x g for 18 h in a Beckman SW41-Ti rotor (Beckman Coulter, Fullerton, MA). All experimental steps were performed at 4 °C and using ice cold buffer. After ultracentrifugation, the top 3.5 mL was discarded and nine 825 μL fractions were collected, starting from the top of the gradient.
Acknowledgements
We would like to thank Dr Xinxin Gao for help with sucrose density gradient fractionation. We would also like to thank Drs Susan S. Taylor, Chinten J. Lim, and Mark H. Ginsberg for providing the mitoAKB constructs. This work is funded by NIH R01 DK073368 and 3M (to J.Z.) and F31 GM087079 (to C.D.).References
- L. J. Pike, J. Lipid Res., 2003, 44, 655–667 CAS.
- D. A. Brown and E. London, Annu. Rev. Cell Dev. Biol., 1998, 14, 111–136 CrossRef CAS.
- R. G. Parton and A. A. Richards, Traffic, 2003, 4, 724–738 CrossRef CAS.
- L. J. Pike, Biochem. J., 2004, 378, 281–292 CrossRef CAS.
- C. Salaun, D. J. James and L. H. Chamberlain, Traffic, 2004, 5, 255–264 CrossRef.
- K. Simons and D. Toomre, Nat. Rev. Mol. Cell Biol., 2000, 1, 31–39 CrossRef CAS.
- B. S. Skalhegg and K. Tasken, Front. Biosci., 2000, 5, d678–93 CrossRef CAS.
- K. Tasken and E. M. Aandahl, Physiol. Rev., 2004, 84, 137–167 CrossRef CAS.
- M. L. Dell'Acqua and J. D. Scott, J. Biol. Chem., 1997, 272, 12881–12884 CrossRef CAS.
- D. L. Beene and J. D. Scott, Curr. Opin. Cell Biol., 2007, 19, 192–198 CrossRef CAS.
- A. Ruppelt, R. Mosenden, M. Gronholm, E. M. Aandahl, D. Tobin, C. R. Carlson, H. Abrahamsen, F. W. Herberg, O. Carpen and K. Tasken, J. Immunol., 2007, 179, 5159–5168 CAS.
- S. M. Pontier, Y. Percherancier, S. Galandrin, A. Breit, C. Gales and M. Bouvier, J. Biol. Chem., 2008, 283, 24659–24672 CrossRef CAS.
- M. D. Allen and J. Zhang, Biochem. Biophys. Res. Commun., 2006, 348, 716–721 CrossRef CAS.
- R. N. Day and M. W. Davidson, Chem. Soc. Rev., 2009, 38, 2887–2921 RSC.
- M. A. Rizzo, G. H. Springer, B. Granada and D. W. Piston, Nat. Biotechnol., 2004, 22, 445–449 CrossRef CAS.
- X. Gao and J. Zhang, Mol. Biol. Cell, 2008, 19, 4366–4373 CrossRef CAS.
- A. J. Morris and C. C. Malbon, Physiol. Rev., 1999, 79, 1373–1430 CAS.
- L. M. DiPilato and J. Zhang, Mol. BioSyst., 2009, 5, 832–837 RSC.
- L. L. Burns-Hamuro, Y. Ma, S. Kammerer, U. Reineke, C. Self, C. Cook, G. L. Olson, C. R. Cantor, A. Braun and S. S. Taylor, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4072–4077 CrossRef CAS.
- C. J. Lim, K. H. Kain, E. Tkachenko, L. E. Goldfinger, E. Gutierrez, M. D. Allen, A. Groisman, J. Zhang and M. H. Ginsberg, Mol. Biol. Cell, 2008, 19, 4930–4941 CrossRef CAS.
- S. Seino and T. Shibasaki, Physiol. Rev., 2005, 85, 1303–1342 CrossRef CAS.
- S. Hilfiker, A. J. Czernik, P. Greengard and G. J. Augustine, J. Physiol., 2001, 531, 141–146 CAS.
- T. Koga and M. Takahashi, Neurosci. Lett., 2004, 360, 145–148 CrossRef CAS.
- T. Lang, D. Bruns, D. Wenzel, D. Riedel, P. Holroyd, C. Thiele and R. Jahn, EMBO J., 2001, 20, 2202–2213 CrossRef CAS.
- J. Seong, S. Lu, M. Ouyang, H. Huang, J. Zhang, M. C. Frame and Y. Wang, Chem. Biol., 2009, 16, 48–57 CrossRef CAS.
Footnote |
| † This article is part of a themed issue of Molecular BioSystems on Post-translational modifications |
| This journal is © The Royal Society of Chemistry 2011 |