Predicting protein–protein interactions in the context of protein evolution
Anna C. F.
Lewis
a,
Ramazan
Saeed
b and
Charlotte M.
Deane
*c
aDepartment of Statistics and Systems Biology DTC, University of Oxford, UK
bDepartment of Statistics, University of Oxford, UK
cDepartment of Statistics, University of Oxford, UK. E-mail: deane@stats.ox.ac.uk
First published on 28th September 2009
Abstract
Here we review the methods for the prediction of protein interactions and the ideas in protein evolution that relate to them. The evolutionary assumptions implicit in many of the protein interaction prediction methods are elucidated. We draw attention to the caution needed in deploying certain evolutionary assumptions, in particular cross-organism transfer of interactions by sequence homology, and discuss the known issues in deriving interaction predictions from evidence of co-evolution. We also conject that there is evolutionary knowledge yet to be exploited in the prediction of interactions, in particular the heterogeneity of interactions, the increasing availability of interaction data from multiple species, and the models of protein interaction network growth.
 Anna C. F. Lewis | Anna Lewis is studying for her DPhil through the Systems Biology Doctoral Training Centre, and is supervised by Drs Charlotte Deane, Mason Porter and Nick Jones. Her thesis will be in the area of applied biological networks. Her undergraduate degree was in Physics and Philosophy at the University of Oxford. |
 Ramazan Saeed | Dr Ramazan Saeed completed his DPhil in 2009 in the Department of Statistics, University of Oxford, supervised by Dr Charlotte Deane. His thesis was on investigating protein structure and evolution through the protein interaction network. He was a student in the Life Sciences Interfaces Doctoral Training Centre. He did his undergraduate degree in software engineering at Manchester University, and has worked as a business consultant at Fujitsu Services. |
 Charlotte M. Deane | Dr Charlotte Deane is a University Lecturer in Statistics at Oxford. She is currently the Director of both the Systems Biology and the new industry focussed Systems Approaches to Biomedical Sciences Doctoral Training Centres (DTCs). She is also Deputy Director of the Doctoral Training Centre at Oxford. In addition she is a member of the management team for Oxford’s Centre for Integrative Systems Biology. All of her research is interdisciplinary and involves application of mathematical and computational techniques to problems across biology and biochemistry, in particular in protein structure, function and interaction. She aims to understand how proteins evolved their properties as well as to develop software to predict these properties. |
1. Introduction
Here we are interested in the physical interactions of proteins in cellular conditions. Proteins carry out their cellular functions through their concerted interactions with other proteins, and it is therefore very important to know which proteins interact with which. Recently, high-throughput assays that detect interactions have been developed. This has led to a continuous rapid expansion of available data.1–9† These assays have known problems associated with them, and have only been applied to a few species (see section ‘Errors in the Network’). It is thus important to be able to predict protein interactions, a problem which is closely tied to the issue of assessing the quality of the data that we do have. Evolutionary knowledge is of great relevance, and has been exploited by different interaction prediction methods (see section ‘Methods for Predicting Protein Interactions’).In this review we have two broad aims. The first is to outline those aspects of protein evolution which are of potential relevance in predicting protein interactions. Our understanding of protein evolution is thus a very broad one, covering sequence evolution, structural evolution, evolution of a protein’s context within and across genomes and location within the protein interaction network (PIN). Our second aim is to clearly state the evolutionary assumptions underlying some of the interaction prediction methods, and ask how accurate and general these assumptions are.
As a motivating example, consider the common practice of transferring protein interactions by orthology from an organism where we have a lot of protein interaction data (e.g. yeast), to one where we have considerably less (e.g. mouse). The assumption is that if proteins A and B interact in yeast, then orthologous proteins A′ and B′ in mouse probably also interact. So, below some threshold of sequence divergence, we can predict an interaction. The threshold at which we can do this with confidence has been shown to be far higher (70% sequence identity) than that which is used for transferring other properties such as function (16–30%),11 demonstrating the care needed in making use of evolutionary assumptions in making interaction predictions.
There is a vast literature on protein interaction networks, notably models for how they evolve, which has not fully engaged with the literature on the properties of individual proteins and how they evolve. As a second motivating example, consider that network evolution models have yet to be fully exploited for making protein interaction predictions.
This review is divided into three main sections. In the first, we summarise the protein interaction data we currently have, including the known problems associated with the different types of data. We discuss the methods that are used to assess the quality of protein interaction data sets and the ability of protein interaction prediction algorithms. The second section reviews some of the aspects of protein evolution most relevant to protein–protein interactions. We organise this literature around three broad themes: how protein interactions are thought to constrain evolution, the co-evolution of interacting proteins, and the evolution of the whole PIN. The third section reviews several different protein interaction prediction methods, and the extent to which they rely on and allow for evolutionary considerations. We conclude by discussing how the available evolutionary information has been utilised in predicting protein–protein interactions, whether this is always justified, and whether further applications of this information are possible.
2. Data and model assessment
2.1 Errors in the network
Despite the growth in protein interaction data, the information is not fully representative of the entire protein interaction network.12,13 Current interaction data are also plagued by a high rate of false positives, some of which may arise from experimental artefacts.14–16The two most common high throughput assays are Yeast Two-hybrid (Y2H) and tandem affinity purification (TAP). In the classical Y2H system, a bait is expressed as a DNA binding domain fusion and the prey expressed as a transcriptional activation domain fusion. The interaction is measured when the two domains join to form a transcriptor which activates a reporter gene.17 TAP methods infer an interaction when a bait protein is affinity captured from cell extracts by either polyclonal antibody or epitope tag and the associated interaction partner is identified, usually by mass spectrometric methods.18 A number of variations on both assays exist.19,20
Errors in both methodologies arise when the protein fusions can lead to restricted mobility and in some cases increase the affinity of a protein for certain targets.21 Both approaches are also very poor at identifying self-interactions.22 In Y2H the baits interact with each other and the preys interact with each other resulting in reduced concentrations of bait/prey interactions. In TAP assays no self interactions are reported due to the lack of untagged baits.
False positives in the Y2H approach arise as the bait and prey are over-expressed, often in a non-native cellular localisation.15 Consequently any observed interaction may not be present in the wild type of the cells where the concentrations are often significantly lower and where the bait and prey may not normally meet. Auto-activation can also lead to false positives as the reporter gene is activated by other factors, in some cases the bait protein itself.23 This shortcoming however has been addressed in more recent Y2H assays 1 where the individual fusions are tested to see if they cause auto-activation.24
TAP methods are also afflicted by false positives, particularly as a reported complex does not actually imply any direct physical interactions between the complex partners. This issue is addressed, but not resolved, by adopting a ‘spoke’ or ‘matrix’ model,25 where the spoke model directly pairs proteins with others in the complex and a matrix model assumes interactions between all.
There are several low throughput technologies that offer more accurate but far fewer interactions, such as crystallised complexes found in the protein data bank.26
The abundance of proteins has some effect on the number of interactions that they participate in, and experimental methods have been shown to be biased towards counting more interactions for abundant proteins.27 Fraser and Hirsh suggest that this relationship with expression is an intrinsic characteristic of yeast rather than an experimental bias.28
2.2 Assessing data quality and predictive accuracy
A number of methods have been developed to assess the error in protein interaction data. The simplest of such methods is to use a gold standard reference set and measure its overlap with a set of interactions and use that overlap as an indicator of the true positive rate.15,29,30 Another approach is to use biological features of proteins such as protein function, localisation and expression to assess how likely the interaction is to occur.16,31–33 A number of methods that simultaneously use such biological features in a combinatorial fashion have also been proposed.34–36 Sequence homology between two interacting proteins and their interologs has also been used to verify interactions.16,37 Graph based methods that use only the topology of the network to make an assessment have also been proposed.38–40 These methods can also be used for verifying the predictive ability of computationally predicted interactions. Most commonly however, interaction prediction methods are judged by using the reference methodology where an overlap between a true positive gold standard set and a true negative gold standard set is employed in a Receiver Operating Characteristic (ROC) analysis.41–43 A common way of obtaining such true positive gold-standard sets is to select interactions observed in multiple assays 25 or manually curated datasets.44,45 However, such sets do not exist for all species due to low experimental coverage and can lead to some degree of bias in results.37,46When using such assessment methodologies on computationally predicted interactions it may be that biased results are obtained due to the possible circular nature of the prediction method and the assessment methodology.
3. Protein interactions in the context of protein evolution
In this section we discuss the aspects of protein evolution relevant to the prediction of protein interactions. There are three main research areas that are of relevance here. The first investigates to what extent protein interactions constrain evolution. The second looks for evidence of co-evolution. These two areas investigate proteins at multiple scales: the proteins themselves, their domains, their interacting interfaces, their genomic context, their position within the PIN. The third area of relevance concerns models for the evolution of the whole PIN. This is a body of literature which started and is still to a large extent rooted in the physics and maths research communities. The motivation is that any patterns found are potentially exploitable in making interaction predictions.For a review of protein evolution in more general contexts see Pál et al.,47 and for a review of the evolution of protein interactions see Levy and Pereira-Leal.48
3.1 Constraints on evolution from interactions
Many models of evolution assume that the more constraints something is under, the slower its process of adaptation.49,50 Specifically in the context of protein evolution, Zuckerkandl proposed the notion of fitness density: the rate of evolution should be inversely proportional to the fraction of residues engaged in specific functions.51 We might therefore expect that (a) proteins with more interactions would evolve more slowly and (b) residues at interaction interfaces should evolve more slowly than other surface residues.Initial work appeared to confirm (a),52 but since then this claim has been challenged, on the basis that it relies on biases in interaction data,27 on confounding independent variables (notably expression rate53), is not particular to number of interactions but to other network features,54 and does not stand up to the data.32,55,56 A review of the methodologies used concluded that the number of translation events (which is well indicated by expression level, protein abundance and codon adaptation index), rather than the number or patterns of protein interactions, was the key determinant of evolutionary rate.57
Work based on structures of proteins crystallised together has been more conclusive with regard to (b): interface residues are more evolutionary conserved than other surface residues,58,59 although this effect is moderate. Considering an individual interface, some residues, known as hot spots, are thought to dominate the interaction binding energy,60,61 and these are found to be even more conserved than other interface residues.62 Comparing protein interfaces, we can distinguish two broad classes: transient and obligate, where the latter describes two proteins that are never found out of complex with each other.63Proteins involved in obligate interactions are more evolutionarily conserved than those involved in transient interactions, which are in turn more evolutionarily conserved than those not known to be involved in any interactions.64
3.2 Co-evolution
In a recent review of co-evolution Pazos and Valencia65 stress the necessity of distinguishing between co-evolution, the existence of mutual selective pressure inferred from similarity of evolutionary histories, and co-adaptation, the molecular mechanisms that would explain co-evolutionary changes. Evidence of co-adaptation would be needed to infer direct physical interactions. Not all cases of co-evolution will be from co-adaptation, due to confounding factors such as similar expression patterns or common function.The genomes of different organisms can be compared to give us information about likely functional association between proteins. If the same genes tend to occur as neighbours in multiple organisms, then we can infer functional association between them (e.g.ref. 66): if two proteins cannot perform their cellular function without each other, then when one is lost, there will be no evolutionary advantage to keeping the other, and they will be lost from the genome as a pair. Patterns of presence and absence of genes in different organisms, termed phylogenetic profiles, are the simplest clue of protein co-evolution. Similarly, profiles encoding the presence and absence of protein domains can be used to detect functional associations.67 Additional patterns in phylogenetic profiles, such as anti-correlation,68 and correlations between triplets of proteins69 can also give information about functional associations.
Interacting proteins are often transcribed as a single unit (operon) in bacteria, whereas in eukaryotes they tend to be co-regulated or regulated by the same protein (regulon). Studies have shown that the products of 63–75% of co-regulated genes tend to interact physically.70 This suggests that the regulation of proteins co-evolves with the proteins and their interactions.
It is also possible to compare the phylogenetic trees of proteins. The motivation for the approach is that the phylogenetic trees of, for example, ligands and their receptors are more similar than would be expected under the standard molecular clock hypothesis, which indicates some degree of co-evolution.71 This relationship is even more striking when the phylogenetic trees are built from the sequence of the interacting interfaces, rather than the whole protein.72 The same study shows that residues at the interface of obligate complexes tend to evolve slowly, allowing co-evolution of the partner interface, whereas transient interfaces tend to have an increased rate of residue substitution, leaving little evidence of correlated mutations across the interface.72 It is also possible to concentrate on the domains within proteins, and this can be used to infer which domains are responsible for a given interaction.73 As these approaches rely on generating reliable phylogenetic trees, they are well placed to take advantage of the growing amount of sequence information available.
A generalisation of this approach comes from the acknowledgement that, as proteins can interact with many different partners, considering only pairwise interactions will never give us the whole picture when it comes to protein co-evolution. Rather, this depends on all the different interactions a protein may be engaged in, so comparing proteins not only pair-wise, but against all other proteins can give a better idea of the co-evolution of a given pair.74
Generalisations in a different direction come from investigating the similarities of whole protein structures, not just sequences. In Williams and Lovell75 the authors offer an integrated view of sequence and structural divergence, claiming that both co-evolution following sequence changes and structural accommodation of non-compensated substitutions can be accommodated in the same framework. The methods above assess similarity based on nucleotide substitutions, but there is some evidence that the role of insertions and deletions of short stretches of nucleotide (indels) is important. Indels are particularly common on the surfaces of proteins (e.g.ref. 76), and are thus suspected to play a large role in ‘re-wiring’ the protein interaction network. The importance of indels has been highlighted by a study that finds that proteins that participate in indel alignments have high centrality measures within the network.77
It is not clear under what circumstances we can infer direct physical interactions from the observation of co-evolution.65 In Hakes et al.78 the authors argue that there is no evidence that co-evolution is due to co-adaptation, arguing for the importance of common evolutionary forces, notably expression levels, as responsible for the co-evolution. In Kann et al.79 it is argued that there is some evidence for co-adaptation, alongside more general evolutionary forces. A better understanding of the molecular mechanisms underpinning compensatory changes will help distinguish these correlations, and perhaps work out the conditions under which co-adaptation can be inferred. A better understanding of how other protein features can influence functional association will be vital.
3.3 Evolution of the PIN
There have been many models proposed for the growth of the protein interaction network. A recent review of this area is found in Stumpf et al.14The field was re-vitalised in 1999 with the proposal of the preferential attachment model of Barabasi and Albert,80 which picked up on ideas dating back to the 1950s.81,82 It was observed that, if new nodes attached themselves to old nodes with a probability proportional to the number of interactions of the old nodes, a power law distribution of node degree would result (often called a scale free distribution in the literature). Such distributions were, at the time, being found in many different types of network,83 including PINs.84 This would fit the observation that older proteins have more interactions.85 The problem with the model of growth by preferential attachment as a model of PIN growth is that it does not obviously correspond to a known biological mechanism, with the possible exception of horizontal gene transfer in the prokaryotic case.
What are the likely factors underlying network evolution? Errors in replication can result in a change in copy number of proteins, from individual genes being duplicated or lost (reviewed in e.g. Zhang86), to the whole genome being duplicated (reviewed in e.g. Kasahara,87 and Scannell et al.88). After a geneduplication event, divergence of function is possible. There are two main competing models for such divergence: sub-functionalisation (partitioning of ancestral function between gene duplicates) and neo-functionalisation (the de novo acquisition of function by one duplicate).86Geneduplication was hypothesised to be disadvantageous in complexes in particular, and evidence for fewer single geneduplication events in gene families encoding complexes has been found in support of this.89
Many PIN evolution models have been based on this idea of duplication followed by divergence90,91 (see Fig. 1(a)). There are many different variants, but all share in common that a node is selected to be copied (duplication), some fraction of the nodes links are replicated in the duplicated node, and some more added (divergence). Recently, models which allow for whole genome duplication events have also been proposed.92 Both preferential-attachment and duplication-divergence models match the power law degree distributions found in PINs, and also both match the data in that nodes of high degree tend to connect to nodes of low degree (node disassortativity).93Duplication-divergence models generate some level of hierarchical modularity (whereby small densely connection groups of proteins, termed modules, join together into larger modules), though not as much as observed in the data.93
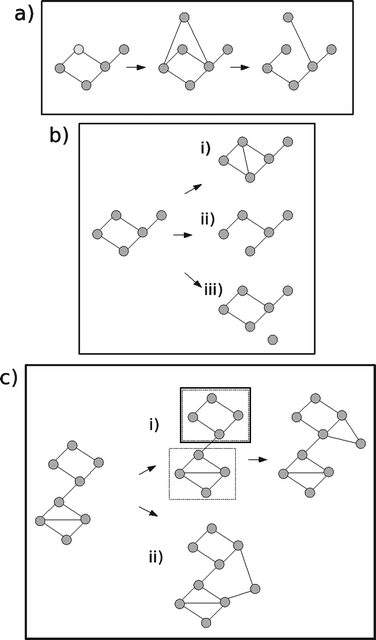 | ||
| Fig. 1 PIN evolution models. (a) Duplication-divergence. A node is chosen to be copied. The new node is given links to the same set of nodes as the chosen node (duplication). Some fraction of links are then lost (divergence). (b) Asymmetric gain and loss of interactions. Three move types are possible. (i) Addition of a link. A link is made between one node chosen at random and another chosen proportional to node degree. (ii) Removal of link. A protein is chosen uniformly at random, and one of its links is chosen uniformly at random to be removed. (iii) A new node is added with zero links. The probabilities of these three moves are chosen such that the mean node degree stays the same and the network grows at some empirically inferred rate. (c) Crystal growth model. After an initial seeding phase, either (i) modules are computed, one is chosen, and a new node is added to this chosen module or (ii) a new node is put into its own module, and connects to other modules which have few links (anti-preferential attachment rule). | ||
Despite the successes of duplication-divergence models in capturing many aspects of the empirical PIN, it has been claimed that geneduplication and divergence may in fact have played only a limited role in the evolution of PINs, as the dynamics of the gain and loss of individual interactions is thought to happen at a much faster time-scale.94,95 In Berg et al.,96 the authors propose a model based on the addition and loss of individual links. They found that the rate of addition and loss of links depends on the number of connections of both interacting partners asymmetrically, which is to be expected because when a new link is formed, typically only one node undergoes a mutation with the other remaining unchanged (see Fig. 1(b)). By building a stochastic model based on these observations they are able to match the degree distribution and node disassortativity found in the data.
In a separate critique of the popular models, Kim and Marcotte93 investigate the age-dependent evolution of proteins, and claim this cannot be accounted for by duplication-divergence or preferential attachment models. Instead, they propose a crystal growth model based on (a) interaction probability increasing with availability of unoccupied interaction surface, (b) tightly connected groups of proteins developing as the network grows, (c) once a protein is committed to such a group, further connections tend to be made with other members of that group (see Fig. 1(c)). In this model, proteins are more likely to link to proteins of a similar age, as observed in real PINs. The model uses network modularity as a key idea in PIN evolution, an idea that is not that well explored elsewhere (though see Li and Maini97 for an abstract model of network growth based on modules).
It is not clear how best to assess different models of network evolution and growth, though it is clear that these have matured significantly beyond matching degree distributions.93,98–100
As more and more PIN data become available, models that use and explain the patterns of interaction found in different organisms will become increasingly prevalent. The alignment of PINs is a rapidly growing field (for examples see Guo and Hartemink,101 Liao et al.,102 and Zaslavskiy et al.,103 for an early review see Sharan and Ideker104). Such alignments can enable the location of evolutionarily conserved sets of interactions, which is of great potential in the field of predicting interactions.
4. Methods for predicting protein interactions
Protein interactions can be predicted computationally by employing various sources of information, including protein features, evolutionary knowledge and network information. In this section we explain the ideas behind many of the most popular interaction-prediction methods. We do not aim to give any algorithmic details, but to highlight the evolutionary ideas used. Fig. 2 illustrates some of the methods discussed.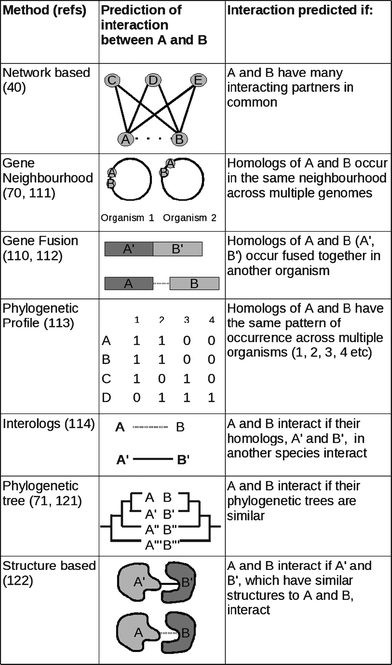 | ||
| Fig. 2 Different interaction prediction methods. | ||
The main category of prediction methods we do not cover are those based on machine learning approaches, as these tend not to be underpinned by biological ideas, explicit or implicit. Two reviews of protein interaction prediction which include these approaches are given in Shoemaker and Panchenko105 and Pitre et al.106
4.1 Network based methods
An early example of using the topology of the network to assess the quality of interactions was proposed by Saito et al.38 who suggested that the greater the number of isolated interaction partners two proteins had, the more unreliable the interaction. This relationship has been used to predict putative protein interactions in an existing network.40 In particular they were able to rank the reliability of a predicted interaction using a measure of the shortest alternative pathway between the two interactors. Chen et al.107 also exploited the high level of clustering in the network to predict interactions based upon triangular motifs in the network. This work demonstrated that there was information encoded in triangles of interactions in the PIN, which suggests that to understand the evolution of the PIN as a whole it is necessary to look beyond pairwise relationships. This statement has been surprisingly hard to demonstrate.107Clauset et al.108 used the hierarchical structure (that is, the structures present at different scales) of networks to predict ‘missing’ links between nodes. Interaction probabilities were assigned between hierarchical groups and a pair of nodes were suggested to be possibly linked if they possessed a high average probability of connection within these hierarchical groups, but were observed as unconnected. Whilst this method was tested on various different networks, including a metabolic network, it has yet to be applied to PINs. It could potentially be successful, if PINs are found to have biologically meaningful hierarchical structure (in Yook et al.109 the authors claim this is the case). This in turn rests on our understanding of the evolution of hierarchical modularity.
A common shortcoming of such network based methodologies is that the results can vary depending on the state of the network, which in some species is sparse and error prone.
4.2 Genome based methods
Genomic information methods rely on the context of the gene/protein in an organism’s genome , and the context of its homologs in other genomes . For a review see Marcotte et al.110Here we assume that there is enough selective pressure that if two proteins interact to perform their cellular function, and one is lost, the other protein will also be lost (for example, this would be the case for some complexes). In the case of horizontal gene transfer in bacteria, genes will only be kept if they are transferred with other genes that they need to interact with to perform a fitness enhancing function.
4.3 Sequence based methods
Matthews et al.115 exploited this principle to predict novel interactions in the worm C. elegans based upon homology inference from high throughput assays of S. cereivisae. This method was later developed by Jonsson and Bates42 to include a scoring scheme based upon the different experimental sets that an interaction was sourced from.
Interologs are often used in species where there is little PIN data but much genetic data, for example in mouse and human.42,116 A comparison of the PINs from different organisms found that such inferences are surprisingly inaccurate.117 Whereas the degree of sequence similarity needed before an interolog is inferred is usually in the range 16–30%,115,118 this study showed that a much larger degree of sequence similarity, about 70%, is required to make an accurate prediction using an interolog. This and more recent studies have shown that paralogous interactions tend to be more conserved than orthologous interactions.11,37,119
The evolutionary assumption behind interologs is clear: if the two proteins have not diverged considerably in sequence from their common ancestor, then the interaction is likely to have been preserved. However, it is often claimed that the small amount of molecular divergence between proteins found in multiple species is not enough to explain the phenotypic divergence between species.120 Re-wiring of the networks, caused by small changes in protein sequence, is hypothesised to make the difference. If this is right, then it is a further reason to use interologs with utmost care.
As discussed above in section 3.2, the main issue is in the inference from functional association to physical interaction (from co-evolution to co-adaptation).
4.4 Structure based methods
A further category of interaction prediction methods consists of approaches that exploit structural similarities and make predictions based upon structural models.In analogy with sequence based interologs, structure based interologs have been investigated. Aloy et al.123 found that proteins with the same folds or structural domains tended to participate in similar interactions if the sequence identity of the proteins was above approximately 30%. Below this percentage, there is a twilight zone where proteins may or may not share similar interactions. The evolutionary assumptions and potential problems are similar to those for sequence based interologs (see above). Their advantage is that they are more accurate, their disadvantage is the relative paucity of structural information.
Protein-docking methods,124,125 where two structural proteins are rigidly combined and then refined can also be used to predict interactions. However, due to the computational cost of such methods they are in general more useful for providing information on the interacting interface of two structurally defined subunits.126 Other methods predict interactions based on surface patch comparison127 and oligomeric protein structure networks.128 Carugo and Franzot127 divided atoms on the surface of each protein into small partially overlapping sets (patches). The shapes of each pair of patches belonging to different proteins were compared, and a statistical analysis of the shape complementarity values was used to discriminate interacting and non-interacting protein pairs with an accuracy of up to 80%. Brinda and Vishveshwara128 attempted to understand the factors involved in protein interactions by analysing interactions between amino acids based on the number of non-covalent bonds, which are known to play a role in mediating protein interactions.129
One lesson from the field of protein docking in general is that supplementing physics and chemistry considerations with information deduced from sequence and structural databases can improve predictions greatly.130 The use of these databases rests on the assumption that similar structures imply similar interactions because the proteins are related to each other through evolution.
4.5 Domain based methods
A range of methods have been devised that attempt to predict which of the domains in a protein interact. The methods annotate protein sequences with domains defined by Pfam, SCOP, CDD and other domain databases.131–133Association methods are a group of prediction methods that look for blocks of sequence or structural motifs that distinguish interacting proteins from non-interacting proteins. In one such study, Sprinzak and Margalit134 looked for sequence domains that were found to interact more often than expected by chance. They used such domains as signatures to predict new interactions.
A problem with association models is that they only consider one domain at a time and ignore the affect of other domains on the interaction. This was addressed by Deng et al.,135 who estimated the probabilities of interactions between every pair of domains, and used these to predict interactions between proteins. Rare interactions between two domains can be missed by this method. To compensate for this, Riley et al.136 developed measures based on the reduction in the likelihood of the protein–protein interactions network, caused by disallowing a given domain–domain interaction. This can give some indication of which domain–domain interaction is more likely to be responsible for a given protein–protein interaction.
In all these approaches domains are assumed to interact independently, though this can depend on other domains within a protein pair, and remains a severe limitation of these methods.
These methods could potentially be powerful in predicting whole PINs of organisms for which interaction data is not available.106 This is because surprisingly few domains have been duplicated and recombined to form proteins across the tree of life: 50% of domain structure annotations in each organism are to fewer than 200 domain families common to all kingdoms. There is cause for caution here, however, as we need to ascertain that domain interactions are not organism specific, remembering that domain combinations tend to be specific to organisms.137 In Basu et al.,138 the authors demonstrate both that domains that occur in diverse domain architectures tend to have more interactions, and that which domains end up in diverse architectures is organism specific.
5. Conclusions
There are two main areas where caution is needed in inferring protein interactions from evolutionary assumptions. The first is the use of interologs. There is wide-spread belief in the bioinformatics community that transfer of biological function, including protein interactions, should be possible from sequence homology. The evolutionary assumption is that close sequence similarity implies little functional divergence. This fundamental assumption has been bought under suspicion in the case of protein interactions.11 The second is separating out the effects of actual molecular co-adaptation from the observation of co-evolution.Key to the reliable use of interologs and co-evolution will be a better understanding of the molecular mechanisms underpinning the evolution of interactions. There is evolutionary knowledge that has yet to be exploited in this regard, notably the large documented differences between transient and obligate interactions. These differences can potentially be detected on the basis of sequence alone.139 An investigation of the different sequence cut-offs that should be employed for interolog prediction for transient and obligate interactions would be a useful starting point.
The theories that state that indels have a key role to play in the re-wiring of the network could be tested, and if found to hold, methods which include the effect of indels could be used both in predicting interologs and for separating out co-adaptation from co-evolution.
Another step for improving interolog and co-evolution based predictions is to make use of the comparative PIN data that is now becoming available. Alignments of entire PINs, rather than just pairs of proteins, can give us additional information as to when an interolog inference is acceptable. If proteins A, B and C all interact in species S, and proteins A′ and B′ and A′ and C′ interact in species S′, then we have better evidence to predict that B′ and C′ also interact.
There is potential for models of network evolution to be used in the prediction of protein interactions. At present, we have no clear consensus as to which are the good models of network evolution. It is clear that there is evolutionary information that can be incorporated into these models to make them more realistic. For example, no model proposed, to our knowledge, distinguishes between transient and obligate interactions, which is perhaps surprising given the known differences between the evolution of these different interaction types. Good models of PIN growth, which have been assessed against multiple datasets, will be helpful in predicting new interactions. These models could be used to generate ensembles of interaction networks matching the observed statistics of the empirical PIN. The frequency with which a given pair of proteins interact in the ensemble can be used to predict likely interactors,108 although this has yet to be put into practice.
An important observation is that it is not easy to assess the relative success of different prediction methods. It is likely that different methods are more successful on certain types of proteins, which would be related to the different evolutionary assumptions underlying the different methods. Through direct comparison of which methods do best on which proteins, interaction prediction could be tailored to what else we know about the proteins involved.
Glossary
PIN, Protein interaction network
Proteins are nodes of the network, with experimentally determined interactions between them as links.ROC curves
In a Receiver Operating Characteristic Curve, the sensitivity of a binary classifier is plotted against (1 − specificity), for different values of the discrimination parameter. The larger the area under the ROC curve for a binary classifier, the better that classifier is.Loops
Proteins have structural loops, secondary structure elements that connect together alpha-helices and beta-sheetsDomains
Proteins are made up of building blocks known as domains. These can be defined on the basis of sequence, structure or function.Indels
Insertions or deletions of short chains of amino acid. These are detected through sequence alignments.Homologs
Homologs are proteins which are believed to share characteristics because of shared ancestry. They are usually detected through sequence alignment. Proteins that are homologs due to a speciation events are referred to as orthologs. Proteins that are homologs due to a geneduplication event are known as paralogs.Interologs
Interologs are predicted homologous interactions: if proteins A and B interact in species S, then proteins A′ and B′ in species S′, which are orthologs of A and B, are predicted to interact.Obligate and transient interactions
Proteins that must interact in order to carry out their cellular role are said to partake in an obligate interaction. Other interactions are termed transient interactions.Acknowledgements
We would like to thank Mason Porter, Rhodri Saunders, Sumeet Agarwal, Rebecca Hamer, Yoon-Joo Choi, Cheung Li and Mirielle Gomes for their helpful comments. A. C. F. Lewis thanks the Systems Biology DTC for her funding.References
- H. Yu, P. Braun, M. A. Yildirim, I. Lemmens, K. Venkatesan, J. Sahalie, T. Hirozane-Kishikawa, F. Gebreab, N. Li, N. Simonis, T. Hao, J. F. Rual, A. Dricot, A. Vazquez, R. R. Murray, C. Simon, L. Tardivo, S. Tam, N. Svrzikapa, C. Fan, A. S. de Smet, A. Motyl, M. E. Hudson, J. Park, X. Xin, M. E. Cusick, T. Moore, C. Boone, M. Snyder, F. P. Roth, A. L. Barabási, J. Tavernier, D. E. Hill and M. Vidal, Science, 2008, 322, 104–110 CrossRef CAS.
- K. Tarassov, V. Messier, C. R. Landry, S. Radinovic, M. M. Serna Molina, I. Shames, Y. Malitskaya, J. Vogel, H. Bussey and S. W. Michnick, Science, 2008, 320, 1465–1470 CrossRef CAS.
- S. R. Collins, P. Kemmeren, X. C. Zhao, J. F. Greenblatt, F. Spencer, F. C. Holstege, J. S. Weissman and N. J. Krogan, Mol. Cell. Proteomics, 2006, 6, 439–450 CrossRef.
- N. J. Krogan, G. Cagney, H. Yu, G. Zhong, X. Guo, A. Ignatchenko, J. Li, S. Pu, N. Datta, A. P. Tikuisis, T. Punna, J. M. Peregrín-Alvarez, M. Shales, X. Zhang, M. Davey, M. D. Robinson, A. Paccanaro, J. E. Bray, A. Sheung, B. Beattie, D. P. Richards, V. Canadien, A. Lalev, F. Mena, P. Wong, A. Starostine, M. M. Canete, J. Vlasblom, S. Wu, C. Orsi, S. R. Collins, S. Chandran, R. Haw, J. J. Rilstone, K. Gandi, N. J. Thompson, G. Musso, P. St Onge, S. Ghanny, M. H. Lam, G. Butland, A. M. Altaf-Ul, S. Kanaya, A. Shilatifard, E. O’Shea, J. S. Weissman, C. J. Ingles, T. R. Hughes, J. Parkinson, M. Gerstein, S. J. Wodak, A. Emili and J. F. Greenblatt, Nature, 2006, 440, 637–643 CrossRef CAS.
- A. C. Gavin, P. Aloy, P. Grandi, R. Krause, M. Boesche, M. Marzioch, C. Rau, L. J. Jensen, S. Bastuck, B. Dümpelfeld, A. Edelmann, M. A. Heurtier, V. man, C. Hoefert, K. Klein, M. Hudak, A. M. Michon, M. Schelder, M. Schirle, M. Remor, T. Rudi, S. Hooper, A. Bauer, T. Bouwmeester, G. Casari, G. Drewes, G. Neubauer, J. M. Rick, B. Kuster, P. Bork, R. B. Russell and G. Superti-Furga, Nature, 2006, 440, 631–636 CrossRef CAS.
- A. Gavin, M. Bosche, R. Krause and P. Grandi, Nature, 2002, 415, 141–147 CrossRef CAS.
- Y. Ho, A. Gruhler and A. Heilbut, Nature, 2002, 415, 180–183 CrossRef CAS.
- T. Ito, T. Chiba, R. Ozawa, M. Yoshida, M. Hattori and Y. Sakaki, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 4569–4574 CrossRef CAS.
- P. Uetz, L. Giot, T. A. Cagney and G. Mansfield, Nature, 2000, 403, 623–627 CrossRef CAS.
- G. D. Bader, M. P. Cary and C. Sander, Nucleic Acids Res., 2006, 34, D504–D506 CrossRef CAS.
- S. Mika and B. Rost, PLoS Comput. Biol., 2006, 2, 0698–0709 Search PubMed.
- G. T. Hart, A. K. Ramani and E. M. Marcotte, Genome Biol., 2006, 7, 120 CrossRef.
- C. Tucker, J. Gera and P. Uetz, Trends Cell Biol., 2001, 11, 102–106 CrossRef CAS.
- M. P. Stumpf, W. P. Kelly, T. Thorne and C. Wiuf, Trends Ecol. Evol., 2007, 22, 366–373 CrossRef.
- C. von Mering, R. Krause, B. Snel, M. Cornell, S. G. Oliver, S. Fields and P. Bork, Nature, 2002, 417, 399–403 CrossRef CAS.
- C. Deane, L. Salwinski, L. Xenarios and D. Eisenberg, Mol. Cell. Proteomics, 2002, 1, 349–356 CrossRef CAS.
- S. Fields and O. Song, Nature, 1989, 340, 245–246 CrossRef CAS.
- O. Puig, F. Caspary, G. Rigaut, B. Rutz, E. Bouveret, E. Bragado-Nilsson, M. Wilm and B. Séraphin, Methods, 2001, 24, 218–229 CrossRef CAS.
- A. Brückner, C. Polge, N. Lentze, D. Auerbach and U. Schlattner, Int. J. Mol. Sci., 2009, 10, 2763–2788 Search PubMed.
- M. O. Collins and J. S. Choudhary, Curr. Opin. Biotechnol., 2008, 19, 324–330 CrossRef CAS.
- J. Mackay, M. Sunde, J. Lowry, M. Crossley and J. Matthews, Trends Biochem. Sci., 2007, 32, 530–531 CrossRef CAS.
- T. A. Gibson and D. S. Goldberg, PLoS Comput. Biol., 2009, 5, 1–11 Search PubMed.
- V. Schachter, Comput. Proteomics Suppl., 2002, 32, S16–S27 Search PubMed.
- P. O. Vidalain, M. Boxem, H. Ge, S. Li and M. Vidal, Methods, 2004, 32, 363–370 CrossRef CAS.
- G. D. Bader and C. W. Hogue, Nat. Biotechnol., 2002, 20, 991–997 CrossRef CAS.
- B. A. Shoemaker and A. R. Panchenko, PLoS: Comput. Biol., 2007, 3, 337–334 Search PubMed.
- J. D. Bloom and C. Adami, BMC Evol. Biol., 2003, 3, 21 CrossRef.
- H. Fraser and A. Hirsh, BMC Evol. Biol., 2004, 4, 13 CrossRef.
- A. Edwards, B. Kus, R. Jansen, D. Greenbaum, J. Greenblatt and M. Gerstein, Trends Genet., 2002, 18, 529–536 CrossRef CAS.
- P. D’haeseleer and G. Church, Proc. IEEE Comput. Soc. Bioinformatics Conf., 2004, 216–223 Search PubMed.
- B. Schwikowski, P. Uetz and S. Fields, Nat. Biotechnol., 2000, 18, 1257–1261 CrossRef CAS.
- R. Saeed and C. M. Deane, BMC Bioinformatics, 2006, 7, 128 CrossRef.
- M. Deng, F. Sun and T. Chen, Pacific Symp. Biocomput., 2003, 140–151 Search PubMed.
- J. S. Bader, A. Chaudhuri, J. M. Rothberg and J. Chant, Nat. Biotechnol., 2004, 22, 78–85 CrossRef CAS.
- R. Sharan, S. Suthram, R. Kelley, T. Kuhn, S. McCuine, P. Uetz, T. Sittler, R. Karp and T. Ideker, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1974–1979 CrossRef CAS.
- X. Lin, M. Liu and X. W. Chen, BMC Bioinformatics, 2009, 10(Suppl 4) Search PubMed.
- R. Saeed and C. Deane, Bioinformatics, 2008, 24, 689–695 CrossRef CAS.
- R. Saito, H. Suzuki and Y. Hayashizaki, Nucleic Acids Res., 2002, 30, 1163–1168 CrossRef CAS.
- R. Saito, H. Suzuki and Y. Hayashizaki, Bioinformatics, 2003, 19, 756–763 CrossRef CAS.
- J. Chen, W. Hsu, M. L. Lee and S. K. Ng, Bioinformatics, 2006, 22, 1998–2004 CrossRef CAS.
- T. Fawcett, Pattern Recogn. Lett., 2006, 27, 861–874 CrossRef.
- P. F. Jonsson and P. A. Bates, Bioinformatics, 2006, 22, 2291–2297 CrossRef CAS.
- A. Patil and H. Nakamura, BMC Bioinformatics, 2005, 6 Search PubMed.
- U. Güldener, M. Münsterkötter, G. Kastenmüller, N. Strack, J. van Helden, C. Lemer, J. Richelles, S. Wodak, J. García-Martínez, J. Pérez-Ortín, H. Michael, A. Kaps, E. Talla, B. Dujon, B. André, J. Souciet, J. De Montigny, E. Bon, C. Gaillardin and H. Mewes, Nucleic Acids Res., 2004, 33, D364–D368 CrossRef.
- T. Reguly, A. Breitkreutz, L. Boucher, B. J. Breitkreutz, G. C. Hon, C. L. Myers, A. Parsons, H. Friesen, R. Oughtred, A. Tong, C. Stark, Y. Ho, D. Botstein, B. Andrews, C. Boone, O. G. Troyanskya, T. Ideker, K. Dolinski, N. N. Batada and M. Tyers, J. Biol., 2006, 5, 11 CrossRef.
- A. Ben-Hur and W. S. Noble, BMC Bioinformatics, 2006, 7(suppl 1), S2 CrossRef.
- C. Pál, B. Papp and M. J. Lercher, Nat. Rev. Genet., 2006, 7, 337–348 CrossRef CAS.
- E. D. Levy and J. B. Pereira-Leal, Curr. Opin. Struct. Biol., 2008, 18, 349–357 CrossRef CAS.
- R. Fisher, The Genetical Theory of Natural Selection, Dover, New York, 1958 Search PubMed.
- H. A. Orr, Nat. Rev. Genet., 2005, 6, 119–127 CrossRef CAS.
- E. Zuckerkandl, J. Mol. Evol., 1976, 7, 167–183 CrossRef CAS.
- H. B. Fraser, A. E. Hirsh, L. M. Steinmetz, C. Scharfe and M. W. Feldman, Science, 2002, 296, 750–752 CrossRef CAS.
- I. Agrafioti, J. Swire, J. Abbott, D. Huntley, S. Butcher and M. P. Stumpf, BMC Evol. Biol., 2005, 5, 23 CrossRef.
- M. W. Hahn and A. D. Kern, Mol. Biol. Evol., 2005, 22, 803–806 CrossRef CAS.
- M. W. Hahn, G. C. Conant and A. Wagner, J. Mol. Evol., 2004, 58, 203–211 CrossRef CAS.
- I. K. Jordan, Y. I. Wolf and E. V. Koonin, BMC Evol. Biol., 2003, 3 Search PubMed.
- D. A. Drummond, A. Raval and C. O. Wilke, Mol. Biol. Evol., 2005, 23, 327–337 CrossRef.
- D. R. Caffrey, S. Somaroo, J. D. Hughes, J. Mintseris and E. S. Huang, Protein Sci., 2004, 13, 190–202 CrossRef CAS.
- W. S. Valdar and J. M. Thornton, Proteins: Struct., Funct., Genet., 2001, 42, 108–124 CrossRef CAS.
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS.
- I. S. Moreira, P. A. Fernandes and M. J. Ramos, Proteins: Struct., Funct., Bioinf., 2007, 68, 803–812 Search PubMed.
- O. Keskin, B. Ma and R. Nussinov, J. Mol. Biol., 2005, 345, 1281–1294 CrossRef CAS.
- I. M. Nooren and J. M. Thornton, EMBO J., 2003, 22, 3486–3492 CrossRef CAS.
- S. A. Teichmann, J. Mol. Biol., 2002, 324, 399–407 CrossRef CAS.
- F. Pazos and A. Valencia, EMBO J., 2008, 27, 2648–2655 CrossRef CAS.
- S. V. Date and E. M. Marcotte, Nat. Biotechnol., 2003, 21, 1055–1062 CrossRef CAS.
- P. Pagel, P. Wong and D. Frishman, J. Mol. Biol., 2004, 344, 1331–1346 CrossRef CAS.
- E. Morett, J. O. Korbel, E. Rajan, G. Saab-Rincon, L. Olvera, M. Olvera, S. Schmidt, B. Snel and P. Bork, Nat. Biotechnol., 2003, 21, 790–795 CrossRef CAS.
- P. M. Bowers, S. J. Cokus, D. Eisenberg and T. O. Yeates, Science, 2004, 306, 2246–2249 CrossRef CAS.
- M. Huynen, B. Snel, W. Lathe and P. Bork, Genome Res., 2000, 10, 1204–1210 CrossRef CAS.
- F. Pazos and A. Valencia, Protein Eng., Des. Sel., 2001, 14, 609–614 CrossRef CAS.
- J. Mintseris and Z. Weng, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10930–10935 CrossRef CAS.
- R. Jothi, P. F. Cherukuri, A. Tasneem and T. M. Przytycka, J. Mol. Biol., 2006, 362, 861–875 CrossRef CAS.
- D. Juan, F. Pazos and A. Valencia, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 934–939 CrossRef CAS.
- S. G. G. Williams and S. C. C. Lovell, Mol. Biol. Evol., 2009, 26, 1055–1065 CrossRef CAS.
- S. A. Benner, M. A. Cohen and G. H. Gonnet, J. Mol. Biol., 1993, 229, 1065–1082 CrossRef CAS.
- F. Hormozdiari, R. Salari, M. Hsing, A. Schönhuth, S. K. Chan, S. C. Sahinalp and A. Cherkasov, J. Comput. Biol., 2009, 16, 159–167 CrossRef CAS.
- L. Hakes, S. C. Lovell, S. G. Oliver and D. L. Robertson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7999–8004 CrossRef CAS.
- M. G. Kann, B. A. Shoemaker, A. R. Panchenko and T. M. Przytycka, J. Mol. Biol., 2009, 385, 91–98 CrossRef CAS.
- A. L. Barabasi and R. Albert, Science, 1999, 286, 509–512 CrossRef.
- H. A. Simon, Biometrika, 1955, 42, 425–440.
- D. J. d. S. Price, J. Am. Soc. Inf. Sci., 1976, 27, 292–306 CrossRef.
- M. E. J. Newman, SIAM Rev., 2003, 45, 167–256 CrossRef.
- E. Eisenberg and E. Levanon, Phys. Rev. Lett., 2003, 91, 138701 CrossRef.
- S. Wuchty, Genome Res., 2004, 14, 1310–1314 CrossRef CAS.
- J. Zhang, Trends Ecol. Evol., 2003, 18, 292–298 CrossRef.
- M. Kasahara, Curr. Opin. Immunol., 2007, 19, 547–552 CrossRef CAS.
- D. R. Scannell, G. Butler and K. H. Wolfe, Yeast, 2007, 24, 929–942 CrossRef CAS.
- B. Papp, C. Pál and L. Hurst, Nature, 2003, 424, 194–197 CrossRef CAS.
- A. Vazquez, A. Flammini, A. Maritan and A. Vespignani, ComPlexUs, 2002, 1, 38–44 Search PubMed.
- I. Ispolatov, P. L. Krapivsky and A. Yuryev, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2005, 71, 061911 CrossRef CAS.
- K. Evlampiev and H. Isambert, BMC Syst. Biol., 2007, 1, 49 CrossRef.
- W. K. Kim and E. M. Marcotte, PLoS Comput. Biol., 2008, 4, e1000232 Search PubMed.
- A. Wagner, Mol. Biol. Evol., 2001, 18, 1283–1292 CAS.
- P. Beltrao and L. Serrano, PLoS Comput. Biol., 2007, 3, 258–267 Search PubMed.
- J. Berg, M. Lässig and A. Wagner, BMC Evol. Biol., 2004, 4, 51–63 CrossRef.
- C. Li and P. K. Maini, J. Phys. A: Math. Gen., 2005, 38, 9741–9749 CrossRef.
- M. Middendorf, E. Ziv and C. Wiggins, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 3192–3197 CrossRef CAS.
- C. Wiuf, M. Brameier, O. Hagberg and M. P. Stumpf, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 7566–7570 CrossRef CAS.
- O. Ratmann, O. Jorgensen, T. Hinkley, M. Stumpf, S. Richardson and C. Wiuf, PLoS Comput. Biol., 2007, 3, 2266–2278 Search PubMed.
- X. Guo and A. J. Hartemink, Bioinformatics, 2009, 25, i240–i246 CrossRef CAS.
- C. S. Liao, K. Lu, M. Baym, R. Singh and B. Berger, Bioinformatics, 2009, 25, i253–i258 CrossRef CAS.
- M. Zaslavskiy, F. Bach and J. P. Vert, Bioinformatics, 2009, 25, i259–i267 CrossRef CAS.
- R. Sharan and T. Ideker, Nat. Biotechnol., 2006, 24, 427–433 CrossRef CAS.
- B. A. Shoemaker and A. R. Panchenko, PLoS Comput. Biol., 2007, 3, 0595–0601 Search PubMed.
- S. Pitre, M. Alamgir, J. R. Green, M. Dumontier, F. Dehne and A. Golshani, Adv. Biochem. Eng./Biotechnol., 2008, 110, 247–267 Search PubMed.
- P. Chen, C. Deane and G. Reinert, Bioinformatics, 2007, 23, 2314–2321 CrossRef CAS.
- A. Clauset, C. Moore and M. E. Newman, Nature, 2008, 453, 98–101 CrossRef CAS.
- S. Yook, Z. Oltvai and A. Barabási, Proteomics, 2004, 4, 928–942 CrossRef CAS.
- C. Marcotte, M. Pellegrini, H. Ng, D. Rice, T. Yeates and D. Eisenberg, Science, 1999, 285, 751–753 CrossRef CAS.
- R. Overbeek, M. Fonstein, M. D’Souza, G. Pusch and N. Maltsev, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 2896–2901 CrossRef CAS.
- A. Enright, I. Iliopoulos, N. Kyrpides and C. A. Ouzounis, Nature, 1999, 402, 86–90 CrossRef CAS.
- M. Pellegrini, E. Marcotte, M. Thompson, D. Eisenberg and T. Yeates, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4285–4288 CrossRef CAS.
- A. J. Walhout, R. Sordella, X. Lu, J. L. Hartley, G. F. Temple, M. A. Brasch, N. Thierry-Mieg and M. Vidal, Science, 2000, 287, 116–122 CrossRef CAS.
- L. R. Matthews, P. Vaglio, J. Reboul, H. Ge, B. P. Davis, J. Garrels, S. Vincent and M. Vidal, Genome Res., 2001, 11, 2120–2126 CrossRef CAS.
- T. W. Huang, C. Y. Lin and C. Y. Kao, BMC Bioinformatics, 2007, 8 Search PubMed.
- S. Mika and B. Rost, PLoS Comput. Biol., 2006, 2, 698–709 Search PubMed.
- H. Yu, N. M. Luscombe, H. X. Lu, X. Zhu, Y. Xia, J. D. Han, N. Bertin, S. Chung, M. Vidal and M. Gerstein, Genome Res., 2004, 14, 1107–1118 CrossRef CAS.
- C. Frech, M. Kommenda, V. Dorfer, T. Kern, H. Hintner, J. Bauer and K. Oender, BMC Bioinformatics, 2009, 10, 21 CrossRef.
- L. Kiemer and G. Cesareni, Trends Biotechnol., 2007, 25, 448–454 CrossRef CAS.
- C. S. Goh, A. A. Bogan, M. Joachimiak, D. Walther and F. E. Cohen, J. Mol. Biol., 2000, 299, 283–293 CrossRef CAS.
- A. Valencia and F. Pazos, Curr. Opin. Struct. Biol., 2002, 12, 368–373 CrossRef.
- P. Aloy, H. Ceulemans, A. Stark and R. B. Russell, J. Mol. Biol., 2003, 332, 989–998 CrossRef CAS.
- G. R. Smith and M. J. Sternberg, Curr. Opin. Struct. Biol., 2002, 12, 28–35 CrossRef CAS.
- S. J. Cockell, B. Oliva and R. M. Jackson, Bioinformatics, 2007, 23, 573–581 CrossRef CAS.
- R. B. Russell, F. Alber, P. Aloy, F. P. Davis, D. Korkin, M. Pichaud, M. Topf and A. Sali, Curr. Opin. Struct. Biol., 2004, 14, 313–324 CrossRef CAS.
- O. Carugo and G. Franzot, Proteomics, 2004, 4, 1727–1736 CrossRef CAS.
- K. V. Brinda and S. Vishveshwara, BMC Bioinformatics, 2005, 6, 296 CrossRef CAS.
- J. A. Loo, Mass Spectrom. Rev., 1997, 16, 1–23 CrossRef CAS.
- R. Méndez, R. Leplae, M. F. Lensink and S. J. Wodak, Proteins: Struct., Funct., Bioinf., 2005, 60, 150–169 Search PubMed.
- A. Andreeva, D. Howorth, S. Brenner, T. Hubbard, C. Chothia and A. Murzin, Nucleic Acids Res., 2004, 32, 226D–D229 CrossRef.
- R. D. Finn, J. Mistry, B. Schuster-Böckler, S. Griffiths-Jones, V. Hollich, T. Lassmann, S. Moxon, M. Marshall, A. Khanna, R. Durbin, S. R. Eddy, E. L. Sonnhammer and A. Bateman, Nucleic Acids Res., 2006, 34, D247–D251 CrossRef CAS.
- A. Marchler-Bauer, J. B. Anderson, M. K. Derbyshire, C. DeWeese-Scott, N. R. Gonzales, M. Gwadz, L. Hao, S. He, D. I. Hurwitz, J. D. Jackson, Z. Ke, D. Krylov, C. J. Lanczycki, C. A. Liebert, C. Liu, F. Lu, S. Lu, G. H. Marchler, M. Mullokandov, J. S. Song, N. Thanki, R. A. Yamashita, J. J. Yin, D. Zhang and S. H. Bryant, Nucleic Acids Res., 2007, 35, D237–D240 CrossRef CAS.
- E. Sprinzak and H. Margalit, J. Mol. Biol., 2001, 311, 681–692 CrossRef CAS.
- M. Deng, S. Mehta, F. Sun and T. Chen, Genome Res., 2002, 12, 1540–1548 CrossRef CAS.
- R. Riley, C. Lee, C. Sabatti and D. Eisenberg, Genome Biol., 2005, 6, R89 CrossRef.
- C. A. Orengo and J. M. Thornton, Annu. Rev. Biochem., 2005, 74, 867–900 CrossRef CAS.
- M. K. Basu, L. Carmel, I. B. Rogozin and E. V. Koonin, Genome Res., 2008, 18, 449–461 CrossRef CAS.
- Y. Ofran and B. Rost, J. Mol. Biol., 2003, 325, 377–387 CrossRef CAS.
Footnote |
| † These can be found in publicly available databases.10 |
| This journal is © The Royal Society of Chemistry 2010 |