Small-molecule modulators of the Sonic Hedgehog signaling pathway
Benjamin Z.
Stanton
ab and
Lee F.
Peng
*abcd
aDepartment of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, MA 02138, USA
bThe Broad Institute, 7 Cambridge Center, Cambridge, MA 02142, USA
cGI Unit, Department of Medicine, Massachusetts General Hospital, 55 Fruit Street, Blake 4, Boston, MA 02114, USA. E-mail: lpeng@partners.org
dDepartment of Medicine, Harvard Medical School, 25 Shattuck Street, Boston, MA 02115, USA
First published on 27th August 2009
Abstract
Sonic hedgehog (Shh) is the most widely characterized of the three vertebrate Hedgehoghomologs, and is essential for proper embryonic development. Shh binds to its receptor, Patched (Ptch1), resulting in the de-repression of Smoothened (Smo). This leads to the activation of Gli2, which regulates the transcription of target genes that include Gli1 and Ptch1. Several synthetic and naturally occurring small-molecule modulators of Smo have been discovered. Shh-signalingantagonists that bind to Smo include cyclopamine, SANT1, and Cur-61414. Shh signalingagonists that bind to Smo include the synthetic small molecules purmorphamine and SAG. Small molecules that inhibit Shh signalingdownstream of Smo, GANT58 and GANT61 have also been reported. Robotnikinin inhibits the Shh pathway by directly targeting Shh. Although progress has been made in understanding and modulating Shh signaling, fundamental aspects of Shh signal transduction remain obscure, including the mechanism(s) whereby Ptch1 regulates Smo activity. Small-molecule modulators of Shh signaling provide a means to regulate the activity of a pathway implicated in medulloblastoma, basal cell carcinoma (BCC), pancreatic cancer, prostate cancer and developmental disorders. Several Shh inhibitors have not succeeded in the clinic for unknown reasons, but clinical trials in BCC and pancreatic cancer with the promising Smo antagonists GDC-0449 and IPI-926 are currently underway.
 Benjamin Z. Stanton | Benjamin Z. Stanton is a postdoctoral fellow in the Chemical Biology program at the Broad Institute. He worked in the laboratory of Professor Richard B. Silverman at Northwestern University where he synthesized nNOS inhibitors, and received his BA in Chemistry in 2004. Under the guidance of Professor Stuart L. Schreiber, he received his PhD in Chemistry from Harvard University in 2009. During his graduate studies, he developed a new class of small-molecule inhibitors of Sonic Hedgehog signaling. |
 Lee F. Peng | Lee F. Peng was born in New York city. He graduated from Harvard College with an A.B. in chemistry, prior to pursuing graduate studies in the MD–PhD program at Harvard Medical School and Harvard University. His PhD in synthetic organic chemistry was completed under the direction of Professor Yoshito Kishi. Upon completing his joint degree program, he pursued clinical training in internal medicine and gastroenterology at the Massachusetts General Hospital (MGH), as well as postdoctoral research in the laboratories of Professor Stuart Schreiber (Harvard University) and Dr Raymond T. Chung (MGH). He currently is a clinician-scientist with faculty appointments at the Massachusetts General Hospital and Harvard Medical School. His clinical practice focuses on gastroenterology, and his basic research interests include signal transduction (including the Sonic Hedgehog pathway), hepatitis C, and chemical biology. |
1. The Sonic Hedgehog signaling pathway
1.1 Hedgehog proteins and signal transduction
The Hedgehog (Hh) pathway is involved in the development of every major organ in most animals.1 The Hhgene was first identified in Drosophila in 1980,2 and the three mammalian orthologs, Sonic Hedgehog (Shh), Desert Hedgehog, (Dhh), and Indian Hedgehog (Ihh), were identified thereafter. Shh regulates anterior/posterior patterning in limb development, the induction of polarity in the central nervous system, embryonic development, and the differentiation of many different cell types.3–6 Shh binds to its 12-pass transmembrane receptor, Patched (Ptc, Ptch1 in mammals), which then releases its inhibitory effect on the pseudo-G-protein coupled receptor Smoothened (Smo).1,7 This leads to the activation of Gli2 in the cytoplasm, which travels to the nucleus and regulates the transcription of Shh-pathway target genes, which include Gli1 and Ptch1 (Fig. 1).8 The expression of both an activator (Gli1) and a repressor (Ptch1) reduces the possibility of inappropriate pathway diminution or over-activation.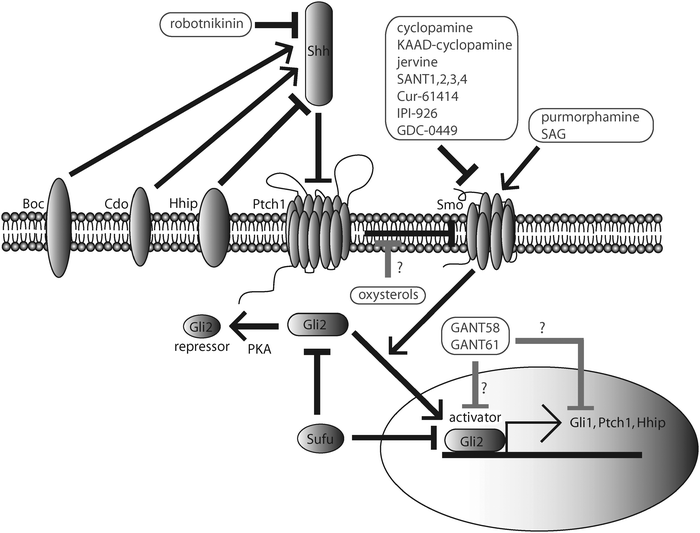 | ||
| Fig. 1 The Sonic Hedgehog (Shh) pathway in vertebrates. Shh binds to Patched1 (Ptch1) and reverses repression of Smoothened (Smo). This activates Gli2, which translocates into the nucleus and participates in the activation of target genes, including Gli1, Ptch1 and Hhip. In the absence of Shh, Gli2 is phosphorylated and cleaved into a truncated peptide that represses transcription of Shh-target genes. Hhip acts at the cell surface to repress Shh. Boc and Cdo act at the cell surface to activate Shh. Sufu is an endogenous Gli-repressor. Small molecules including cyclopamine, KAAD-cyclopamine, jervine, SANT1-4, Cur-61414, IPI-926, and GDC-0449 inhibit Shh signaling by targeting Smo. Robotnikinin is a small molecule that binds directly to Shh and inhibits Shh signalingupstream of Smo, at the Shh level. Purmorphamine and SAG are synthetic Shh-signalingagonists that bind directly to Smo and compete with cyclopamine–Smo interactions. GANT58 and GANT61 inhibit Shh-signaling activity downstream of Smo and Sufu. Several oxysterols, including 20α-hydroxycholesterol, 22(S)-hydroxycholesterol, 24(S)-hydroxycholesterol, and 25-hydroxycholesterol, activate the Shh pathway. The Shh signaling pathway contributes to a variety of biologic processes including vertebrate development and oncogenesis (Section 1.2). | ||
The active N-terminal portion of Shh (ShhN) is highly conserved across species, whereas the C-terminal domain (CTD) is conserved to a lesser extent.9 Statistical analyses indicate that amino acid substitutions are approximately fourfold less likely to occur in the NTD than in the CTD. The original Hhgene evolved approximately 900 mya, at the same time as the TGFβgene and other growth factors. As both zebrafish and mouse Shh pathway activation produce similar gene expression patterns, it appears that certain Hh-pathway target genes are widely conserved.9
1.2 The Shh pathway’s significance in development and cancer
Shh signaling plays a role in many developmental processes. The pathway provides a unique example of how the same molecular machinery can lead to different outcomes in developmental patterning in different tissue types. Shh acts as a morphogen, and induces distinct transcriptional programs and cell types based upon its local concentration.1 In vertebrates, Hhsignaling facilitates the patterning of many tissue types, including hematopoietic,10,11 osteoblastic,12,13 retinal,14,15 gastrointestinal,16,17 hair,18,19 neuronal,20,21 limb,22,23 and prostatic tissue.24,25 In the developing neural tube, Shh induces the expression of different transcription factors (TFs), including Pax6, Nkx2.2 and Olig2, depending on its concentration gradient along the dorsal–ventral axis.26,27 Different combinations of Shh-dependent TFs facilitate the specification of various cells types, including motor, floor plate, and dopaminergic neurons in the developing neural tube. Shh expression in the zone of polarizing activity acts in concert with FGF4 in the apical ectodermal ridge to facilitate limb-bud development.22 Recent evidence suggesting that Gli facilitates the transcription of cyclin B1 and cyclin D1, implies that the pathway not only directs tissue specification but also tissue maintenance through regulating cyclin-dependent cellular proliferation.28,29Given the Hh pathway’s significant role in tissue patterning and cell number maintenance, mutations in Shh-pathway genes or events causing misregulation of the pathway are associated with certain cancers. Inappropriate Shh signaling is frequently related to tumor initiation and maintenance in basal cell carcinoma (BCC), medulloblastoma, and pancreatic cancer.30,31 Mutations in Ptch1, which lead to constitutive Smo activation, appear in many BCCs. Patients with Gorlin syndrome have one defective Ptch1allele, and frequently develop BCCs, medulloblastomas, or other developmental defects. Gorlin-syndrome patients develop BCC upon the UV-induced mutation of the one remaining wild-type Ptch1allele in the epidermis.32Gene expression profiling of BCC explants have shown that the majority of BCCs involve increased transcription of Shh-pathway target genes.31 Transgenic mice, with Shh overexpression driven by a constitutive Keratin 14 (K14) promoter, develop BCC-like lesions with no UV exposure.33 Many of the K14-Shh mice also displayed developmental abnormalities like shorter limbs, excess digits, and improperly formed neural tubes.
Medulloblastoma is a brain tumor associated with increased Gli1 expression in the cerebellum. Being the most common type of brain cancer in the juvenile population, it is associated with high levels of morbidity, which are in the range of 50%, even with treatment.31 Mutations in Smo or Ptch1 in neuronal cells in the cerebellum that result in constitutive activation of Smo usually result in medulloblastoma. The majority of transgenic mice with Ptch1+/− and p53−/− develop medulloblastoma.
The Shh pathway is over-activated in pancreatic ductal adenocarcinoma (PDAC).34Gene expression profiling studies in PDAC precursor lesions have demonstrated that there are high expression levels of Shh target genes, including Gli1 and Ptch1, and cyclin D1. Pdx1-driven Shh overexpression may be linked to the initiation of PDAC lesions.35 Shh pathway-driven cyclin D1 expression is associated with pancreatic duct epithelial cell (PDEC) proliferation. Studies have shown that constitutive Shh expression will lead to PDEC proliferation, relative to control PDECs overexpressing green-fluorescent protein (GFP).34 Furthermore, in PDAC precursor lesions, elevated Shh pathway activity is correlated to inhibition of apoptotic regulators, such as caspase 3. PDECs overexpressing Shh demonstrate reduced levels of apoptosis upon exposure to 100 μM cycloheximide (CHX) or UV light.34,36 In the same study, the control PDECs overexpressing GFP displayed higher expression levels of caspase 3 and had significantly higher levels of cell deathviaapoptosis. Increased Shh signaling in PDECs mediates tumor initiation, proliferation, and reduced cell death, and the Shh pathway plays a major role in the pathogenesis of PDAC.
Paracrine Shh signaling plays an important role in certain cancers. When the pancreatic cancer cell lines CFPAC-1 and BxPC-3 were treated with Shh pathway antagonists, a similar level of growth inhibition was observed despite the fact that CFPAC-1 expresses Smo and BxPC-3 does not.37 Additionally, no effect on Gli mRNA was observed when the cell lines were treated with Shh antagonists. However, expression-profiling experiments of tissue samples showed that Shh ligand was overexpressed in a variety of human cancers. It was proposed that the Shh-pathway reliance displayed by human tumor explants and cancer cell lines could be paracrine in nature, with the cancer cells overexpressing Shh ligand and adjacent stromal cells being competent to receive the signal. This hypothesis was tested both in vitro and in vivo. In in vitro co-culture assays, the pancreatic cancer cell lines PANC-1 and ASPC-1 (which overexpress Shh) were able to activate Gli transcription levels in co-cultured 10T1/2 cells.37 When the human pancreatic cancer cell line HPAF-II was implanted into Ptch1-LacZ mice, LacZ staining indicated upregulation of Ptch1 in the stromal cells surrounding the implant, but not in the tumor tissue. When a panel of human tumor explants were implanted into nude mice, rtPCR with species-specific primers for human and mouse Gli1 indicated that stromal levels of Gli1 increased in the presence of tumor tissue that expressed Shh, but there was no correlation between stromal Shh and tumor-associated Gli1. These data support a paracrine signaling mechanism of Shh in the context of certain human cancers and associated stromal tissue.37
Shh signaling also plays a role in the pathogenesis of chronic myelogenous leukemia (CML), gliomas, and multiple myeloma (MM).38–40 When mice were implanted with CML stem cells and subjected to conditional Smo knockouts, the frequency of CML was reduced by nearly 50 percent relative to mice expressing wild-type (WT) Smo.38 Additionally, mice expressing a constitutively activated form of Smo had a fourfold increase in CML stem cell number relative to mice expressing wild-type Smo. These data support a model of Shh pathway-dependent activation of CML progenitor stem cells.
Glioma stem cell growth is inhibited in the presence of the Smo inhibitorcyclopamine.39Cyclopamine treatment of CD133+ glioma stem cells diminished cell number by almost 50 percent relative to mock treatment and reduced Gli1 and Ptch1 mRNA levels by 50 percent and 40 percent, respectively. Levels of Sox2 mRNA were decreased by 57 percent in the presence of cyclopamine, indicating that Shh signaling is integral in the maintenance of stemness in glioma progenitor cells.39
MM is a non-Gorlin cancer affecting plasma cells.40 Transcript levels of Smo, Gli1, and Ptch1 were found to be significantly upregulated in tumorgenic MM cells relative to healthy cells. However, in a population of MM stem cells, transcription levels of Smo and Gli1, but not Ptch1, were found to be strongly upregulated relative to mature MM cells, suggesting that Shh pathway activity resides predominantly in the MM tumor stem cell population. Shh treatment facilitated clonal expansion of MM stem cells relative to mature MM cells, and cyclopamine treatment reduced MM stem cell number and facilitated differentiation of MM stem cells into non-proliferating mature MM cells.40 Thus in CML, glioma, and MM, the Shh pathway activity facilitates the maintenance and proliferation of cancer stem cells.
Table 1 summarizes the tumors that have been linked to the Shh pathway.
| Tumor entity | Experimental evidence | |
|---|---|---|
| Gorlin | Basal cell carcinoma (BCC) | •Epidermal Ptch1 mutations |
| •Upregulation of Shh-target genes in BCC explants | ||
| Medulloblastoma | •Smo, Ptch1 mutations | |
| •Increased Gli1 expression in the cerebellum | ||
| Non-Gorlin | Pancreatic ductal adenocarcinoma (PDAC) | •Increased Gli1, Ptch1, cyclin D1 expression |
| •Pdx1-driven Shh transcription | ||
| Chronic myelogenous leukemia (CML) | •Conditional Smo knockouts reduce CML initiation in mice | |
| •Activating mutations in Smo increase CML initiation in mice | ||
| Glioma | •Increased Gli1, Ptch1, Sox2 expression | |
| •cyclopamine treatment reduces CD133+ glioma progenitor cell number | ||
| Multiple myeloma (MM) | •Increased Smo, Gli1, Ptch1 expression | |
| •Shh treatment resulted in clonal expansion of MM progenitor cells | ||
1.3 Shh structural analysis and localization
The crystal structure of the ShhN protein was first reported by Leahy and co-workers in 1995.41 Originally it was thought that the Zn-binding site in ShhN had a catalytic role in hydrolytic peptide cleavage, especially because of the homology to known peptidases, such as thermolysin and carboxypeptidase A.41 As there was space in the crystal lattice for a peptide substrate, it was hypothesized that the signaling activities of secreted ShhN were related to its putative peptidase activity. However, further investigation indicated that ShhN did not have peptidase activity, but functioned as a ligand for Ptch1.42,43 These studies showed that mutant ShhN proteins which lacked the residues necessary for enzymatic activity still maintained signaling efficacy, as measured by HNF-3β expression.38 These mutant ShhN proteins retained binding affinity for Ptch1 of the order of that demonstrated by the wild-type ligand.Drosophila HhN forms a dimeric complex with Ihog and other associated proteins, including heparin (HP) and heparinsulfate (HS).44 Ihog is a two-domain containing transmembrane protein that is necessary for the interaction between HhN and Ptch. Heparin binds in the cleft where Ihog is associated with HhN,44 and it is not only necessary for the induction of the 2 : 2 HhN–Ihog dimer complex, but also necessary for Ptc-mediated Smo de-repression. The equilibrium dissociation constants for ShhN–HS and HhN–HP were determined to be 32 nM and 48 nM, respectively. The strongest binding affinity measured was the ShhN–HP interaction where KD = 67 nM.45
ShhN acts at the cell surface in association with other associated proteins. Hhip binds directly to Shh, Dhh, and Ihh, and is both a transcriptional target and repressor of Shh signaling.46 Similarly to how Ihog associates with DrosophilaHhN, the vertebrate Ihog orthologs Cdo and Boc bind to and activate ShhN.47,48 The equilibrium dissociation constants of Cdo–ShhN, Boc–ShhN, and Hhip–ShhN were found to be 3.8 nM, 2.9 nM, and 1.1 nM, respectively.48 ShhN–Cdo binding is calcium dependent, whereas HhN–Ihog binding is not.49 Although the equilibrium dissociation constant for ShhN–calcium was >100 μM, the capacity for ShhN to bind Cdo was strongly reduced without calcium present or in the presence of a strong meal chelating agent like EGTA. When the residues necessary for DrosophilaHh to bind to Ihog were substituted into ShhN, the resulting protein bound Ihog in a heparin-dependent manner, but the binding was calcium independent.49 The presence of EGTA also reduced binding capacity between ShhN–Ptch1 and ShhN–Hhip, suggesting that the ShhN–calcium interaction confers binding potential with multiple partners.
The localization of Shh signalingproteins in mammalian cells is vital to signal transduction. Primary cilia are protrusions on most mammalian cells that use intraflagellar transport (IFT) proteins to help receive and transduce extracellular signals.50 Primary cilia are integral in the processes of photoreception, olfaction and in several examples of signal transduction pathways.51 Two proteins involved in IFT were linked to Shh signaling.52 The IFT genes, wimple and polaris, were discovered during a mutational screen in mouse embryos. Mutations in these two genes resulted in phenotypes characteristic of Shh signaling abnormalities,52 and IFTproteins were found to regulate Gli localization in the context of Shh signaling.53 It was hypothesized that the wimple and polarisgene products localized Shh signalingproteins at the non-motile primary cilia of mammalian cells.
Reiter and co-workers reported that Smo localization at the primary cilium is essential for Shh signaling in mammals.54 MDCK cells constitutively expressing myc-tagged Smo were treated with ShhN, leading to increased Smo localization at the primary cilium. McMahon and co-workers, using fluorescent Smo proteins, showed that cyclopamine treatment led to the accumulation of Smo in the primary cilium of NIH3T3 cells.89 Scott and co-workers reported that the Smo antagonists SANT1, SANT2 and cyclopamine have differential effects in the context of subcellular localization of Smo.55 Their findings support a 2-step 3-state model of Smo activation. Smo is first in an inactive state in the cytoplasm (state 1; targeted by SANT1, SANT2). It then relocates to the primary cilium but remains in an inactive state (state 2; targeted by cyclopamine, Ptch1). It is finally activated in the cilium (state 3; induced by Shh or small-molecule Smo agonists).
ShhN treatment prevents the localization of Ptch1 at the primary cilium in NIH3T3 cells.56 Retroviral introduction of Ptch1 into a Ptch1−/− MEF cell line resulted in the trafficking of Smo away from the primary cilium. Treatment with either SAG (a Smo-binding Shh pathway agonist) or 20α-hydroxycholesterol (a non-Smo-binding Shh pathway agonist) resulted in the retention of Ptch1 at the primary cilium in NIH3T3 cells.56 This suggests that ShhN activates the pathway with a different mechanism than is characteristic of several downstream pathway agonists.
2. Small-molecule modulators of the Sonic Hedgehog signaling pathway
2.1 Shh pathway inhibition with naturally occurring products: jervine alkaloids, sesquiterpenes, bisindole alkaloids and physalins as small-molecule antagonists
Cyclopamine and jervine are naturally occurring molecules found in Veratrum californicum (Fig. 2, Table 2).57,58 Both compounds are teratogenic, causing holoprosencephaly or cyclopia in the offspring of animals that ingested V. californicum.59,60 Cyclopic phenotypes include the loss of midline facial features, a single eye, and absent anatomical features from the forebrain.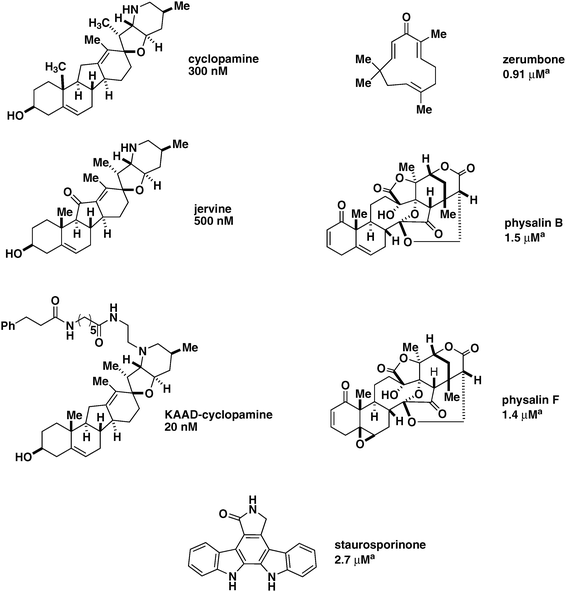 | ||
| Fig. 2 Shh-pathway inhibitors. Cyclopamine, jervine, and KAAD-cyclopamine are examples of jervine alkaloid Shh pathway inhibitors, with EC50 values shown above. KAAD-cylopamine is a chemically-modified form of cyclopamine that displays reduced cytotoxicity. Zerumbone, physalinB, physalinF, and staurosporinone have been shown to repress Gli2-mediated transcription, with EC50 values specific for Gli2 shown above, as denoted with a superscript a. | ||
| Small-molecule modulator | Effects | |
|---|---|---|
| Naturally occurring | Cyclopamine | •Targets Smo |
| •Inhibits transcription of Gli1, Gli2, Ptch1, and other Shh-target genes in a variety of cell types | ||
| Jervine | •Targets Smo | |
| •Inhibits transcription of Gli1, Gli2, Ptch1, and other Shh-target genes in a variety of cell types | ||
| Staurosporinone | •Inhibits Gli1,2-mediated transcription in HaCaTcells | |
| •Inhibits transcription of Ptch1 and BCL2 in HaCaTcells | ||
| Zerumbone | •Inhibits Gli1,2-mediated transcription in HaCaTcells | |
| •Inhibits transcription of Ptch1 and BCL2 in HaCaTcells | ||
| Physalin B | •Inhibits Gli1,2-mediated transcription in HaCaTcells | |
| •Inhibits transcription of Ptch1 and BCL2 in HaCaTcells | ||
| Physalin F | •Inhibits Gli1,2-mediated transcription in HaCaTcells | |
| •Inhibits transcription of Ptch1 and BCL2 in HaCaTcells | ||
| 20α-Hydroxycholesterol | •Does not bind directly to Smo | |
| •Activates Gli-mediated transcription in a variety of cell types | ||
| 22(S)-Hydroxycholesterol | •Does not bind directly to Smo | |
| •Activates Gli-mediated transcription in a variety of cell types | ||
| 24(S)-Hydroxycholesterol | •Does not bind directly to Smo | |
| •Activates Gli-mediated transcription in a variety of cell types | ||
| 25-Hydroxycholesterol | •Does not bind directly to Smo | |
| •Activates Gli-mediated transcription in a variety of cell types | ||
| Synthetic | SANT1,2,3,4 | •Targets Smo |
| •Represses Gli-mediated transcription in a variety of cell types | ||
| Cur-61414 | •Represses Gli-mediated transcription in a variety of cell and tissue types | |
| •Targets Smo | ||
| GANT58 | •Represses Gli-mediated transcription in a variety of cell types | |
| •Acts downstream of Smo and Sufu | ||
| GANT61 | •Represses Gli-mediated transcription in a variety of cell types | |
| •Acts downstream of Smo and Sufu | ||
| Robotnikinin | •Represses Gli-mediated transcription in a variety of cell types, and synthetic human tissue | |
| •Binds directly to Shh | ||
| SAG | •Activates Gli-mediated transcription in a variety of cell types | |
| •Binds Smo and competes directly with cyclopamine–Smo interactions | ||
| Purmorphamine | •Activates Gli-mediated transcription in a variety of cell types | |
| •Binds Smo and competes directly with cyclopamine–Smo interactions | ||
| IPI-926 | •Represses Gli-mediated transcription in a variety of cell types | |
| •Targets Smo | ||
| •Clinical trials for BCC and pancreatic adenocarcinoma are ongoing | ||
| GDC-0449 | •Represses Gli-mediated transcription in a variety of cell types | |
| •Targets Smo | ||
| •Clinical trials for BCC and other solid tumors are ongoing | ||
Cyclopamine inhibits transcription of the Shh pathway target genes, Gli1, Gli2, and Ptch1, with an EC50 of 300 nM. KAAD-cyclopamine is a chemically-modified derivate of naturally occurring cyclopamine that displays higher potency, with an EC50 of 20 nM, and reduced levels of cytotoxicity.62Cyclopamine and KAAD-cyclopamine inhibited Gli activity in NIH3T3 cells with a stably incorporated Gli-luciferase reporter (Shh-LIGHT2 cells), but had decreased efficacy in the same cell line overexpressing Smo. Cyclopamine retained its inhibitory effect in a Ptch1−/−MEFcell line, where both Ptch1alleles were replaced with a Gli-dependent LacZ reporter.62 These data indicate that cyclopamine and KAAD-cyclopamine act downstream of Ptch1, and activity at the Smo level in the pathway was confirmed by photo-affinity labeling experiments.61 Jervine exerts its inhibitory effects on Shh pathway mediated gene expression with an EC50 of 500 nM.59,62
In a preliminary screen for repressors of Gli1-mediated transcription, Ishibashi and co-workers used HaCaTcells with stably incorporated tertacyclin-inducible constructs for Gli1 and Gli2, along with transient transfections of a Gli-dependent luciferase reporter.63 Zerumbone, physalinB, physalinF, and staurosporinone inhibited both Gli1 and Gli2-mediated transcription. Zerumbone, physalinB, physalinF, and staurosporinone were also found to repress the transcription of other Shh signalinggenes, including Ptch1 and BCL2 in HaCaTcells (Fig. 2, Table 2).63
2.2 Shh pathway inhibition with non-naturally occurring small molecules
Cell-based assays with reporter geneconstructs have been widely used for discovering non-naturally occurring small-molecule antagonists and agonists of Shh signaling. Sasaki et al. reported a construct with eight adjacent Gli binding sitesupstream of a firefly luciferase reporter gene, which has been successfully used in cell lines, including the C3H10T1/2 and NIH3T3 (Shh-LIGHT2), to screen for small-molecule inhibitors of Gli activity.64 Among the Shh-pathway antagonists identified using this construct are Cur-61414, SANT1, SANT2, SANT3, SANT4, GANT61 and GANT58 (Fig. 3, Table 2).32,65–67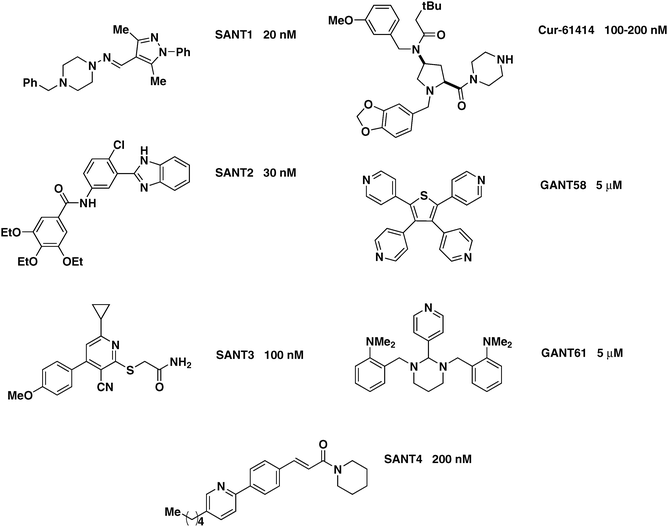 | ||
| Fig. 3 Synthetic Shh-pathway inhibitors. SANT1, SANT2, SANT3, SANT4, Cur-61414, GANT58, and GANT61 are shown, with their respective EC50 values indicated.78,88 | ||
GANT58 and GANT61 act downstream of Smo.67 These two compounds have EC50 values of approximately 5 μM in Shh-LIGHT2 cells. When tested in C3H10T1/2 cells in the presence of Shh, GANT61 and GANT58 were able to repress alkaline phosphatase activity associated with Shh pathway activity. In Ptch1−/− MEFs, which lack Ptch1 function, the compounds retained inhibitory activity, indicating that their respective targets were downstream of Ptch1. When the two inhibitors were tested in MEFcells lacking both alleles of the endogenous downstreaminhibitorSufu, the compounds retained activity, whereas cyclopamine did not, as measured by Gli1 and Hhip1 mRNA levels. Thus, GANT58 and GANT61 act downstream of both Smo and Sufu.67
Robotnikinin is a synthetic small-molecule inhibitor of Shh signaling that acts upstream of Smo.68–74 To date, it is the only small-molecule inhibitor of Shh signaling that acts upstream of Smo. Robotnikinin inhibits Gli transcription in Shh-LIGHT2 cells stimulated with ShhN, but when the Smo agonists purmorphamine or SAG were co-administered, its inhibitory activity was abolished. The compound is therefore able to inhibit Gli transcription by targeting a proteinupstream of Smo in the Shh signaling pathway in Shh-LIGHT2 cells (Fig. 4, Table 2). Robotnikinin reduced alkaline phosphatase activity in a concentration-dependent fashion in C3H10T1/2 cells stimulated with ShhN, but this trend was eliminated when purmorphamine was co-administered. Thus, the activity of robotnikinin is not dependent on a cell line or reporter construct. Robotnikinin was also tested in primary human-derived keratinocytes stimulated with Shh. After 30 hours of treatment, Gli1 and Gli2 mRNA levels were reduced in a dose-dependent fashion, but this inhibition was eliminated with co-treatment with purmorphamine or SAG. Robotnikinin is able to inhibit Shh pathway activity in both mouse and human-derived cellsupstream of Smo. In a Ptch1−/−MEFcell line with both Ptch1 alleles replaced with a LacZ reporter gene, no pathway inhibition was observed using robotnikinin. These data were consistent with a model where the small molecule inhibits the Shh pathway upstream of Ptch1. The compound also binds purified ShhN by SPR. These data provide strong evidence that robotnikinin directly targets the ShhN protein.
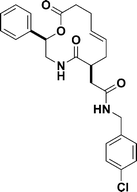 | ||
| Fig. 4 The structure of robotnikinin. | ||
2.3 Shh pathway agonists: naturally occurring products
Cholesterol and other certain oxysterols participate in the activation of the Shh signaling pathway.75–77 Beachy and co-workers reported that when intracellularsterol levels were depleted, but still remained sufficient for Shh post-translational modification, cells became less responsive to ShhN.77 Corcoran and Scott co-workers demonstrated that cholesterol, 20α-hydroxycholesterol, 22(S)-hydroxycholesterol, 24(S)-hydroxycholesterol, and 25-hydroxycholesterol activated Shh pathway activity, as measured by Ptch1–LacZ reporter activity in PZp53MEDcells, while 7β-hydroxycholesterol had no effect (Fig. 5, Table 2).76 In another study, 20α-hydroxycholesterol and 22(S)-hydroxycholesterol were found to induce Shh target gene upregulation as measured by rtPCR for Gli1 and Ptch1 in M2-10B4 cells.75Cholesterol depletion therefore affects Shh signaling directly, not merely through inhibition of autoprocessing.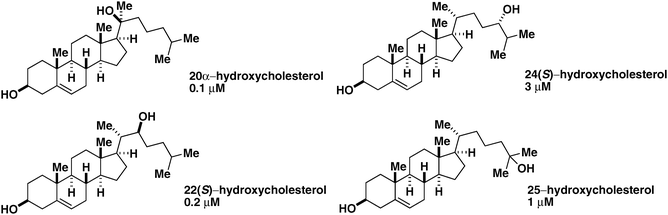 | ||
| Fig. 5 Naturally occurring small-molecule Shh pathway agonists. Oxysterols are shown. 20α-Hydroxycholesterol, 22(S)-hydroxycholesterol, 24(S)-hydroxycholesterol, and 25-hydroxycholesterol along with their respective EC50 values derived from Ptch1–LacZ reporter gene activity in PZp53MEDcells are shown. | ||
The exact mechanism of action of oxysterol Shh pathway agonists remains unknown, despite investigation. 20α-Hydroxycholesterol and 22(S)-hydroxycholesterol incubated with HEK293 cells overexpressing Smo did not compete with boron-dipyrromethene (BODIPY)–cyclopamine for Smo binding.75 A 1 : 1 combination of 20α-hydroxycholesterol and 22(S)-hydroxycholesterol (SS) was not able to shift the EC50 for cyclopamine in Ptch1−/− MEFs, while Smo targeting agonists like SAG and purmorphamine can increase the cyclopamine’s EC50 dramatically. Furthermore, Smo−/− MEFs were not sensitive to SS, but cyclopamine was able to override the effect of SS in C3H10T1/2 cells. Although the oxysterols do not bind directly to Smo, they still may indirectly affect Smo, perhaps by stabilizing it in a conformation where it is less sensitive to Ptch1-mediated repression.75,76
2.4 Shh pathway agonists: non-naturally occurring products
SAG, discovered during a high-throughput screen in Shh-LIGHT2 cells, activates Shh signaling by binding to Smo (Fig. 6, Table 2).78SAG induces Gli activity well above the level of 2 nM ShhN induction (approximately 40-fold vs. 25-fold) in Shh-LIGHT2 cells. SAG retains its stimulatory activity in a Ptch1−/−MEFcell line, demonstrating that it acts downstream of Ptch1. In Shh-LIGHT2 cells, KAAD-cyclopamine, titrated in increasing concentrations, ablates SAG-mediated Gli induction in the presence of 200 nM SAG.78 Similarly, increasing concentrations of SAG induces Gli activity in the presence of 100 nM KAAD-cyclopamine. SAG therefore acts on Smo.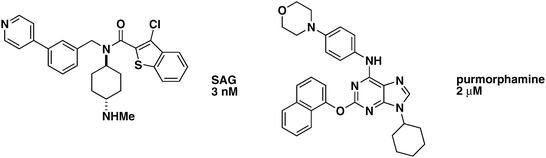 | ||
| Fig. 6 Non-naturally occurring small-molecule Shh pathway agonists. SAG and purmorphamine along with their respective EC50 values are shown. | ||
Purmorphamine is a synthetic Shh pathway agonist (Fig. 6, Table 2),79 discovered by Schultz and co-workers. When C3H10T1/2 cells differentiate into osteoblasts, or bone precursors, alkaline phosphatase (AP), a marker of Shh pathway activation, is upregulated. Initially, purmorphamine was found to induce osteoblast formation in this cell line. Subsequent gene expression profile studies showed that purmorphamine upregulates Gli1 and Ptch1, but not Ihh, Dhh, or Shh, confirming purmorphamine’s role as a Shh pathway agonist.80
Purmorphamine targets Smo.81 When Ptch1−/− MEFs were treated with purmorphamine, its activity was unchanged. In Smo−/− MEFs purporphamine had no measurable activity. In NIH3T3 cells transfected with the Sasaki construct, purmorphamine was found to diminish the activity of KAAD-cyclopamine, and KAAD-cyclopamine was found to reduce the activity of purmorphamine. Purmorphamine was able to compete with BODIPY–cyclopamine for Smo binding. Taken together, these data show that both purmorphamine and cyclopamine act on Smo.
2.5 Therapeutic implications of naturally occurring and non-naturally occurring Shh pathway inhibitors
In BCC, medulloblastoma and other cancer models, Shh pathway inhibitors reduce the tumorigenic effects resulting from pathway over-activation (see Section 1.3). Clinical trials in BCC and other solid tumors involving the Smo antagonists GDC-0449 (Phase II: NCT00636610, NCT00739661, NCT00833417, NCT00887159; Phase I: NCT00822458) and IPI-926 (Phase I: NCT00761696) are currently ongoing.82,83Cur-61414 (Fig. 3, Table 2) has been shown to reduce aberrant Shh signaling in BCC models.16 Skin explants from 17.5-day-old Ptch1+/−–LacZ embryos showed dramatically reduced levels of Ptch1 expression when treated with Cur-61414 and ShhN, relative to ShhN treatment alone. In skin explant assays from Ptch1+/− mice exposed to UV light to produce BCC-like lesions, Cur-61414 increased apoptotic levels by nearly 26% relative to mock treatment. The mitotic index was reduced by about 26% with 1 μM Cur-61414 and about 28% with 5 μM Cur-61414, relative to mock treatment in the same skin explant assay.Cyclopamine reduces proliferation in medulloblastoma cell lines derived from patient cultures, as well as in medulloblastoma tumor transplants (allografts).6,30,84Cyclopamine can reduce Shh pathway target gene expression in transformed tumor cell lines with a Ptch1–lacZ reporter.30 Medulloblastoma-derived cell lines transformed with a Gli1 overexpression construct were not affected by cyclopamine, while mock transfected cell lines showed dramatic losses in tumor volume. KAAD-cyclopamine at approximately 1 μM reduced the viability of medulloblastoma-derived tumor explant cultures significantly within 48 hours, whereas cyclopamine had the same effect at approximately 3 μM.30
Cyclopamine reduces Shh-mediated pancreatic adenocarcinoma cell growth, which is accelerated with ShhN treatment.31 Shh pathway over-activation acts in concert with K-Ras signaling in pancreatic cancer.85 When PDEC-derived tumor cell lines overexpressing K-Ras and Shh were treated with cyclopamine alone, there was no inhibition of proliferation. When PDEC-derived tumor cell lines expressing K-Ras alone were treated with cyclopamine, there was moderate inhibition of proliferation. Total growth inhibition was observed only when both cyclopamine and LY294002 (a PI3K inhibitor which blocks K-Ras signalingdownstream) were used. K-Ras therefore induces Shh pathway activation in PDEC-derived tumorigenic cell lines, and maximal growth inhibition was observed only when Shh and K-Ras pathways were inhibited.
2.6 Outlook: future directions for Shh chemical biology
There remains ample room for further exploration of the Shh pathway. Mechanistic approaches to studying Shh signaling may strengthen the community’s knowledge about existing small-molecule modulators of the Shh pathway, while the discovery of new protein targets that transduce the Shh signal will be useful in better understanding how the pathway functions in physiologically meaningful ways. The discovery of small-molecule Shh signaling modulators can provide further insight into the workings of the signaling pathway, especially if they target previously-undiscovered components of the pathway. There are many unanswered questions in the realm of Shh-chemical biology, and potentially great rewards to medicine and society for further exploration.There are a growing number of compounds in the arsenal of small-molecule Shh modulators. In the future, researchers may take advantage of different classes of small-molecule Shh inhibitors to gain greater mechanistic understanding of the processes of Shh recognition at the cell membrane, Ptch1 inactivation, Ptch1–Smo interactions, and Sufu–Smo interactions. Furthermore, because of the signaling cross-talk between the Shh and Ras/PI3K pathways (Section 2.5), explorations of synergy between Akt, PI3K, inhibitors, and non-Smo targeting Shh inhibitors may be useful in providing a new medical context for established inhibitors of Ras signaling.
Most of the aforementioned small-molecule modulators of Shh signaling act at the level of Smo. The discovery of more potent Smo antagonists and agonists is always both important and welcome. However, other Shh-related protein targets are also being explored. Recent reports have illustrated the targeting of downstream elements (GANT58, GANT61) and upstream elements (robotnikinin). Strikingly, robotnikinin was found to have efficacy in primary human cells and synthetic human skin, which suggests that it may be of potential medical use after further optimization. Furthermore, the examples of robotnikinin and the GANT compounds suggest that the community can indeed take a broader approach and need not limit itself to Smo to attain meaningful results in the context of Shh pathway modulation.
Although several clinical trials and pre-clinical investigations are underway (see Section 2.5), small-molecule Shh pathway modulators have yet to come into clinical use. The exploration of the therapeutic potentials of Shh pathway inhibitors is extremely important, especially given the prominent role that the Shh pathway plays in several disease processes and cancers. Small-molecule, Shh-pathway modulators are now undergoing clinical evaluation. GDC-0449 is a Smo inhibitor co-developed by Curis and Genentech that has activity in patients with BCC (see Section 2.5). BMS-833923 is a Shh-signalinginhibitor that is currently in phase I clinical trials for metastatic cancers, and was co-developed by Bristol-Myers Squibb and Exelixis. Infinity pharmaceuticals developed IPI-926. The compound is a Smo inhibitor that has activity in pre-clinical medulloblastoma and small-cell lung cancer models, and is currently in phase I clinical trials.86 A promising clinical candidate, the Smo inhibitorCur-61414, was unsuccessful in clinical trials for the treatment of BCC (http://www.curis.com/news/php).87 Because it is not known why the excellent pre-clinical data for Cur-6141432 did not translate into the clinic, medicinalizing existing small-molecule probes of Shh signaling and better characterizing their respective mechanisms will be of paramount importance to the successful development of Shh-based pharmaceuticals.
Some remaining challenges in the field of Shh chemical biology include finding Shh-targeting pathway agonists, agonistsdownstream of Smo, or selective antagonists of interactions between IFT-proteins and Smo or Gli. Finding selective Ihh and Dhh small-molecule antagonists would also be a challenge that could provide a means to regulate Hhsignaling in a tissue-specific manner. As more advanced and physiologically-representative assays are developed which take into account the relevance of stromal interactions, as more becomes known about the precise mechanisms of action of existing Shh small-molecule probes, and as more Shh signaling modulators begin to be examined in clinical trials, there will surely be many exciting discoveries to look forward to in the coming years.
Acknowledgements
The authors would like to thank James K. Chen, Stuart L. Schreiber, and Angela N. Koehler for their careful editing and helpful suggestions. L.F.P. is supported by funding from the National Pancreas Foundation, the American Gastroenterological Association, and the American Liver Foundation.References
- P. W. Ingham and A. P. McMahon, Genes Dev., 2001, 15, 3059–3087 CrossRef CAS.
- C. Nusslein-Volhard and E. Wieschaus, Nature, 1980, 287, 795–801 CrossRef CAS.
- Y. Echelard, D. J. Epstein, B. St.-Jacques, L. Shen, J. Mohler, J. A. McMahon and A. P. McMahon, Cell (Cambridge, Mass.), 1993, 75, 1417–1430 CrossRef CAS.
- J. B. Weitzman, J. Biol., 2002, 7, 1–5.
- R. B. Pepinsky, P. Rayhorn, E. S. Day, A. Dergay, K. P. Williams, A. Galdes, F. Taylor, A. Boriack-Sjodin and E. A. Garber, J. Biol. Chem., 2000, 275, 10995–11001 CrossRef CAS.
- B. Stecca and A. R. Altaba, J. Biol., 2002, 9, 1–4.
- D. M. Stone, M. Hynes, M. Armanini, T. A. Swanson, Q. Gu, R. Johnson, M. P. Scott, D. Pennica, A. Goddard, H. Phillips, M. Noll, J. E. Hooper, F. de Sauvage and A. Rosenthal, Nature, 1996, 384, 129–133 CrossRef CAS.
- J. A. Goetz, S. Singh, L. M. Suber and D. J. Robbins, J. Biol. Chem., 2006, 281, 4087–4093 CrossRef CAS.
- S. Kumar, K. A. Balczarek and Z. –C. Lai, Genetics, 1996, 142, 965–972 CAS.
- G. Bhardwaj, B. Murdoch, D. Wu, D. Baker, K. Williams, K. Chadwick, L. Ling, F. Karanu and M. Bhatia, Nat. Immunol., 2001, 2, 172–180 CrossRef CAS.
- M. Gering and R. Patient, Dev. Cell, 2005, 8, 389–400 CrossRef CAS.
- H. Hu, M. J. Hilton, X. Tu, K. Yu, D. M. Ornitz and F. Long, Development (Cambridge, UK), 2005, 132, 49–60 CAS.
- F. Long, U. Chung, S. Ohba, J. McMahon, H. M. Kronenberg and A. P. McMahon, Development (Cambridge, UK), 2004, 131, 1309–1318 CrossRef CAS.
- Y. P. Wang, G. Dakubo, P. Howley, K. D. Campsall, C. J. Mazarolle, S. A. Shiga, P. M. Lewis, A. P. McMahon and V. A. Wallace, Nat. Neurosci., 2002, 5, 831–832 CAS.
- G. D. Dakubo, Development (Cambridge, UK), 2003, 130, 2967–2980 CrossRef CAS.
- M. Ramalho-Santos, D. A. Melton and A. P. McMahon, Development (Cambridge, UK), 2000, 127, 2763–2772 CAS.
- J. Zhang, A. Rosenthal, F. de Sauvage and R. A. Shivdasani, Dev. Biol., 2001, 229, 188–202 CrossRef CAS.
- P. Mill, R. Mo, H. Fu, M. Grachtchouk, P. W. C. Kim, A. A. Dlugosz and C. C. Hui, Genes Dev., 2003, 17, 282–294 CrossRef CAS.
- K. Michno, K. Boras-Granic, P. Mill, C. C. Hui and P. A. Hamel, Dev. Biol., 2003, 264, 153–165 CrossRef CAS.
- Q. Ding, J. Motoyama, S. Gasca, R. Mo, H. Sasaki, J. Rossant and C. C. Hui, Development (Cambridge, UK), 1998, 125, 2533–2543 CAS.
- C. Bai, D. Stephen and A. Joyner, Dev. Cell, 2004, 6, 103–115 CrossRef CAS.
- L. Niswander, S. Jeffrey, G. R. Martin and C. Tickle, Nature, 1994, 371, 609–612 CrossRef CAS.
- V. Marigo, R. L. Johnson, A. Vortkamp and C. J. Tabin, Dev. Biol., 1996, 180, 273–283 CrossRef CAS.
- J. Doles, C. Cook, X. Shi, J. Valosky, R. Lipinski and W. Bushman, Dev. Biol., 2006, 295, 13–25 CrossRef CAS.
- P. Sanchez, A. M. Hernandez, B. Stecca, A. J. Kahler, A. M. DeGueme, A. Barrett, M. Beyna, M. W. Datta, S. Datta and A. Ruiz i Altaba, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12561–12566 CrossRef CAS.
- V. Marigo and C. Tabin, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9346–9351 CrossRef CAS.
- E. Dessaud, A. P. McMahon and J. Briscoe, Development (Cambridge, UK), 2008, 135, 2489–2503 CrossRef CAS.
- J. P. Morton and B. C. Lewis, Cell Cycle, 2007, 6, 1553–1557 Search PubMed.
- E. A. Barnes, K. J. Heidtman and D. J. Donaghue, Oncogene, 2005, 24, 902–915 CrossRef CAS.
- D. A. Berman, S. S. Karhadkar, A. R. Hallahan, J. I. Pritchard, C. G. Eberhart, D. N. Watkins, J. K. Chen, M. K. Cooper, J. Taipale, J. M. Olson and P. A. Beachy, Science, 2002, 297, 1559–1561 CrossRef CAS.
- L. L. Rubin and F. J. de Sauvage, Nat. Rev. Drug Discovery, 2006, 5, 1026–1033 CrossRef CAS.
- J. A. Williams, O. M. Guichert, B. I. Zaharian, Y. Xu, L. Chai, H. Wichterle, C. Kon, C. Gatchalian, J. A. Porter, L. L. Rubin and F. Y. Wang, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4616–4621 CrossRef CAS.
- A. E. Oro, K. M. Higgins, H. Zhilan, J. M. Bonifas, E. H. Epstein Jr. and M. P. Scott, Science, 1997, 276, 817–820 CrossRef CAS.
- J. P. Morton, M. E. Mongeau, D. S. Klimstra, J. P. Morris, Y. C. Lee, Y. Kawaguchi, C. Wright, M. Hebrok and B. C. Lewis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5103–5108 CrossRef CAS.
- S. P. Thayer, M. P. di Magliano, P. W. Heiser, C. M. Nielsen, D. J. Roberts, G. Y. Lauwers, Y. P. Qi, S. Gysin, C. Fernandez del-Castillo, V. Yajnik, B. Antoniu, M. McMahon, A. L. Warshaw and M. Hebrok, Nature, 2003, 425, 851–856 CrossRef CAS.
- J. Gong, X. Li and Z. Darzynkiewicz, J. Cell. Physiol., 1993, 157, 263–270 CrossRef CAS.
- R. L. Yauch, S. E. Gould, S. J. Scales, T. Tang, H. Tian, C. P. Ahn, D. Marshall, L. Fu, T. Janaurio, D. Kallop, M. Nannini-Pepe, K. Kotkow, J. C. Marsters, L. L. Rubin and F. J. de Sauvage, Nature, 2008, 455, 406–410 CrossRef CAS.
- C. Zhao, A. Chen, C. H. Jameieson, M. Fereshteh, A. Abrahamsson, J. Blum, H. Y. Kwon, J. Kim, J. P. Chute, D. Rizzieri, M. Munchhof, T. Vanarsdale, P. A. Beachy and T. Reya, Nature, 2009, 458, 776–779 CrossRef CAS.
- V. Clement, P. Sanchez, N. de Tribolet, I. Radovanovic and A. R. i Altaba, Curr. Biol., 2007, 17, 165–172 CrossRef CAS.
- C. D. Peacock, Q. Wang, G. S. Gesell, I. M. Corcoran-Schwartz, E. Jones, J. Kim, W. L. Devereux, J. T. Rhodes, C. A. Huff, P. A. Beachy, D. N. Watkins and W. Matsui, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4048–4053 CrossRef CAS.
- T. M. T. Hall, J. A. Porter, P. A. Beachy and D. J. Leahy, Nature, 1995, 378, 212–216 CrossRef CAS.
- V. Marigo, R. A. Davey, Y. Zuo, J. T. Cunningham and C. J. Tabin, Nature, 1996, 384, 176–179 CrossRef CAS.
- N. Fuse, T. Maiti, B. Wang, J. Porter, T. M. T. Hall, D. J. Leahy and P. A. Beachy, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10992–10999 CrossRef CAS.
- J. S. McLellan, S. Yao, X. Zheng, B. V. Geisbrecht, R. Ghirlando, P. A. Beachy and D. J. Leahy, Proc. Natl. Acad. Sci. U. S. A., 2007, 46, 17208–17213.
- F. Zhang, J. S. McClellan, A. M. Ayala, D. J. Leahy and R. J. Linhardt, Biochemistry, 2007, 46, 3933–3941 CrossRef CAS.
- P.-T. Chuang and A. P. McMahon, Nature, 1999, 397, 617–621 CrossRef CAS.
- S. Yao, L. Lum and P. A. Beachy, Cell (Cambridge, Mass.), 2006, 125, 343–357 CrossRef CAS.
- T. Tenzen, B. L. Allen, F. Cole, J. S. Kang, R. S. Krauss and A. P. McMahon, Dev. Cell, 2006, 10, 647–656 CrossRef CAS.
- J. S. McLellan, X. Zheng, G. Hauk, R. Ghirlando, P. A. Beachy and D. J. Leahy, Nature, 2008, 455, 979–982 CrossRef CAS.
- E. J. Michaud and B. K. Yoder, Cancer Res., 2006, 66, 6463–6467 CrossRef CAS.
- V. Singla and J. Reiter, Science, 2006, 313, 629–633 CrossRef CAS.
- D. Huangfu, A. Liu, A. S. Rakeman, N. S. Murcia, L. Niswander and K. V. Anderson, Nature, 2003, 426, 83–87 CrossRef CAS.
- A. Liu, B. Wang and L. A. Niswander, Development (Cambridge, UK), 2005, 132, 3103–3111 CrossRef CAS.
- K. C. Corbit, P. Aanstad, V. Signla, A. R. Norman, D. Y. R. Stainier and J. F. Reiter, Nature, 2005, 437, 1018–1021 CrossRef CAS.
- R. Rohatgi, L. Milenkovic, R. B. Corcoran and M. P. Scott, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 3196–3201 CrossRef CAS.
- R. Rohatgi, L. Milenkovic and M. P. Scott, Science, 2007, 317, 372–376 CrossRef CAS.
- W. F. Binns, L. F. James, J. L. Shupe and G. Everett, Am. J. Vet. Res., 1963, 24, 1164–1175 CAS.
- R. F. Keeler and W. Binns, Teratology, 1968, 1, 5–10 CrossRef CAS.
- M. K. Cooper, J. A. Porter, K. E. Young and P. A. Beachy, Science, 1998, 280, 1603–1607 CrossRef CAS.
- P. A. Beachy, M. K. Cooper, K. E. Young, D. P. von Kessler, W. J. Park, T. M. Hall, D. J. Leahy and J. A. Porter, Cold Spring Harbor Symp. Quant. Biol., 1997, 62, 191–204 CAS.
- J. K. Chen, J. Taipale, M. K. Cooper and P. A. Beachy, Genes Dev., 2002, 16, 2743–2748 CrossRef CAS.
- J. Taipale, J. K. Chen, M. K. Cooper, B. Wang, R. K. Mann, L. Milenkovic, M. P. Scott and P. A. Beachy, Nature, 2000, 406, 1005–1009 CrossRef CAS.
- T. Hosoya, M. A. Arai, T. Koyano, T. Kowithayakorn and M. Ishibashi, ChemBioChem, 2008, 9, 1082–1092 CrossRef CAS.
- H. Sasaki, C. Hui, M. Nakafuku and H. Kondoh, Development (Cambridge, UK), 1997, 124, 1313–1322 CAS.
- M. Frank-Kamenetsky, X. M. Zhang, S. Bottega, O. Guicherit, H. Wichterle, H. Dudek, D. Bumcrot, F. Y. Wang, S. Jones, J. Shulok, L. Rubin and J. Porter, J. Biol., 2002, 10, 1–19.
- S. Ding and P. G. Schultz, Nat. Biotechnol., 2004, 22, 833–840 CrossRef.
- M. Lauth, A. Bergstrom, T. Shimokawa and R. Toftgard, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 8455–8460 CrossRef CAS.
- B. Z. Stanton, L. F. Peng, N. Maloof, K. Nakai, X. Wang, J. L. Duffner, K. M. Taveras, J. M. Hyman, S. W. Lee, A. N. Koehler, J. K. Chen, J. L. Fox, A. Mandinova and S. L. Schreiber, Nat. Chem. Biol., 2009, 5, 154–156 CrossRef CAS.
- G. MacBeath, A. N. Koehler and S. L. Schreiber, J. Am. Chem. Soc., 1999, 121, 7967–7968 CrossRef CAS.
- F. G. Kuruvilla, A. F. Shamji, S. M. Sternson, P. J. Hergenrother and S. L. Schreiber, Nature, 2002, 416, 653–657 CrossRef CAS.
- D. S. Tan, Nat. Biotechnol., 2002, 20, 561–563 CrossRef CAS.
- M. D. Burke, E. M. Berger and S. L. Schreiber, Science, 2003, 302, 613–618 CrossRef CAS.
- J. C. Wong, S. M. Sternson, J. B. Louca, R. Hong and S. L. Schreiber, Chem. Biol., 2004, 11, 1279–1291 CrossRef CAS.
- J. E. Bradner, O. M. McPherson, R. Mazitschek, D. Barnes-Seeman, J. P. Shen, J. Dhaliwal, K. E. Stevenson, J. L. Duffner, S. B. Park, D. S. Neuberg, P. Nghiem, S. L. Schreiber and A. N. Koehler, Chem. Biol., 2006, 13, 493–504 CrossRef CAS.
- J. R. Dwyer, N. Sever, M. Carlson, S. F. Nelson, P. A. Beachy and F. Parhami, J. Biol. Chem., 2007, 282, 8959–8968 CrossRef CAS.
- R. B. Corcoran and M. P. Scott, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8408–8413 CrossRef CAS.
- M. K. Cooper, C. A. Wassif, P. A. Krakowiak, J. Taipale, R. Gong, R. I. Kelley, F. D. Porter and P. A. Beachy, Nat. Genet., 2003, 33, 508–513 CrossRef CAS.
- J. K. Chen, J. Taipale, K. E. Young, T. Maiti and P. A. Beachy, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14071–14076 CrossRef CAS.
- X. Wu, Q. Ding, N. Gray and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 14520–14521 CrossRef CAS.
- X. Wu, J. Walker, J. Zhang, S. Ding and P. G. Schultz, Chem. Biol., 2004, 11, 1229–1238 CrossRef CAS.
- S. Sinha and J. K. Chen, Nat. Chem. Biol., 2006, 2, 29–30 CrossRef CAS.
- D. Von Hoff, C. Rudin, P. LoRusso, M. Borad, R. Korn, E. Heath, R. Yauch, W. Darbonne, E. Kadel, K. Zerivitz, L. Nelson, H. Mackey, J. Marsters, F. de Sauvage and J. Low, Efficacy data of GDC-0449, a systemic Hedgehog pathway antagonist, in a first-inhuman, first-in-class Phase I study with locally advanced, multifocal or metastatic basal cell carcinoma patients, Proceedings of the 99th Annual Meeting of the American Association for Cancer Research, 2008, LB-138 Search PubMed.
- E. H. Epstein, Nat. Rev. Cancer, 2008, 8, 743–754 CrossRef CAS.
- N. Dahmane, P. Sanchez, Y. Gitton, V. Palma, T. Sun, M. Beyna, H. Weiner and A. R. i Altaba, Development (Cambridge, UK), 2001, 128, 5201–5212 CAS.
- S. R. Hingorani, L. Wang, A. S. Multani, C. Combs, T. B. Deramaudt, R. H. Hruban, A. K. Rustgi, S. Chang and D. A. Tuveson, Cancer Cell, 2005, 7, 469–483 CrossRef CAS.
- N. Mahindroo, C. Punchichewa and N. Fujii, J. Med. Chem., 2009, 52, 3829–3845 CrossRef CAS.
- S. J. Scales and F. J. de Sauvage, Trends Pharmacol. Sci., 2009, 30, 303–312 CrossRef CAS.
- Adapted from L. L. Rubin and F. J. de Sauvage, Nat. Rev. Drug Discovery, 2006, 5, 1026–1033 Search PubMed; S. Ding and P. G. Schultz, Nat. Biotechnol., 2004, 22, 833–840 CrossRef CAS.
- W. Wang, Z. Zhou, C. T. Walsh and A. P. McMahon, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2623–2628 CrossRef.
| This journal is © The Royal Society of Chemistry 2010 |