Incorporation of unnatural amino acids for synthetic biology
Natalya
Voloshchuk
a and
Jin Kim
Montclare
ab
aDepartment of Chemical and Biological Sciences, Polytechnic Institute of New York University, Brooklyn, NY 11201, USA
bDepartment of Biochemistry, SUNY-Downstate Medical Center, Brooklyn, NY 11203, USA. E-mail: jmontcla@poly.edu; Fax: +1 718-260-3676; Tel: +1 718-260-3679
First published on 4th September 2009
Abstract
The challenge of synthetic biology lies in the construction of artificial cellular systems. This requires the development of modular “parts” that can be integrated into living systems to elicit an artificial, yet programmed, response or function. The development of methods to engineer proteins bearing unnatural amino acids (UAAs) provides essential components that may address this challenge. Here we review the emerging strategies for incorporating UAAs into proteins with the endgame of engineering artificial cells and organisms.
1. Introduction
Synthetic biology aims to engineer artificial biological systems with the ultimate goal of programming novel cell and organism behaviour.1–5 This is being achieved via a bottom-up approach in which the key “parts” are nucleic acids, metabolites and proteins. With the advances in nucleic acid synthesis and recombinant DNA technology driving down the cost of constructing genes,6,7 the fabrication of cells to stimulate pattern formation,8 produce drugs,9 target cancer cells10 and disperse biofilms11 (among many others) have been realized. To meet the challenge inherent in expanding the function of biological systems, the development of orthogonal “parts” capable of functioning in the context of living matter is becoming increasingly important.12,13Proteins are essential components of synthetic cellular networks. Synthesis of cellular pathways with the ultimate goal of creating functional artificial cells can benefit greatly from the generation of proteins with desirable properties that can be effectively regulated. Proteins capable of functioning independently of the host organism’s endogenous circuitry require further developments in “parts” design.
The recent advances in reassigning the genetic code to unnatural amino acids (UAAs)12,14–16 expand the molecular toolbox of protein “parts”. In fact, introduction of UAA can impart new functions that are difficult or impossible to create with proteins comprised of the natural 20 amino acid building blocks such as photo-induced switching,17IR probe active18 and redox sensitive19proteins. Moreover, proteins bearing UAAs may result in improved properties such as hyperstability20,21 and protease resistance.22 Such features may be useful for tailoring orthogonal proteins in the context of synthetic biological networks. Here we discuss approaches that focus on protein engineering and that can potentially expand synthetic biology methods by UAA incorporation for the creation of artificial proteins.
2. Proteins in synthetic biology
Proteins play an important role in synthetic biology especially since the cellular pathways are mediated by protein–nucleic protein–protein and protein–small molecule interactions. The earliest example of synthetic genetic circuits relied on stitching various protein–DNA regulators in specified arrangements.23,24 Elowitz and Leibler engineered a repressilator comprised of a series of 3 well-studied transcriptional repressors PLlac01, PLtet01 and λPR in a single construct that resulted in oscillations akin to a biological clock.23 The Collins group also exploited the use of repressors and promoters to assemble a genetic toggle switch in which each promoter was inhibited by the repressor produced from the opposing promoter (Fig. 1A).24 This allowed the use of inducers to control the switch. The role of proteins as regulatory elements in genetic networks designs remain important for oscillating systems and genetic toggle switch engineering (Fig. 1A).23–25 Changes in half-life of proteins and cooperative regulatory protein–DNA as well as protein–protein (vida infra) interactions represent valuable aspects for tuning oscillations in synthetic genetic systems.25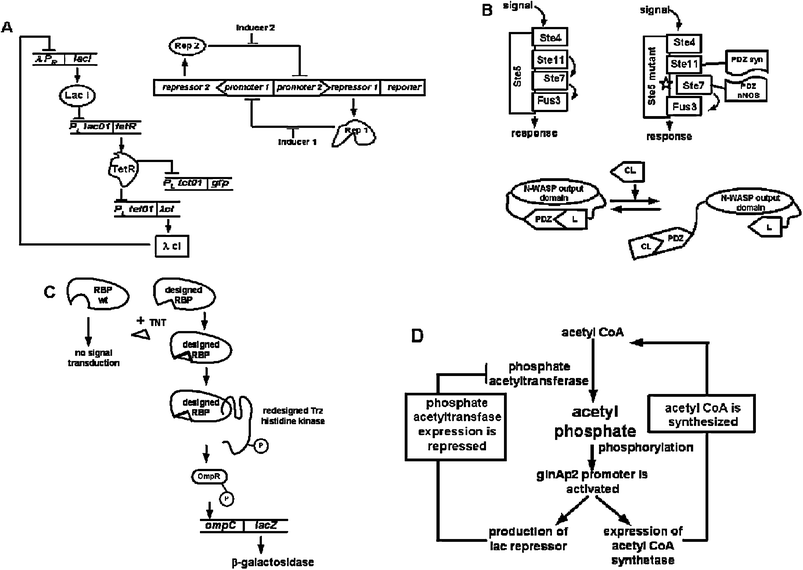 | ||
| Fig. 1 Proteins in synthetic biology. (A) Repressilator23 (left) is a synthetic network under transcriptional regulation where repressor protein, LacI, inhibits the transcription of tetR repressor gene. The product of the tetRgene, in turn, inhibits the transcription of λcIgene. To complete the cycle, λCI inhibits lacI expression. TetR also inhibits transcription of the gfp reporter gene. Oscillations of the repressilator network are observed in E. coli cells by monitoring GFP fluorescence. Genetic toggle switch24 (right) is constructed of two repressors and two constitutive promoters, resulting in oscillations produced by transient inducers. In the network, promoters are inhibited by repressors that are under transcriptional control of the opposing promoter, i.e. Rep 1 inhibits expression of repressor 2gene. (B) Non-native protein–protein interactions (top)28 are designed to rewire cellular pathway. Ste5 mutant contains mutations that destroy recruitment of Ste7 kinase resulting in non-functional mating pathway in yeast. Replacement of defective recruitment interaction with a designed heterologous protein–protein interactions rewires the signal transduction pathway and alleviates mating deficiency. A single input protein switch26 (bottom) is turned on and off by introduction of competitive ligand (CL). In the “on” position, N-WASP domain is able to carry out actin polymerization. (C) The RBP receptor is computationally designed to bind non-biological ligand (TNT)32 and initializes signal transduction pathway through interaction with transmembrane histidine kinase, Trz, resulting in autophosphorylation and phosphorylation of transcription regulator OmpR. Phosphorylated OmpR activates the lacZ reporter gene expression. (D) Signalling metabolite acetyl phosphate is used to regulate an oscillating synthetic circuit.33 The circuit is comprised of two enzymes that are under the transcriptional control of acetyl phosphate and two metabolites (acetyl CoA and acetyl phosphate) to generate oscillations. | ||
In addition to protein–nucleic acid interactions, protein–protein interactions have also been manipulated to re-wire signal transduction cascades.26–31 As the first example of engineering protein–protein networks, Lim et al. inserted a heterologous protein interaction within the yeast MAP kinase pathway and demonstrated that simple tethering can produce the appropriate function or output.28 Known mutations that disabled the kinase function in the mating pathway in Saccharomyces cerevisiae were rescued by introduction of alternative interactions in the scaffold. Most remarkably, a synthetic pathway was designed where native interactions were destroyed by mutagenesis and non-native interactions were established through tethering a different kinase set, allowing for a non-natural input-output scheme (Fig. 1B).28 This confirmed that proteins could form functional pathways that were “modular and flexible”.28 Moreover, using the actin regulatory protein N-WASP as the model system, Lim and coworkers designed a synthetic switch where the output of N-WASP was essentially spliced into a PDZ domain-ligand pair.26 This resulted in the switch being in the “off” state and upon addition of PDZ ligand, the switch was turned “on” (Fig. 1B). To further expand the ability of these switches to process multiple inputs, a combinatorial library was synthesized.
Four parameters were varied: (1) domain type; (2) domain–ligand interaction; (3) linker length and (4) domain architecture.26 The library resulted in synthetic switches with a variety of novel gating behaviours to non-physiological inputs. In order to engineer artificial pathways and functions like the ones described,26–31,34 a method to systematically identify common protein motifs and protein–protein interactions is needed. Programs and databases are being developed to recognize, classify, and organize productive protein–protein interactions with the overall objective for construction and reprogramming of intracellular interaction for synthetic biosystems.35–37
Signal transduction cascades as well as metabolic pathways often rely on protein–small molecule interactions. Key elements of molecular recognition for effective receptor–ligand interaction can be extracted for building highly specific cellular pathways. An initial example of engineering proteins for new ligands focused on employing computational methods to alter the specificities of periplasmic binding protein (PBP) receptors that were experimentally tested for binding artificial ligands (Fig. 1C).32 When the re-engineered receptors for trinitrotoluene (TNT) and seratonin were integrated into a 2-component synthetic signal transduction pathway with a reporter gene in Escherichia coli, expected dose response signals were observed.32
In the case of metabolic pathway engineering,9,33,38 the small molecule and how it interacts with various proteins in a pathway are critical. The Liao group designed an oscillatory circuit that coupled metabolite inputs and outputs with transcriptional regulation.33 The metabolite acetyl coenzyme A (AcCoA) was designed to be converted into acetyl phosphate (AcP) by phosphate acetyltransferase (Pta) and acetate by acetate kinase (Ack). The enzyme acetyl coenzyme A synthetase (Acs) could then process acetate back into AcCoA (Fig. 1D). This two-metabolite system was connected to transcription by engineering the acsgene under the glnAp2 promoter, which could be activated by AcP. The glnAp2 promoter was also engineered to control the lac repressor to repress the ptagene.33 The oscillatory circuit demonstrated the importance of small molecules in the network connectivity, in addition to protein–protein and protein–DNA interactions within the network.
Because of the integral role proteins play in synthetic biology as described above, protein engineering methods are increasingly in demand for the design of transcription networks,39–41 novel input-output signal transduction cascades,27,42 and metabolic pathways.38,43 As the applications for synthetic biology become focused on generating an “artificial” or “non-natural” function or product, approaches that can enhance orthogonality within the system will become vital. Protein modifications such as UAA incorporation provides a valuable mode of integrating orthogonality and may enable tuning of a complex circuit beyond the constrains of nature.44
3. Existing cellular pathways of unnatural amino acid incorporation
3.1 Selenocysteine and pyrrolysine
Until very recently, the standard genetic code was thought to encode for a set of 20 amino acids. Two amino acids that have always existed in natural organisms, selenocysteine and pyrrolysine, have been the latest additions to this set.45Selenocysteine is genetically encoded in prokaryotes and eukaryotes by the UGA stop codon.46 A selenocystenyl-tRNA[Ser]Sec is aminoacylated with serine by the seryl-tRNA synthetase and serine is converted into selenocysteine by selenocysteine synthase (SelA) in the presence of a selenium monophosphate donor provided by SelD46,47 (Fig. 2). Incorporation of selenocysteine depends on the existence of a selenocysteine insertion sequence (SECIS) adjacent to the UGA codon in prokaryotes or in the 3′-untranslated region of the selenoprotein mRNA in eukaryotes.46 The presence of the SECIS element allows suppression of UGA and cotranslational selenocysteine incorporation. In addition, selenocysteine incorporation requires the presence of several specialized factors, such as selenocysteinyl tRNA[Ser]Sec-specific elongation factor (SelB) in prokaryotes.46 The EFsec (eSelB), selenocysteinyl tRNA[Ser]Sec-specific elongation factor and the SECIS binding protein (SBP2) together form a functional equivalent of SelB in eukaryotes.46 An additional factor, L30 ribosomal protein (rpL30), enhances Sec incorporation into proteins in eukaryotes.46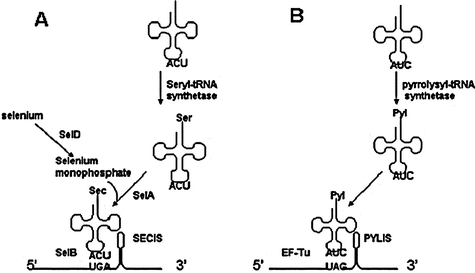 | ||
| Fig. 2 Cotranslational insertion mechanism of selenocysteine and pyrrolysine.46,47 (A) An opal suppressor tRNA is charged with serine by seryl-tRNA synthetase. In the presence of selenium monophosphate, serine is converted into selenocysteine in a reaction catalyzed by selenocysteine synthase (SelA). SelB mediates selenocysteine insertion at the UGA codon adjacent to the SECIS element on the mRNA. (B) Pyrrolysine is charged to amber suppressor tRNA by pyrrolysyl-tRNA synthetase and inserted at the amber codon next to the PYLIS element. EF-Tu aids in the cotranslational insertion. | ||
Pyrrolysine is determined to be genetically encoded by Methanosarcinaceae by the UAG stop codon in the methylamine methyltransferasegenes required for methylamine metabolism as well as the trimethylamine methyltransferase homolog from the gram-positive Desulfitobacterium hafniense.48,49 Similar to selenocysteine, pyrrolysine incorporation is reported to depend on a specific structural element, termed pyrrolysine insertion sequence (PYLIS) in the mRNA.48 Whereas selenocysteine is synthesized on the tRNA, pyrrolysine is directly charged to tRNAPyl (pylT) by the designated pyrrolysyl-tRNA synthetase (PylS) (Fig. 2).48 Elongation factor EF-Tu binds tRNAPyl and is able to decode UAG as pyrrolysine as long as the pylT and pylSgenes are also provided.48 Because both selenocysteine and pyrrolysine utilize a stop codon for incorporation, it has been a challenge to identify these amino acids until recently.
4. Unnatural amino acid incorporation methods
4.1 Site-specific incorporation
Methods designed specifically for UAA incorporation in vitro and in vivo rely on engineering changes into the cellular protein translation pathways. Based on our understanding of the selenocysteine and pyrrolysine pathways, the use of stop codonvia nonsense suppression appears to be a suitable strategy that can be applied to other UAAs. In particular, UAA incorporation by this method employs: (i) introduction of a stop codon at the position of interest into mRNA; (ii) isolation of a suppressor tRNA that recognizes the stop codon; and (iii) selection of aminoacyl-tRNA synthetase (AARS) to charge the suppressor tRNA with the particular analog. This would allow for the site-specific incorporation of amino acid analogs.Although the first two steps can be achieved by site-directed mutagenesis and use of suppressor tRNAs, the aminoacylation of the tRNA with UAA requires more effort. As an alternative to employing the AARS, tRNA aminoacylation with UAA can be accomplished with T4 RNA ligasein vitro.50 The UAA can be aminoacylated chemically upon reaction with pdCpA dinucleotide and then ligated to the truncated supressor tRNA lacking pCpA dinucleotide. Such aminoacylated tRNAs have been initially used in a cell-free translation mixture to incorporate UAA.51In vitro translation systems use cellular extracts that contain all the required components for efficient translation. The site of UAA incorporation is based on a readthrough of an amber (UAG) codon in mRNA by a suppressor tRNA aminoacylated with the analog. From initial in vitro experiments, it was recognized that the suppressor tRNA should not be charged by the endogenous AARSs from the cellular extracts since this would result in reduced efficiency of UAA incorporation. Thus, S. cerevisiae tRNACUAPhe is utilized in the in vitro E. coli expression system because S. cerevisiaetRNAs are orthogonal and not recognized by E. coli AARS.51 Although this allows for efficient incorporation of UAA, the chemically aminoacylated tRNA is not regenerated, limiting the amount of protein that can be produced.
4.1.1 In vivo UAA incorporation in Escherichia coli
Engineered AARS/suppressor tRNA pairs have been developed in which the AARS is optimized to charge suppressor tRNA with a particular UAA.52 Efficient enzymatic charging of the tRNA requires optimization or engineering of the AARS for specific recognition of a particular UAA. Initial experiments relied on using the nonessential suppressor tRNACUAPhe/phenylalanyl-tRNA synthetase pair from S. cerevisiae (SctRNACUAPhe/PheRS) by Furter for site-specific incorporation of p-fluorophenylalanine in E. coli.53 Since then, tremendous progress had been made in improving the AARS/tRNA pair so that they are completely non-cross reactive and orthogonal to the endogenous AARS/tRNA pairs within the cellular translational machinery. The Schultz group used tRNACUATyr/tyrosyl-tRNA synthetase pair from Methanococcus jannaschii (MjtRNACUATyr/TyrRS) for UAA incorporation in E. coli.18,54–61 Orthogonality of the pair was established through rounds of negative and positive screening of the MjtRNACUATyr mutant library15,61 (Fig. 3A). Suppressor tRNA variants in which eleven nucleotides of MjtRNACUATyr, that did not directly interact with MjTyrRS, were targeted by mutagenesis. For negative selection, the suppressor MjtRNACUATyr library was co-transformed into E. coli with a plasmid encoding the barnase gene bearing TAG codons at permissive sites within the gene. Cell death occurred when the suppressor MjtRNACUATyr was recognized and charged by endogenous E. coli AARS, resulting in barnase production. Truncated nonfunctional barnase was produced due to inability of endogenous AARSs to charge MjtRNACUATyr, yielding orthogonal or nonfunctional tRNAs. For positive selection, a plasmid containing the MjtRNACUATyrgene was purified from cells that survived negative selection and transformed into E. coli already bearing a plasmid encoding the cognate MjTyrRS and β-lactamase with an in-frame TAG codon. The transformed cells were grown on ampicillin allowing selection of survivors with β-lactamase transcribed, indicating successful charging of MjtRNACUATyr by cognate MjTyrRS. Cells died when cognate MjTyrRS could not charge MjtRNACUATyr or suppressor MjtRNACUATyr was not functional.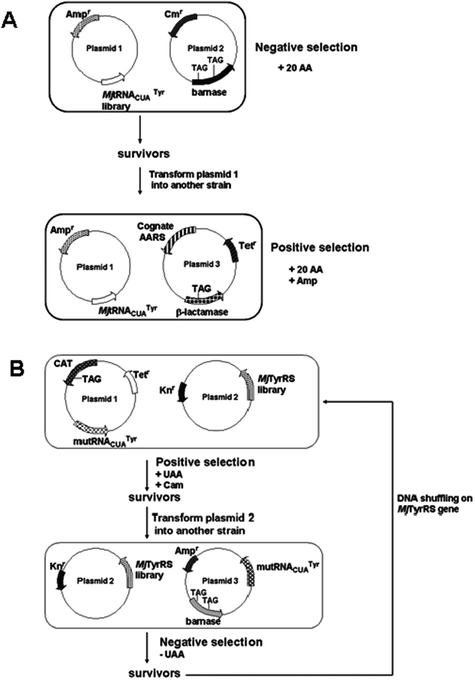 | ||
| Fig. 3 Selection strategies of orthogonal AARS/tRNA pairs for in vivo, site-specific incorporation using nonsense codons. (A) Negative and positive selection using barnase and β-lactamase as reporter genes for orthogonal amber suppressor MjtRNA in E. coli.15,61 When MjtRNA is charged by endogenous AARS, barnase is produced and cells die. Orthogonal MjtRNA cannot be recognized by cellular AARS and cells survive. Cognate AARS was identified by ability of cells to grow on ampicillin. (B) Positive and negative selection scheme for MjTyrRS specific for UAA. The selected mutRNACUATyr as shown in (A) is used for the following selections.54MjTyrRS library variants capable of activating UAA survive on chloramphenicol in presence of UAA, but cannot survive in media containing no UAA due to read through of the catgene. | ||
MjTyrRS variants that are able to charge suppressor MjtRNACUATyr with a particular UAA were identified from a library.18,54,57–61 Five residues in the active site of the MjTyrRS were randomized via saturation mutagenesis to generate a library of 1.6 × 109 variants.15,61 The residues were identified from the crystal structure of a TyrRS homolog from Bacillus stearothermophilus. A MjTyrRS variant capable of charging the suppressor MjtRNACUATyr with a selected UAA with high fidelity was identified through several rounds of positive and negative selection (Fig. 3B).
As a positive selection, the MjTyrRS library was transformed into E. coli bearing a plasmid with a gene for the orthogonal suppressor MjtRNACUATyr and chloramphenicol acetyltransferase (CAT) with an in-frame TAG codon. Surviving cells grown on chloramphenicol in the presence of UAA indicated that MjTyrRS was able to charge MjtRNACUATyr with UAA or natural amino acids. This positive selection was followed by negative selection, where plasmid containing MjTyrRS variants from survived cells was transformed into E. coli bearing a plasmid encoding barnase with several in-frame nonsense codons. Cells were grown in the media depleted of UAA. Cells expressing MjTyrRS variants capable of charging suppressor MjtRNACUATyr only with UAA survived because barnase was not produced. Genes for MjTyrRS variants that charged MjtRNACUATyr with UAA could be isolated and serve as a base for a new library created by DNA shuffling. The cycle of positive and negative selections could be repeated several times to identify the most effective and efficient MjTyrRS variants for UAA incorporation. Another selection method for synthetase specificity involved combination of chloramphenicol resistance and a fluorescent reporter allowing use of a single E. coli strain for positive and negative screens.54–55,63 The MjtRNACUATyr/TyrRS pair was used to incorporate a variety of UAA into proteins in E. coli15,18,54–60,63–65 (Table 1). In addition, other orthogonal tRNA/AARS pairs were developed for in vivo incorporation in E. coli66,67 (Table 1). Recently, a single plasmid selection system for the evolution of AARSs was reported by Schultz and coworkers.68 The fusion genechloramphenicol acetyltransferase (cat) and uracil phosphoribosyltransferase (uprt) was used as a positive/negative selection marker.68 Cells that were able to incorporate UAA in response to the amber codon were selected for in the presence of chloramphenicol and cells that harbored AARS variants that incorporated endogenous amino acids were selected against by adding 5-fluorouracil.68 The creation of orthogonal pairs further expands the molecular toolbox of UAAs that can be cotranslationally inserted into proteins.
| Organism | tRNA/AARS pair | Derived from | Analog |
|---|---|---|---|
| E. coli | tRNACUAPhe/PheRS | S. cerevisiae | p-fluorophenylalanine 53 |
| tRNACUATyr/TyrRS | M. jannaschii | p-aminophenylalanine 55,60,63 | |
| naphthylalanine 55 | |||
| p-benzoylphenylalanine 55,65 | |||
| p-methyoxyphenylalanine55 | |||
| p-aminomethylphenylalanine 55 | |||
| p-methylphenylalanine 55 | |||
| p-trifluoromethylphenylalanine55,56 | |||
| p-nitrophenylalanine 55 | |||
| p-isopropylphenylalanine 63 | |||
| O-allyl-tyrosine 63 | |||
| p-acetylphenylalanine 57,65 | |||
| p-azidophenylalanine 54,65 | |||
| p-iodophenylalanine 58,65,68 | |||
| β-N-acetylglucosamine-serine59 | |||
| p-cyanophenylalanine 18 | |||
| p-hydroxyphenyllactic acid 64 | |||
| O-methyltyrosine74 | |||
| phenyl-selenocysteine 75 | |||
| (2,2′-bipyridin-5-yl)alanine76,77 | |||
| (8-hydroxyquinolin-3-yl)alanine 78 | |||
| p-iodophenylalanine 68 | |||
| 2-amino-3-(8-hydroxyquinolin-3-yl)propanoic acid 78 | |||
| tRNACUAPyl/PylRS | M. maize | N ε -cyclopentyloxycarbonyl-L-lysine79 | |
| O-nitrobenzyl-oxycarbonyl-Nε-L-lysine79 | |||
| tRNAAAAPhe/PheRS T415G | S. cerevisiae | 3-(2-naphthyl)alanine 80 | |
| tRNACUAPhe/PheRS T415G | S. cerevisiae | p-bromophenylalanine 67 | |
| benzothienylalanine66 | |||
| 6-bromotryptophan 66 | |||
| 6-chlorotryptophan 66 | |||
| tRNACUAPyl/PylRS | M. barkeri | N ε -acetyllysine81 | |
| S. cerevisiae | tRNACUATyr/TyrRS | E. coli | p-benzoylphenylalanine 62,69 |
| p-azidophenylalanine 62,69 | |||
| p-acetylphenylalanine 62,69 | |||
| p-propargyloxyphenylalanine69 | |||
| O-methyltyrosine62 | |||
| p-iodotyrosine 62 | |||
| 4,5-dimehtoxy-2-nitrobenzylserine62 | |||
| tRNACUALeu/LeuRS | E. coli | methionine and cysteine homologs82 | |
| 2-aminocaprylic acid82 | |||
| 2-aminononanoic acid 82 | |||
| 2-aminodecanoic acid 82 | |||
| P. pastoris | tRNACUATyr/TyrRS | E. coli | p-benzoylphenylalanine 83 |
| p-azidophenylalanine 83 | |||
| p-(propargyloxy)-phenylalanine83 | |||
| p-methoxyphenylalanine83 | |||
| p-iodophenylalanine 83 | |||
| tRNACUALeu/LeuRS | E. coli | 4,5-dimethoxy-2-nitrobenzylserine 83 | |
| dansylalanine83 | |||
| Mammalian cells | tRNACUATyr/TyrRS | B. stearothermophilus | 3-iodotyrosine 70 |
| E. coli | p-acetylphenylalanine 73 | ||
| p-benzoylphenylalanine 73 | |||
| tRNACUATrp/TrpRS | B. subtilis | 5-hydroxytryptophan 71 | |
| tRNACUATyr/TyrRS | E. coli | O-methyltyrosine72 | |
| p-benzoylphenylalanine 72 | |||
| tRNACUALeu/LeuRS | E. coli | 2-amino-3-(5-(dimethylamino)naphthalene-1-sulfoamido) | |
| propanoic acid 72 | |||
| tRNACUAPyl/PylRS | M. maize | o-nitrobenzyl-oxycarbonyl-Nε-L-lysine79 | |
| N ε -tert-butyloxycarbonyl-L-lysine84 | |||
| N ε -benzyloxycarbonyl-L-lysine84 | |||
| N ε -acetyllysine84 |
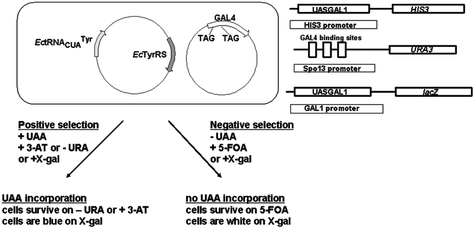 | ||
| Fig. 4 Positive and negative selection of orthogonal EcTyrRS/mutRNACUATyr pair specific for UAA in S. cerevisiae.62 Only S. cerevisiae cells carrying genes for orthogonal EcTyrRS/mutRNACUATyr pair that mediate UAA incorporation can survive on 3-AT or without uracil when grown in presence of UAA. In the absence of UAA, cells that contain active EcTyrRS/mutRNACUATyr pair cannot survive in presence of 5-FOA. When GAL4 amber mutations are suppressed with natural amino acids, URA3 is expressed and cells are converting 5-FOA to a toxic product.62 | ||
Exploiting the orthogonal pairs from previous selections, the Wang group utilized the EctRNACUATyr/TyrRS pair selected from yeast.72 Several expression constructs were tested in HeLa cell lines . In order to overcome the challenge of expressing sufficient quantities of functional tRNAs, the H1 promoter was utilized to drive the functional biosynthesis of EctRNACUATyr. Moreover, the 3′-flanking sequence of human tRNAfMet was also needed for efficient expression of the EctRNACUATyr in mammalian cells. The genes for EctRNACUATyr and EcTyrRS was engineered on a single tRNA/AARS expression plasmid. This plasmid was coexpressed with another plasmid containing the GFP gene with an in-frame TAG codon. Those EctRNACUATyr that were expressed and correctly processed was aminoacylated, yielding a fluorescent cell through suppression of the TAG codon in GFP. Orthogonality of EctRNACUATyr was tested by removing EcTyrRS gene from the expression construct. Transfection of the resultant plasmid into HeLa cells did not change fluorescent intensity, indicating inability of the endogenous synthetases to aminoacylate the EctRNACUATyr. In addition, they proved that the H1 promoter could efficiently express E. colitRNAs by demonstrating fluorescence using a different orthogonal EctRNACUALeu/EcLeuRS pair. For UAA incorporation, the EcTyrRS gene in the tRNA/AARS expression plasmid were replaced by the OmeTyrRS gene, a synthetase specific for O-methyltyrosine (OmeTyr) previously evolved in yeast.72
Successful suppresssion of TAG in GFP was demonstrated in HeLa cells grown in media containing UAA, with a 41% incorporation efficiency. Expression of BraRS, a synthetase specific for p-benzoylphenylalanine (Bra) evolved in yeast, led to 13% incorporation of the UAA. EctRNACUALeu and mutant synthetase specific for 2-amino-3-(5-(dimethylamino)-naphthalene-1-sulfonamido)propanoic acid (DanAla) were expressed in HeLa cells with a 13% incorporation efficiency. Similarly, incorporation of OmeTyr and Bra was achieved in mouse hippocampal neurons. While OmeTyr was incorporated into position 19 of the inactivation peptide of potassium channel Kv1.4 expressed in HEK293T cells using the EctRNACUATyr/OmeTyrRS pair, DanAla was incorporated using the EctRNACUALeu/DanAlaRS pair. The presence of different sized UAAs allowed insight into the inactivation mechanism of Kv1.4.72 RajBhandary, Sakmar and coworkers employed amber suppressor tRNA derived from B. stearothermophilus tRNACUATyr, and E. coli TyrRS variants developed previously to incorporate p-acetylphenylalanine (Acp) and p-benzoylphenylalanine (Bzp) into CCR5 and rhodopsin in HEK293T cells.73 In this case, the firefly luciferase reporter was used to screen for orthogonality and specificity of UAA incorporation. The luciferase reporter contained a TAG codon at position Y70, which in the presence of the appropriate orthogonal tRNA/TyrRS pair and UAA produced full length luciferase.
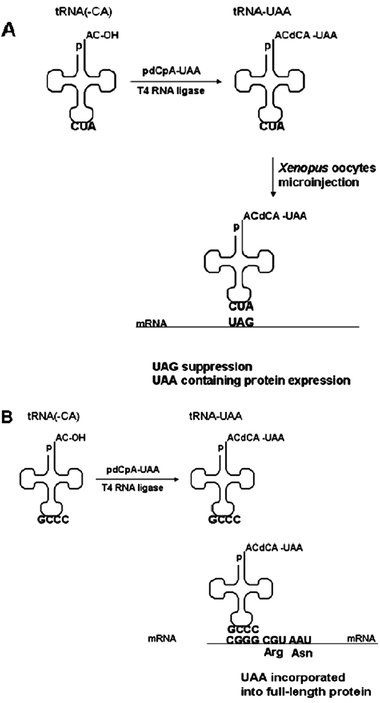 | ||
| Fig. 5 Alternative approaches for site-specific incorporation. (A) UAA incorporation through nonsense suppression in Xenopusoocytes.88 Designed tRNA with a chemically ligated UAA is introduced into Xenopusoocytes by microinjection and translates the mRNA bearing the stop codon. (B) Frameshift method for site-specific incorporation of UAA using four-base codons in cell-free expression systems. When CGGG is translated by UAA-charged tRNA, full length protein is produced with the UAA inserted at the CGGG site.89 When CGGG is read as CGG by endogenous tRNAprotein synthesis is terminated. | ||
| Expression system | tRNA | Analog |
|---|---|---|
| Xenopus oocytes | S. cerevisiae tRNA CUA Phe | 3-N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)2,3-diaminopropionic acid86 |
| T. thermophila tRNACUA | over 10088 | |
| T. thermophila tRNACUAGln | 5-fluorotryptophan 90 | |
| T. thermophila tRNAACUGln | 5-fluorotryptophan 85 | |
| S. cerevisiae tRNACCCGPhe | 5-fluorotryptophan 85 | |
| S. cerevisiae tRNAACCCPhe | α-aminobutyric acid85 | |
| norvaline 85 | ||
| E. coli | Archae tRNAAGGALys/LysRS | L-homoglutamine93 |
| Archae tRNAAGGALys/LysRS and Mj tRNACUATyr/TyrRS | L-homoglutamine and O-methyltyrosine93 | |
| E. coli cell-free expression system | S. cerevisiae tRNAACCUPhe | p-biphenylalanine94 |
| S. cerevisiae tRNAACCCPhe | p-benzoylphenylalanine 94 | |
| S. cerevisiae tRNAACCGPhe | p-phenylazophenylalanine94 | |
| p-naphthylalanine 94 | ||
| S. cerevisiae tRNACCCGPhe | BODIPY FL p-aminophenylalanine95 | |
| S. cerevisiae tRNAUACCGPhe | p-nitrophenylalanine 96 | |
| ε-nitrobenzoxadiazolyllysine96 | ||
| Eukaryotic rabbit reticulocyte cell-free expression system | S. cerevisiae tRNAACCGPhe | p-nitrophenylalanine 97 |
| S. cerevisiae tRNAAGCGPhe | p-nitrophenylalanine 97 | |
| S. cerevisiae tRNAAGGGPhe | p-nitrophenylalanine 97 | |
| S. cerevisiae tRNAAGAGPhe | p-nitrophenylalanine 97 | |
| S. cerevisiae tRNAAUAGPhe | p-nitrophenylalanine 97 | |
| S. cerevisiae tRNAACCCPhe | p-nitrophenylalanine 97 | |
| Sf21 insect cell-free expression system | M. acetivorans tRNAPylCCCG or | p-nitrophenylalanine 98 |
| M. acetivorans tRNAPylUCCG | 1-naphthylalanine 98 | |
| 2-naphthylalanine 98 |
A selection system for efficient frameshift suppressor tRNAs with high decoding activity was developed recently.95 Synthetic tRNAs were ligated with 5′-phosphodeoxycytidyl-phospho-puromycin (pdCp-puromycin) by T4 RNA ligase. These puromycin-tRNAs were added to the cell-free translation system with streptavidin-linked mRNA that contained the CGGG codon at the C-terminus. Puromycin-tRNAs that bound to the ribosome produced steptavidin-tRNA fusions that could be released from the ribosome complex through reaction with EDTA and recovered with biotin-covered beads. To create a DNA library that encoded functional tRNAs, recovered steptavidin-tRNAs were subjected to RT-PCR. Several rounds of selection were performed through reaction with EDTA and recovered with biotin-covered beads. To create a DNA library that encoded functional tRNAs, recovered steptavidin-tRNAs were subjected to RT-PCR. Several rounds of selection were carried out to identify frameshift tRNACGGG with high efficient decoding activity in E. coli cell-free expression system.85 Frameshift suppression was also used in vivo to introduce L-homoglutamine in response to the four-base codon AGGA in E. coli.93 The orthogonal LysRS/tRNA pair derived from Archaea was used in the system.93 Simultaneous incorporation of two amino acids (L-homoglutamine and O-methyltyrosine) at two different positions in myoglobin was demonstrated in vivo in E. coli using both the developed frameshift suppression (Archae tRNAAGGALys/LysRS) and amber suppression (MjtRNACUATyr/TyrRS).93
In vivo UAA incorporation in Xenopusoocytes using frameshift suppression was demonstrated with SctRNACCCGPhe for 5-fluorotryptophan and SctRNAACCCPhe for α-aminobutyric acid or norvaline.85 Simultaneous incorporation of 5-fluorotryptophan and α-aminobutyric acid (or norvaline) was done by microinjection of both tRNACUA (THG73, described above) and SctRNACCCGPhe (or SctRNAACCCPhe) with a neuroreceptor mRNA. For incorporation of three amino acids, three tRNAs were microinjected into oocytes: 5-fluorotryptophan-tRNACUA/THG73, α-aminobutyric acid-SctRNACCCGPhe and norvaline-tRNAACCCPhe. Expression levels were adequate for electric current measurements of the functional neuroreceptor.89
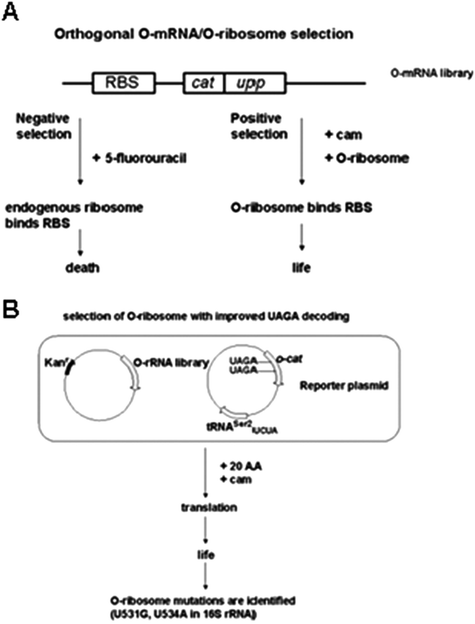 | ||
| Fig. 6 Orthogonal O-mRNA/O-ribosome selection in E. coli for improved UAA incorporation.99 (A) The ribosome binding site (RBS) on the mRNA is modified to eliminate endogenous ribosome binding. Uracil phosphoribosyltransferase (UPRT) is encoded by uppgene. 5-fluorouracil is converted by UPRT into an inhibitor of thymidylate synthase, causing cell death. Translation of cat allows O-ribosome containing cells survive on chloramphenicol. (B) Selection of O-ribosome with improved UAGA decoding in E. coli.100 Cells are transformed with O-rRNA library and the reporter plasmido-cat. Survival of cells on chloramphenicol indicates efficient UAGA decoding. | ||
The ribosome library was co-transformed with a reporter plasmid bearing the catgene with 2 UAGA stop codons fused downstream of the orthogonal ribosome-binding site (o-cat) to isolate ribosomes capabe of translating the O-mRNA.100 Cells containing o-cat (UAGA103,UAGA146)/tRNAser2UCUA and O-ribosome library were grown in the presence of chloramphenicol. The ribosome binding site sequence of the O-mRNA was altered so that it was not recognized by the endogenous ribosome.99,100 Through this selection U531G, U534A 16S rRNA mutant ribosome was identified for efficient UAGA decoding. This resulted in an O-ribosome variant that could only carry out translation of the O-mRNA, and not the cellular mRNAs.100 The O-ribosome/O-mRNA pair was capable of functioning in parallel to the cellular translation.100 The system offered the unique advantage of not interrupting cellular translation that could consequently cause cell death.100
Using a previously developed p-benzoylphenylalanyl-tRNA synthetase/tRNACUA (BraRS/tRNACUA) pair evolved from the MjTyrRS/tRNACUA pair in E. coli,63 in conjunction with the evolved O-ribosome with corresponding O-mRNA, they reported an increase in efficiency of 62% for site-specific p-benzoylphenylalanine (Bpa) incorporation into protein expressed in E. coli100,102Bpa incorporation at two amber stop codon sites in the test protein resulted in a slightly lower efficiency of 38%.100 Thus O-ribosome/O-mRNA pair was shown to effectively read through the amber codon for UAA incorporation. Essentially, the evolved O-ribosome decreased the functional interaction with E. coli RF-1, allowing suppressor tRNA to compete more efficiently for A-site binding in the presence of a UAG codon on the mRNA.100,102
4.2 Residue-specific incorporation
Residue-specific UAA incorporation employs the organism’s cellular translational system to replace a natural amino acid with an analog. Since all the amino acids are substituted with an UAA, this often results in alteration of the global properties of proteins.105 Initially, UAAs have been incorporated into proteins expressed in E. coli strains auxotrophic for a particular amino acid. The first experiments to demonstrate this was performed by Cowie and Cohen in 1957 in which methionine was replaced with selenomethonine.106,107 This provided a heavy atom replacement that would be used subsequently as a tool for X-ray crystallography. By residue-specific incorporation, structurally similar analogs to the natural amino acids such as difluoromethionine,108trifluoromethionine,1092-amino-5-hexenoic acid,1102-amino-5-hexynoic acid,110,111norleucine,110,112 structurally diverse set of proline analogs,113–115 trifluoroisoleucine,116,117 2-amino-3- methyl-4-pentenoic acid,118trifluorovaline,116,119azidohomoalanine,120,121fluorophenylalanine isomers,122aminotryptophan isomers,123fluorotryptophan isomers,124–1265-hydroxytryptophan,126 7-azatryptophan,126 homopropargylglycine121,127 and trifluoroleucine128,129 was incorporated in vivo into proteins expressed in E. coli (Table 3). Expression of proteins bearing UAA such as azidohomoalanine,130,131 homopropargylglycine,132 photo-eucine133 and photo-methionine133 in mammalian cells has been accomplished as well (Table 3).In order to expand the structural diversity of UAA incorporated globally, modifications to the cellular biosynthetic machinery have been made. Of the components involved in protein translational, the AARS serves as the gatekeeper for accepting or rejecting a particular amino acid to be coupled with its tRNA. Specifically, the methods implemented to shepherd UAA incorporation relies on: (i) introducing additional copies of the natural AARS; (ii) enlargening the AARS binding pocket; and (iii) shrinking the editing domain of the AARS.
Substrate recognition specificity and editing function of the AARS have been modified in order to expand incorporation of non-isosteric amino acid analogs. In order to introduce bulkier UAAs, Henneke and coworkers created an A294G mutation in the E. coliphenylalanyl-tRNA synthetase (PheRS).136,137 This increased the size of the binding pocket to allow charging of tRNAPhe with the para-halogenated phenylalanine analogs: p-chlorophenylalanine and p-bromophenylalanine (Table 3).136–139 The same mutant PheRS was used by the Tirrell group to incorporate additional phenylalanine analogs p-azidophenylalanine, p-iodophenylalanine, p-ethynylphenylalanine and p-cyanophenylalanine in E. coli (Table 3).127,140,141 The introduction of a T251G in addition to the A294G mutation in PheRS enabled the incorporation of ketone functionality of p-acetylphenylalanine into recombinant dihydrofolate reductase (DHFR) (Table 3).143 As an alternative to altering the binding pocket of the AARS, others have made changes to disable the editing domain to facilitate UAA incorporation. First demonstrated by Schimmel and coworkers, a single amino acid substitution in the editing pocket of valyl-tRNA synthetase (ValRS) from threonine to proline resulted in the misacylation of tRNAVal.144 This enabled aminobutyrate incorporation in response to valinecodons with 20% efficiency.144 Subsequently Tang and Tirrell illustrated that substitution of T252 in the editing pocket of leucyl-tRNA synthetase (LeuRS) with larger amino acids led to misacylation of tRNALeu, allowing the incorporation of norvaline, norleucine, allylglycine, homoallylglycine, homopropargylglycine and 2-butynylglycine (Table 3).145
Recently, the development of screening methods to identify variants of AARS capable of charging amino acid analogs has allowed for the rapid expansion of residue-specific UAA incorporation.103,104,120 A saturation mutagenesis library of MetRS variants was screened to identify mutants that enable azidonorleucine incorporation by Tirrell et al.103 (Fig. 7A, Table 3). The library was cloned into a vector that also contained the gene for outer membrane protein C (OmpC). Reactive UAA, such as azidohomoalanine or azidonorleucine, was introduced into OmpC in E. coli. Since the OmpC was expressed on the outer surface of the E. coli, cells displaying UAA could be covalently biotinylated by a Cu-catalyzed azide-alkyne ligation and labeled with fluorescent avidin.120Fluorescence-activated cell sorting (FACS) was employed to sort and differentiate labeled from unlabeled cells. Selected cells could undergo another round of sorting viaFACS to identify MetRS variants able to charge the azide-bearing UAAs. Through this screen a MetRS variant with a L13G mutation was identified to incorporate azidonorleucine.127
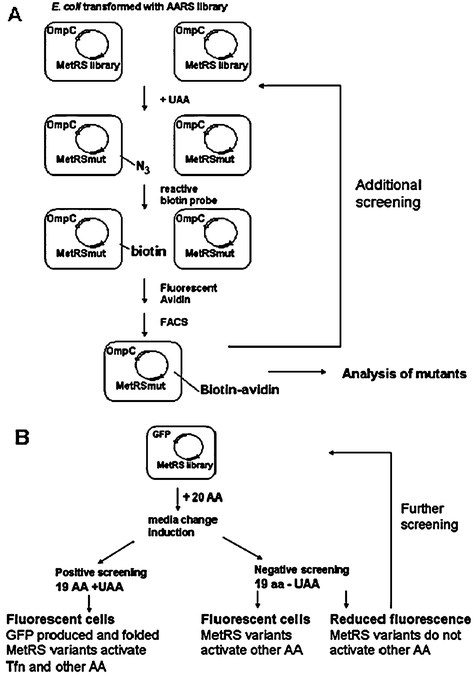 | ||
| Fig. 7 Screening methods of AARS mutants for residue-specific incorporation of UAAs. (A) Identification of MetRS variants in E. coli for incorporation of azidonorleucine by exploiting its reactive side chain via bio-orthogonal 3 + 2 cycloaddition.103 Cells are transformed with plasmid bearing a MetRS library and the reporter OmpCgene. OmpC expression is induced in presence of UAA. Successful incorporation of UAA results in cell surface display of reactive side chain that forms covalent bond with reactive biotin in the presence of Cu(II). Biotin on the cell surface is subsequently bound to fluorescent avidin. Employing fluorescence-activated cell sorting (FACS), labelled cells can be isolated. Sorted cells can be analyzed to identify MetRS mutants or subject to additional screening. (B) Screening for MetRS variants that activate trifluoronorleucine for residue-specific incorporation in E. coli.104 Cells transformed with a construct containing a MetRS library and the GFP reporter gene are grown in media containing 19 amino acids in the presence (positive screening) or absence (negative screening) of UAA. Cells collected in negative screen are subjected to further positive and negative screening to identify cells that grow in the presence of UAA and exhibit high fluorescence. | ||
For this type of screen, the UAA must be limited to a bioorthogonal chemical handle for reaction with the label. To overcome this limitation, OmpC was replaced with a GFP reporter (Fig. 7B).104 All the methionine residues in the β-barrel region of GFP were randomized first to generate a mutant that retained its fluorescence even upon methionine replacement. A GFP mutant in which all the methionine residues were replaced and exhibited fluorescence comparable to wild type was identified. Five methionine residues were then introduced into this mutant at positions that did not alter fluorescence of the folded GFP. This resulting mutant GFP was used for the screening process. Cells were transformed with a plasmid bearing the MetRS library and mutant GFP were grown in media supplemented with 19 amino acids in the presence of 6,6,6-trifluoronorleucine (Tfn) and sorted by FACS as a positive screen (Table 3). A negative screen was perfomed in which the selected cells were expressed in 19 amino acids in the absence of methionine to remove any MetRS variants that could activate natural amino acids other than methionine The postive and negative screening was repeated several times to enrich for clones active towards UAA incorporation. The resulting MetRS variant (L13S, Y260L, H301L) was identified to demonstrate Tfn incorporation into the recombinant test protein DHFR with near complete replacement.
4.3 Multi-site-specific incorporation
In the prior cases of site-specific incorporation, the ability to include a second or additional UAAs in response to the same stop codon is limited due to lower read through and competition with release factors. Residue-specific incorporation, on the other hand, does not suffer from low read through or yield but is restricted to complete replacement of a designated natural amino acid with UAAs. Although the development of orthogonal ribosomes and mRNAs is improving the suppression efficiency, an alternative approach to accomplish multi-site-specific incorporation relies on exploiting the degeneracy of codons.Reassignment of degenerate sense codons was used to incorporate L-3-(2-naphthyl)alanine (Nal) at five positions in DHFR by Tirrell and coworkers80 (Table 1, Fig. 8). Similar to site-specific incorporation, an orthogonal SctRNA/PheRS pair was introduced into the E. coli expression system. Rather than utilizing a suppressor tRNA, they employed the tRNAGAAPhe from yeast and altered the anticodon to produce tRNAAAAPhe. This allowed for exact Watson–Crick pairing with the UUU minor codon such that Nal could be incorporated in response to their presence. The yeast PheRS T415G mutant was selected to accommodate the bulky Nal. This mutant ScPheRS/ tRNAAAAPhe pair was used for the expression of DHFR in an E. coliphenylalanine auxotrophic strain.80 Although the tRNAAAAPhe was shown to compete with endogenous tRNA for UUU decoding, the codons were assigned to Nal with 80% efficiency.105 The efficiency of incorporation was comparable to the efficiency of site-specific incorporation.55 Moreover, by this method, five residues were replaced with the UAA.
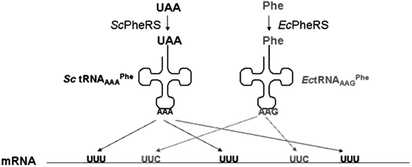 | ||
| Fig. 8 Reassignment of degenerate sense codonsin vivo in E. coli.80,105 A modified orthogonal tRNAAAA/PheRS pair from S. cerevisiae are introduced into E. coli that enable incorporation of naphthylalanine in response to the UUU codon. At the same time, endogenous E. colitRNA/PheRS pair mediates phenylalanine incorporation in response to the UUC codon. | ||
5. Organismal UAA incorporation
In all the aforementioned work, the UAAs have been incorporated into target proteins that are overexpressed15,16 or are being newly synthesized and later isolated or tagged.130,131,133 In addition to tailoring specific proteins in the cell, the ability to design most or all proteins with UAA in the cell would advance synthetic biology by providing a whole organism comprised of unnatural “parts”. This may lead to the development of truly artificial cells that will potentially possess different properties from their natural counterparts. Initial experiments were performed by Rennert and Anker in 1963.146 They demonstrated that E. coli was adaptable to growth on medium containing the UAA by the use of a chemostat. Normal growth rates and 5′,5′,5′-trifluoroleucine incorporation into all the proteins in E. coli were reported.146 More recently, E. coli variants capable of surviving in media containing 4-fluorotryptophan were evolved by Bacher and Ellington.147 Instead of using a chemostat, they gradually increased the amount of amino acid analog in the growth media until complete replacement was accomplished. This yielded global UAA incorporation throughout E. coli proteome. To identify mutations that contributed to the ability of E. coli to grow on 4-fluorotryptophan (Flt), genes from evolved clones were compared to the parental strain. They found that three proteins were mutated: tryptophanyl-tRNA synthetase (Q109P), aromatic amino acid permease (T30S, I47V, V365G), and tyrosine repressor (R380P).147 This suggested that these proteins and their mutations were critical for the organism to grow and integrate the Flt into E. coli. Moreover through this process, one might be able to generate strains capable of surviving and functioning in the presence of other UAAs.6. Engineered pathways for biosynthesis of UAA
In the previous case, the UAA is added externally by combining it with the medium that is used to culture the cells. Without UAA supplementation the cells are not capable of surviving, which may be problematic for synthetic biology applications that aim to interface artificial cell circuits with natural organisms and tissue. Schultz and coworkers engineered E. coli capable of biosynthesizing and incorporating p-aminophenylalanine (pAF) into proteins in response to amber nonsense codons60 (Fig. 9). The papA (4-amino-4-deoxychorismate synthase), papB (chorismate mutase), papC (prephenate dehydrogenase) genes involved in the biosynthetic pathway for pAF production from S. venezuelae were introduced into E. coli, along with a plasmid containing the MjtRNACUATyr/pAFRS pair previously isolated by rounds of positive and negative selection from MjTyrRS.60 Cellular synthesis of pAF and substitution into the recombinant sperm whale myoglobin in response to TAG codon (position 4) was detected by sequencing and mass spectroscopy analysis.60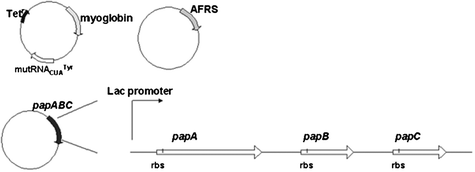 | ||
| Fig. 9 Biosynthesis and site-specific incorporation of p-aminophenylalanine (pAF) in E. coli.60 The genespapA, papB, and papC from S. venezuelae were engineered into the constructs to enable the in vivo production of pAF. | ||
7. Directed evolution of proteins bearing UAA
In order to achieve fully artificial organisms comprised of unnatural protein “parts”, methodology that enables the design of functional proteins bearing UAA is critical. Although the incorporation of UAA has led to functional artificial proteins with improved properties, in many cases, a reduction in function ensues especially when the UAA is integrated without rational design.129,148 Directed evolution provides a solution for the identification of functional proteins and can be applied to proteins comprised of UAAs.129,148 A diverse library of protein variants can be created by error-prone PCR or DNA shuffling and then screened for a particular function.150–152,129,148 In contrast to rational design where mutants are constructed based on known structural information of the target protein, directed evolution relies on the construction of a library of mutants in the absence of structure.150–152,129,148Sequence optimization experiments were reported for GFP containing UAA at one position by Sisido et al. (Fig. 10).149 These experiments were carried out in cell-free E. colitranscription system. Specifically, p-aminophenylalanine (pAF) was incorporated into GFP at Y66 in response to the frameshift CGGG codon, resulting in reduced fluorescence intensity as compared to wild type GFP. Saturation mutagenesis was used to create library of mutants with amino acid substitutions at S65 and Y145 (Fig. 10). From this library, 42 colonies were subjected to colony PCR, in vitro translation and fluorescence measurements. Three mutants, Y145L, Y145F and Y145M, were identified to exhibit improved fluorescence intensity with pAF at position 66. By combining site-specific incorporation with directed evolution, they were able to enhance the fluorescent property of the GFP bearing the UAA.
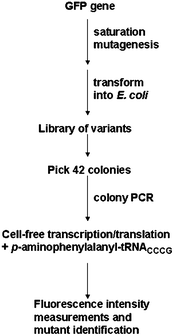 | ||
| Fig. 10 Identification of a variant with improved fluorescence of GFP bearing as site-specific inserted UAA.149 GFP library, created by saturation mutagenesis, was transformed into E. coli. Selected colonies were isolated for GFP gene amplification . These genes were translated in cell-free E. coli expression system in the presence of UAA and expressed variants were analyzed by fluorescence measurements. | ||
Residue-specific incorporation and directed evolution was integrated to recover or enhance the function of proteins bearing UAAs (Fig. 11). The incorporation of 5′,5′,5′-trifluoroleucine (TFL) into chloramphenicol acetyltransferase (CAT) yielded a loss in thermostability.129,148,156 In the case of CAT, Montclare and Tirrell created random mutagenesis libraries by error-prone PCR, transformed them into E. colileucine auxotrophic strains and expressed them in the presence of TFL in 96-well plates.129
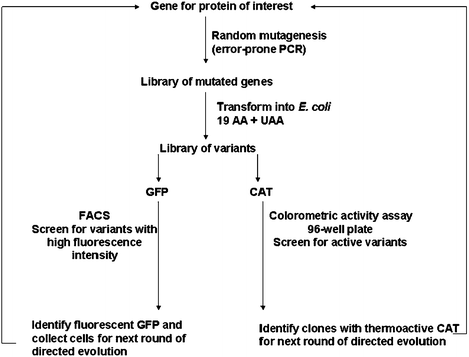 | ||
| Fig. 11 Scheme of directed evolution protocol for CAT129 and GFP148proteins with UAA incorporated in residue-specific manner expressed in E. coli. Gene library generated by error-prone PCR was transformed in E. coli and grown in media containing UAA. Variants were screened for desirable properties: activity for CAT, fluorescence intensity for GFP. The best variants were chosen for additional rounds of directed evolution. | ||
A high-throughput colorimetric screening method was developed for cell lysates bearing the fluorinated variants.129 Most active clones were identified for the additional rounds of random mutagenesis and screening. A fluorinated CAT variant with enhanced thermal stability containing three mutations (K46M, S87N and M142I) was identified.129 Recently, this screen was also employed by the Montclare and coworkers to obtain an activity and thermostability profile of single-TFL-to-isoleucine mutants.157 This protocol of applying directed evolution to proteins bearing UAA was performed on GFP using FACS as a screening method. The incorporation of TFL into GFP resulted in a loss of fluorescence.148 To identify GFP variants with enhanced folding character, a random library was generated via error-prone PCR similar to the CAT work but with a slightly higher mutation rate.129,148 Cells underwent additional rounds of directed evolution resulting in the isolation of a fluorinated GFP mutant bearing twenty mutations with a total of fifteen fluorinated analogs.148 These experiments demonstrate that through combination of residue-specific UAA incorporation and directed evolution, functional proteins can be achieved.129,148
Recently site-specific incorporation in combination with phage display was employed to select a highly functional artificial antibody containing UAA by the Schultz and Smider groups.153 Multivalent hyperphage display was employed in which the C-terminus of the antibody scFv fragment was fused to pIII. Phagemid-encoded scFv was engineered to bear an amber codon within its DNA sequence, resulting in the incorporation of UAA at position 111 in the protein.153 Read-through of the amber codon yielded expression of functional pIII leading to phage assembly and scFv display on the phage surface153 (Fig. 12). By optimizing expression system, the bias of low yield of UAA containing scFv was eliminated and directed evolution experiments were carried out.153 The phage displayed scFv bearing the UAA sulfotyrosine (sTyr) was selected for gp 120 binding.153 Enhanced binding affinity and specificity of gp120 binding by the identified antibody was further confirmed and characterized by ELISA. These results demonstrate that directed evolution of proteins with UAA can offer a selective advantage through the specific functional role it provides beyond the natural set of 20 amino acids.
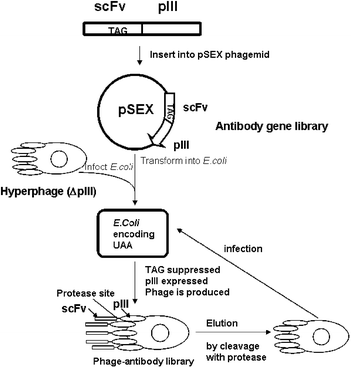 | ||
| Fig. 12 Illustration of phage display integrated with site-specific UAA incorporation.153–155 Phage produced by E.coli displayed scFv antibody that undergoes directed evolution. After cleavage with protease and elution, scFv antibodies were assayed for gp120 binding, and the best variants were selected to undergo another round of directed evolution. | ||
Conclusions
Tremendous strides have been made in the construction of unnatural protein “parts” and much of the advances described here have emanated from exploiting the principle of orthogonality.13,15 The development of elegant screens or selections for molecular evolution has not only accelerated the ability to incorporate UAA, but also has enabled the creation of tailor-made artificial proteins.148,149 This, coupled to advances in organismal evolution to allow for survival on UAA147 as well as engineering pathways for UAA biosynthesis,60 will facilitate the integration of such unnatural proteins into living matter. With these molecular tools now available, the tantalizing prospect of engineering wholly synthetic biological cells and systems may soon be realized.Acknowledgements
We are grateful for support from the NYU:Poly Seed Grant, Wechsler Award, Othmer Institute for Interdisciplinary Studies, National Science Foundation (CBET-072242 and MRSEC program DMR-0820341) and the Air Force Office of Scientific Research Young Investigator Program (FA-9-550-07-1-0060).Notes and references
- E. Andrianantoandro, S. Basu, D. K. Karig and R. Weiss, Mol. Syst Biol., 2006, 2, 0028.
- D. Baker, B. F. Group, G. Church, J. Collins, D. Endy, J. Jacobson, J. Keasling, P. Modrich, C. Smolke and R. Weiss, Scientific American, 2006, 294, 44–51.
- D. A. Drubin, J. C. Way and P. A. Silver, Genes Dev., 2007, 21, 242–254 CrossRef CAS.
- E. L. Haseltine and F. H. Arnold, Annu. Rev. Biophys. Biomol. Struct., 2007, 36, 1–19 CrossRef CAS.
- J. W. Chin, Nat. Chem. Biol., 2006, 2, 304–311 CrossRef CAS.
- J. Tian, H. Gong, N. Sheng, X. Zhou, E. Gulari, X. Gao and G. M. Church, Nature, 2004, 432, 1050–1054 CrossRef CAS.
- P. A. Carr, J. S. Park, Y. J. Lee, T. Yu, S. Zhang and J. M. Jacobson, Nucleic Acids Res., 2004, 32, e162 CrossRef.
- S. Basu, Y. Gerchman, C. H. Collins, F. H. Arnold and R. Weiss, Nature, 2005, 434, 1130–1134 CrossRef CAS.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Y. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940–943 CrossRef CAS.
- J. C. Anderson, E. J. Clarke, A. P. Arkin and C. A. Voight, J. Mol. Biol., 2005, 335, 619–627.
- T. K. Lu and J. J. Collins, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11197–11202 CrossRef CAS.
- N. Budisa, Engineering the Genetic Code. Expanding the Amino Acid Repertoire for the Design of Novel Proteins, edn, Wiley-VCH, Weinheim, 2005 Search PubMed.
- J. W. Chin, Curr. Opin. Struct. Biol., 2006, 16, 551–556 CrossRef CAS.
- J. Xie and P. G. Schultz, Curr. Opin. Chem. Biol., 2005, 9, 548–554 CrossRef CAS.
- J. Xie and P. G. Schultz, Nat. Rev. Mol. Cell Biol., 2006, 7, 775–782 CrossRef CAS.
- R. E. Connor and D. A. Tirrell, Journal of Macromolecular Science, Part C: Polymer Reviews, 2007, 47, 9–28 Search PubMed.
- M. Bose, D. Groff, J. M. Xie, E. Brustad and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 388–389 CrossRef CAS.
- K. C. Schultz, L. Supekova, Y. Ryu, J. Xie, R. Perera and P. G. Schultz, J. Am. Chem. Soc., 2006, 128, 13984–13985 CrossRef CAS.
- L. Alfonta, Z. W. Zhang, S. Uryu, J. A. Loo and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 14662–14663 CrossRef CAS.
- Y. Tang, G. Ghirlanda, W. A. Petka, T. Nakajima, W. F. DeGrado and D. A. Tirrell, Angew. Chem., Int. Ed., 2001, 40, 1494–1496 CrossRef CAS.
- Y. Tang and D. A. Tirrell, J. Am. Chem. Soc., 2001, 123, 11089–11090 CrossRef CAS.
- H. Meng and K. Kumar, J. Am. Chem. Soc., 2007, 129, 15615–15622 CrossRef CAS.
- M. B. Elowitz and S. Leibler, Nature, 2000, 403, 335–338 CrossRef CAS.
- T. S. Gardner, C. R. Cantor and J. J. Collins, Nature, 2000, 403, 339–342 CrossRef CAS.
- M. R. Atkinson, M. A. Savageau, J. T. Myers and A. J. Ninfa, Cell, 2003, 113, 597–607 CrossRef CAS.
- J. E. Dueber, B. J. Yeh, K. Chak and W. A. Lim, Science, 2003, 301, 1904–1908 CrossRef CAS.
- J. E. Dueber, E. A. Mirsky and W. A. Lim, Nat. Biotechnol., 2007, 25, 660–662 CrossRef CAS.
- S. H. Park, A. Zarrinpar and W. A. Lim, Science, 2003, 299, 1061–1064 CrossRef CAS.
- K. E. Prehoda, J. A. Scott, R. D. Mullins and W. A. Lim, Science, 2000, 290, 801–806 CrossRef CAS.
- A. Zarrinpar, S. H. Park and W. A. Lim, Nature, 2003, 426, 676–680 CrossRef CAS.
- J. E. Dueber, B. J. Yeh, R. P. Bhattacharyya and W. A. Lim, Curr. Opin. Struct. Biol., 2004, 14, 690–699 CrossRef CAS.
- L. L. Looger, M. A. Dwyer, J. J. Smith and H. W. Hellinga, Nature, 2003, 423, 185–190 CrossRef CAS.
- E. Fung, W. W. Wong, J. K. Suen, T. Bulter, S. G. Lee and J. C. Liao, Nature, 2005, 435, 118–122 CrossRef CAS.
- T. Schrader and S. Koch, Mol. BioSyst., 2007, 3, 241–248 RSC.
- H. Kitano, Nature, 2002, 420, 206–210 CrossRef CAS.
- J. S. Bader, A. Chaudhuri, J. M. Rothberg and J. Chant, Nat. Biotechnol., 2004, 22, 78–85 CrossRef CAS.
- J. B. Pereira-Leal, A. J. Enright and C. A. Ouzounis, Proteins: Struct., Funct., Bioinf., 2004, 54, 49–57 Search PubMed.
- S. K. Lee, H. Chou, T. S. Ham, T. S. Lee and J. D. Keasling, Curr. Opin. Biotechnol., 2008, 19, 556–563 CrossRef CAS.
- N. Rosenfeld, M. B. Elowitz and U. Alon, J. Mol. Biol., 2002, 323, 785–793 CrossRef CAS.
- A. E. Friedland, T. K. Lu, X. Wang, D. Shi, G. Church and J. J. Collins, Science, 2009, 324, 1199–1202 CrossRef CAS.
- K. Brenner, D. K. Karig, R. Weiss and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17300–17304 CrossRef CAS.
- C. J. Bashor, N. C. Helman, S. Yan and W. A. Lim, Science, 2008, 319, 1539–1543 CrossRef CAS.
- S. Atsumi, T. Hanai and J. C. Liao, Nature, 2008, 451, 86–89 CrossRef CAS.
- J. B. Lucks, L. Qi, W. R. Whitaker and A. P. Arkin, Curr. Opin. Microbiol., 2008, 11, 567–573 CrossRef.
- J. F. Atkins and P. V. Baranov, Nature, 2007, 448, 1004–1005 CrossRef CAS.
- D. L. Hatfield, B. A. Carlson, X. M. Xu, H. Mix and V. N. Gladyshev, Prog. Nucleic Acid Res. Mol. Biol., 2006, 81, 97–142 Search PubMed.
- C. Baron and A. Bock, in tRNA: Structure, Biosynthesis, and Function, ed. D. a. R. Soll and L. Uttam, ASM Press, Washington, D.C., 1995 Search PubMed.
- J. A. Krzycki, Curr. Opin. Microbiol., 2005, 8, 706–712 CrossRef CAS.
- M. Ibba and D. Soll, Curr. Biol., 2002, 12, R464–466 CrossRef CAS.
- S. M. Hecht, B. L. Alford, Y. Kuroda and S. Kitano, J. Biol. Chem., 1978, 253, 4517–4520 CAS.
- C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith and P. G. Schultz, Science, 1989, 244, 182–188 CAS.
- M. C. Hartman, K. Josephson and J. W. Szostak, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4356–4361 CrossRef CAS.
- R. Furter, Protein Sci., 1998, 7, 419–426 CAS.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS.
- J. C. Jackson, S. P. Duffy, K. R. Hess and R. A. Mehl, J. Am. Chem. Soc., 2006, 128, 11124–11127 CrossRef CAS.
- J. C. Jackson, J. T. Hammill and R. A. Mehl, J. Am. Chem. Soc., 2007, 129, 1160–1166 CrossRef CAS.
- L. Wang, Z. Zhang, A. Brock and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 56–61 CrossRef CAS.
- J. Xie, L. Wang, N. Wu, A. Brock, G. Spraggon and P. G. Schultz, Nat. Biotechnol., 2004, 22, 1297–1301 CrossRef CAS.
- Z. Zhang, J. Gildersleeve, Y. Y. Yang, R. Xu, J. A. Loo, S. Uryu, C. H. Wong and P. G. Schultz, Science, 2004, 303, 371–373 CrossRef CAS.
- R. A. Mehl, J. C. Anderson, S. W. Santoro, L. Wang, A. B. Martin, D. S. King, D. M. Horn and P. G. Schultz, J. Am. Chem. Soc., 2003, 125, 935–939 CrossRef CAS.
- L. Wang and P. G. Schultz, Chem. Biol., 2001, 8, 883–890 CrossRef CAS.
- J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. W. Zhang and P. G. Schultz, Science, 2003, 301, 964–967 CrossRef CAS.
- S. W. Santoro, L. Wang, B. Herberich, D. S. King and P. G. Schultz, Nat. Biotechnol., 2002, 20, 1044–1048 CrossRef CAS.
- J. Guo, J. Wang, J. C. Anderson and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 47, 722–725.
- Y. H. Ryu and P. G. Schultz, Nat. Methods, 2006, 3, 263–265 CrossRef CAS.
- I. Kwon and D. A. Tirrell, J. Am. Chem. Soc., 2007, 129, 10431–10437 CrossRef CAS.
- I. Kwon, P. Wang and D. A. Tirrell, J. Am. Chem. Soc., 2006, 128, 11778–11783 CrossRef CAS.
- C. E. Melancon, 3rd and P. G. Schultz, Bioorg. Med. Chem. Lett., 2009, 19, 3845–3847 CrossRef.
- S. Chen, P. G. Schultz and A. Brock, J. Mol. Biol., 2007, 371, 112–122 CrossRef CAS.
- K. Sakamoto, A. Hayashi, A. Sakamoto, D. Kiga, H. Nakayama, A. Soma, T. Kobayashi, M. Kitabatake, K. Takio, K. Saito, M. Shirouzu, I. Hirao and S. Yokoyama, Nucleic Acids Res., 2002, 30, 4692–4699 CrossRef CAS.
- Z. Zhang, L. Alfonta, F. Tian, B. Bursulaya, S. Uryu, D. S. King and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 8882–8887 CrossRef CAS.
- W. Wang, J. K. Takimoto, G. V. Louie, T. J. Baiga, J. P. Noel, K. F. Lee, P. A. Slesinger and L. Wang, Nat. Neurosci., 2007, 10, 1063–1072 CrossRef CAS.
- S. Ye, C. Kohrer, T. Huber, M. Kazmi, P. Sachdev, E. C. Yan, A. Bhagat, U. L. RajBhandary and T. P. Sakmar, J. Biol. Chem., 2008, 283, 1525–1533 CAS.
- L. Wang, A. Brock, B. Herberich and P. G. Schultz, Science, 2001, 292, 498–500 CrossRef CAS.
- J. Guo, J. Wang, J. S. Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 6399–6401 CrossRef CAS.
- H. S. Lee and P. G. Schultz, J. Am. Chem. Soc., 2008, 130, 13194–13195 CrossRef CAS.
- J. Xie, W. Liu and P. G. Schultz, Angew. Chem., Int. Ed., 2007, 46, 9239–9242 CrossRef CAS.
- H. S. Lee, G. Spraggon, P. G. Schultz and F. Wang, J. Am. Chem. Soc., 2009, 131, 2481–2483 CrossRef CAS.
- P. R. Chen, D. Groff, J. Guo, W. Ou, S. Cellitti, B. H. Geierstanger and P. G. Schultz, Angew. Chem., Int. Ed., 2009, 48, 4052–4055 CrossRef CAS.
- I. Kwon, K. Kirshenbaum and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 7512–7513 CrossRef CAS.
- H. Neumann, S. Y. Peak-Chew and J. W. Chin, Nat. Chem. Biol., 2008, 4, 232–234 CrossRef CAS.
- E. Brustad, M. L. Bushey, A. Brock, J. Chittuluru and P. G. Schultz, Bioorg. Med. Chem. Lett., 2008, 18, 6004–6006 CrossRef CAS.
- T. S. Young, I. Ahmad, A. Brock and P. G. Schultz, Biochemistry, 2009, 48, 2643–2653 CrossRef CAS.
- T. Mukai, T. Kobayashi, N. Hino, T. Yanagisawa, K. Sakamoto and S. Yokoyama, Biochem. Biophys. Res. Commun., 2008, 371, 818–822 CrossRef CAS.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8650–8655 CrossRef CAS.
- G. Turcatti, K. Nemeth, M. D. Edgerton, U. Meseth, F. Talabot, M. Peitsch, J. Knowles, H. Vogel and A. Chollet, J. Biol. Chem., 1996, 271, 19991–19998 CrossRef CAS.
- D. L. Beene, K. L. Price, H. A. Lester, D. A. Dougherty and S. C. Lummis, J. Neurosci., 2004, 24, 9097–9104 CrossRef CAS.
- D. A. Dougherty, Curr. Opin. Chem. Biol., 2000, 4, 645–652 CrossRef CAS.
- K. Yamanaka, H. Nakata, T. Hohsaka and M. Sisido, J. Biosci. Bioeng., 2004, 97, 395–399 CAS.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, RNA, 2007, 13, 1703–1714 CrossRef CAS.
- E. A. Rodriguez, H. A. Lester and D. A. Dougherty, RNA, 2007, 13, 1715–1722 CrossRef CAS.
- M. Sisido and T. Hohsaka, Appl. Microbiol. Biotechnol., 2001, 57, 274–281 CrossRef CAS.
- J. C. Anderson, N. Wu, S. W. Santoro, V. Lakshman, D. S. King and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7566–7571 CrossRef CAS.
- N. Muranaka, T. Hohsaka and M. Sisido, Nucleic Acids Res., 2006, 34, e7 CrossRef.
- H. Taira, T. Hohsaka and M. Sisido, Nucleic Acids Res., 2006, 34, e44 CrossRef.
- T. Hohsaka, Y. Ashizuka, H. Murakami and M. Sisido, Nucleic Acids Res., 2001, 29, 3646–3651 CrossRef CAS.
- H. Taira, M. Fukushima, T. Hohsaka and M. Sisido, J. Biosci. Bioeng., 2005, 99, 473–476 CrossRef CAS.
- M. Taki, Y. Tokuda, T. Ohtsuki and M. Sisido, J. Biosci. Bioeng., 2006, 102, 511–517 CrossRef CAS.
- O. Rackham, K. Wang and J. W. Chin, Nat. Chem. Biol., 2006, 2, 254–258 CrossRef CAS.
- K. H. Wang, H. Neumann, S. Y. Peak-Chew and J. W. Chin, Nat. Biotechnol., 2007, 25, 770–777 CrossRef CAS.
- O. Rackham and J. W. Chin, J. Am. Chem. Soc., 2005, 127, 17584–17585 CrossRef CAS.
- O. Rackham and J. W. Chin, Biochem. Soc. Trans., 2006, 34, 328–329 CrossRef CAS.
- A. J. Link, M. K. Vink, N. J. Agard, J. A. Prescher, C. R. Bertozzi and D. A. Tirrell, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10180–10185 CrossRef CAS.
- T. H. Yoo and D. A. Tirrell, Angew. Chem., Int. Ed., 2007, 46, 5340–5343 CrossRef CAS.
- A. J. Link and D. A. Tirrell, Methods, 2005, 36, 291–298 CrossRef CAS.
- G. N. Cohen and D. B. Cowie, C R Hebd Seances Acad. Sci., 1957, 244, 680–683 Search PubMed.
- D. B. Cowie and G. N. Cohen, Biochim. Biophys. Acta, 1957, 26, 252–261 CAS.
- P. Walasek and J. F. Honek, BMC Biochem., 2005, 6, 21 CrossRef.
- H. S. Duewel, E. Daub, V. Robinson and J. F. Honek, Biochemistry, 2001, 40, 13167–13176 CrossRef CAS.
- J. C. van Hest, K. L. Kiick and D. A. Tirrell, J. Am. Chem. Soc., 2000, 122, 1282–1288 CrossRef CAS.
- K. L. Kiick and D. A. Tirrell, Tetrahedron, 2000, 56, 9487–9493 CrossRef CAS.
- C. Wolschner, A. Giese, H. A. Kretzschmar, R. Huber, L. Moroder and N. Budisa, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7756–7761 CrossRef CAS.
- W. Kim, A. George, M. Evans and V. P. Conticello, ChemBioChem, 2004, 5, 928–936 CrossRef CAS.
- N. Budisa, C. Minks, F. J. Medrano, J. Lutz, R. Huber and L. Moroder, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 455–459 CrossRef CAS.
- W. Kim, K. I. Hardcastle and V. P. Conticello, Angew. Chem., Int. Ed., 2006, 45, 8141–8145 CrossRef CAS.
- S. Son, I. C. Tanrikulu and D. A. Tirrell, ChemBioChem, 2006, 7, 1251–1257 CrossRef CAS.
- P. Wang, Y. Tang and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 6900–6906 CrossRef CAS.
- M. L. Mock, T. Michon, J. C. van Hest and D. A. Tirrell, ChemBioChem, 2006, 7, 83–87 CrossRef CAS.
- P. Wang, A. Fichera, K. Kumar and D. A. Tirrell, Angew. Chem., Int. Ed., 2004, 43, 3664–3666 CrossRef CAS.
- A. J. Link and D. A. Tirrell, J. Am. Chem. Soc., 2003, 125, 11164–11165 CrossRef CAS.
- A. Wang, N. Winblade Nairn, R. S. Johnson, D. A. Tirrell and K. Grabstein, ChemBioChem, 2008, 9, 324–330 CrossRef CAS.
- C. Minks, R. Huber, L. Moroder and N. Budisa, Anal. Biochem., 2000, 284, 29–34 CrossRef CAS.
- M. Rubini, S. Lepthien, R. Golbik and N. Budisa, Biochim. Biophys. Acta, Proteins Proteomics, 2006, 1764, 1147–1158 CrossRef CAS.
- Q. S. Zhang, L. Shen, E. D. Wang and Y. L. Wang, J. Protein Chem., 1999, 18, 187–192 CrossRef CAS.
- J. F. Parsons, G. Xiao, G. L. Gilliland and R. N. Armstrong, Biochemistry, 1998, 37, 6286–6294 CrossRef CAS.
- M. Katragadda and J. D. Lambris, Protein Expression Purif., 2006, 47, 289–295 CrossRef CAS.
- K. E. Beatty, F. Xie, Q. Wang and D. A. Tirrell, J. Am. Chem. Soc., 2005, 127, 14150–14151 CrossRef CAS.
- Y. Tang, G. Ghirlanda, W. A. Petka, T. Nakajima, W. F. DeGrado and D. A. Tirrell, Angew. Chem., Int. Ed., 2001, 40, 1494–1496 CrossRef CAS.
- J. K. Montclare and D. A. Tirrell, Angew. Chem., Int. Ed., 2006, 45, 4518–4521 CrossRef CAS.
- D. C. Dieterich, A. J. Link, J. Graumann, D. A. Tirrell and E. M. Schuman, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9482–9487 CrossRef CAS.
- D. C. Dieterich, J. J. Lee, A. J. Link, J. Graumann, D. A. Tirrell and E. M. Schuman, Nat. Protoc., 2007, 2, 532–540 Search PubMed.
- K. E. Beatty, J. C. Liu, F. Xie, D. C. Dieterich, E. M. Schuman, Q. Wang and D. A. Tirrell, Angew. Chem., Int. Ed., 2006, 45, 7364–7367 CrossRef CAS.
- M. Suchanek, A. Radzikowska and C. Thiele, Nat. Methods, 2005, 2, 261–267 CrossRef CAS.
- K. L. Kiick, J. C. van Hest and D. A. Tirrell, Angew. Chem., Int. Ed., 2000, 39, 2148–2152 CrossRef CAS.
- K. L. Kiick, R. Weberskirch and D. A. Tirrell, FEBS Lett., 2001, 502, 25–30 CrossRef CAS.
- M. Ibba and H. Hennecke, FEBS Lett., 1995, 364, 272–275 CrossRef CAS.
- M. Ibba, P. Kast and H. Hennecke, Biochemistry, 1994, 33, 7107–7112 CrossRef CAS.
- P. Kast and H. Hennecke, J. Mol. Biol., 1991, 222, 99–124 CrossRef CAS.
- N. Sharma, R. Furter, P. Kast and D. A. Tirrell, FEBS Lett., 2000, 467, 37–40 CrossRef CAS.
- K. Kirshenbaum, I. S. Carrico and D. A. Tirrell, ChemBioChem, 2002, 3, 235–237 CrossRef.
- I. S. Carrico, S. A. Maskarinec, S. C. Heilshorn, M. L. Mock, J. C. Liu, P. J. Nowatzki, C. Franck, G. Ravichandran and D. A. Tirrell, J. Am. Chem. Soc., 2007, 129, 4874–4875 CrossRef CAS.
- J. K. Montclare, S. Son, G. A. Clark, K. Kumar and D. A. Tirrell, ChemBioChem, 2009, 10, 84–86 CrossRef CAS.
- D. Datta, P. Wang, I. S. Carrico, S. L. Mayo and D. A. Tirrell, J. Am. Chem. Soc., 2002, 124, 5652–5653 CrossRef CAS.
- V. Doring, H. D. Mootz, L. A. Nangle, T. L. Hendrickson, V. de Crecy-Lagard, P. Schimmel and P. Marliere, Science, 2001, 292, 501–504 CAS.
- Y. Tang and D. A. Tirrell, Biochemistry, 2002, 41, 10635–10645 CrossRef CAS.
- O. M. Rennert and H. S. Anker, Biochemistry, 1963, 2, 471–476 CrossRef CAS.
- J. M. Bacher and A. D. Ellington, J. Bacteriol., 2001, 183, 5414–5425 CrossRef CAS.
- T. H. Yoo, A. J. Link and D. A. Tirrell, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13887–13890 CrossRef CAS.
- D. Kajihara, T. Hohsaka and M. Sisido, Protein Eng., Des. Sel., 2005, 18, 273–278 CrossRef CAS.
- C. M. Yuen and D. R. Liu, Nat. Methods, 2007, 4, 995–997 CrossRef CAS.
- H. Zhao, Biotechnol. Bioeng., 2007, 98, 313–317 CrossRef CAS.
- F. H. Arnold, P. L. Wintrode, K. Miyazaki and A. Gershenson, Trends Biochem. Sci., 2001, 26, 100–106 CrossRef CAS.
- C. C. Liu, A. V. Mack, M. L. Tsao, J. H. Mills, H. S. Lee, H. Choe, M. Farzan, P. G. Schultz and V. V. Smider, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17688–17693 CrossRef CAS.
- S. Rondot, J. Koch, F. Breitling and S. Dubel, Nat. Biotechnol., 2001, 19, 75–78 CrossRef CAS.
- M. Pastrnak and P. G. Schultz, Bioorg. Med. Chem., 2001, 9, 2373–2379 CrossRef CAS.
- T. Panchenko, W. W. Zhu and J. K. Montclare, Biotechnol. Bioeng., 2006, 94, 921–930 CrossRef CAS.
- N. Voloshchuk, M. X. Lee, W. W. Zhu, I. C. Tanrikulu and J. K. Montclare, Bioorg. Med. Chem. Lett., 2007, 17, 5907–5911 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |