DOI:
10.1039/B910706A
(Review Article)
Mol. BioSyst., 2010,
6, 81-88
Molecular recognition of poly(A) targeting by protoberberinealkaloids: in vitro biophysical studies and biological perspectives
Received
3rd June 2009
, Accepted 18th August 2009
First published on 22nd September 2009
Abstract
The use of small molecules to specifically control important cellular functions through binding to nucleic acids is an area of major current interest at the interface of chemical biology and medicinal chemistry. The polyadenylic acid [poly(A)] tail of mRNA has been recently established as a potential drug target due to its significant role in the initiation of translation, maturation and stability of mRNA as well as in the production of alternate proteins in eukaryotic cells. Very recently some small molecule alkaloids of the isoquinoline group have been found to bind poly(A) with remarkably high affinity leading to self-structure formation. Plant alkaloids are small molecules known to have important traditional roles in medicinal chemistry due to their extensive biological activity. Especially, noteworthy are the protoberberinealkaloids that are widely distributed in several botanical families exhibiting myriad therapeutic applications. This review focuses on the structural and biological significance of poly(A) and interaction of protoberberinealkaloids with this RNA structure for the development of new small molecule alkaloids targeted to poly(A) structures as futuristic therapeutic agents.

Prabal Giri
| Prabal Giri has just received his PhD in Chemistry in 2009 from Jadavpur University, Kolkata, India. In 2004 he joined as a Junior Research Fellow in the Biophysical Chemistry Laboratory at the Indian Institute of Chemical Biology, Kolkata and in 2006 he became the Senior Research Fellow. His research interests include probing RNA structure and dynamics, small molecule–RNA interaction and photochemistry of small organic molecules. He is a Life member of Indian Biophysical Society and Indian Association for the Cultivation of Science. |

Gopinatha Suresh Kumar
| Gopinatha Suresh Kumar obtained his PhD in Chemistry from the University of Delhi, India in 1987. He then joined the Indian Institute of Chemical Biology, Kolkata, as a Scientist where he worked extensively on polymorphic nucleic acid structures and small molecule interaction to these structures. During 1993–1995 and 1999–2001, he was a Visiting Scientist in the laboratory of Professsor Maria Tomasz at the Department of Chemistry, Hunter College of the City University of New York, New York, USA studying the various aspects of DNA alkylation of the clinically used anticancer agent Mitomycin C and its metabolites. Since 2004, he heads the Biophysical Chemistry Laboratory of the Indian Institute of Chemical Biology. His research interests include design of specific molecular tools to probe nucleic acid structure, RNA structure–function and interactions. |
Introduction
The biological importance of the nucleic acids raises the necessity of investigations on their structural and conformational analysis through interaction with ligands. Recent progress in molecular biology and biotechnology has created many opportunities for the development of nucleic acid-based therapeutics for the treatment of genetic and acquired diseases.1–5DNAs and more recently RNAs have been the focus of such drug targeting. More recent discovery of the many micro-RNAs and emerging knowledge of their critical roles in essential cellular activities have led to a paradigm shift from DNA to RNA as the focus of drug targeting to control genetic activity. The recent anti-viral drug design is based on targeting RNA molecules, especially to unique structural regions in RNA such as mRNA by which new drugs could be potentially designed for regulation of gene expression. Various serious human diseases such as HIV, AIDS, hepatitis C etc. caused by RNA viruses also necessitates immediate research leading to the designing of new RNA binding compounds for therapy. In eukaryotes, the polyadenylic acid [poly(A)] tail consisting of 200–250 adenine bases at the 3′-end of mRNA occupies a significant role for the initiation of translation, maturation and stability of mRNA as well as in the production of alternate forms of protein.6–11 Recent discovery of the role of 3′-end formation in splicing and transcriptional termination also gives a special recognition to poly(A).12–15 A schematic diagram of the transcription process in a eukaryotic cell is given in Fig. 1. Recently, poly(A) has been recognized as a contributor in increasing length and as a platform to recruit mRNA export factors.16 Polyadenylation of mRNA is a critical cellular event in the transcription process for the maturation of all eukaryotic mRNAs. Polyadenylation is catalyzed by the enzyme polyA polymeraze (PAP). Neo-PAP, a recently identified human poly(A) polymerase, is significantly over-expressed in human cancer cells in comparison to its expression in normal or virally transformed cells, and may represent a tumor-specific target.17,18 Thus, molecules capable of recognizing and binding to the poly(A) tail of mRNA might interfere with the full processing of mRNA by PAP and switch off protein synthesis, suggesting the poly(A) tail as a potential malignancy specific target.19 Polymorphic conversion of poly(A) to the double-stranded form from the single-stranded form, depending on pH and salt concentration, also makes it a potential target for chemico-biological investigations.20–23
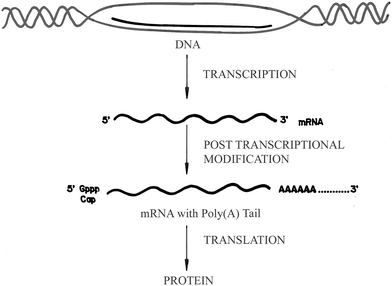 |
| | Fig. 1 A schematic diagram for the transcription process in eukaryotic cells. (Reprinted from Yadav et al.,52Bioorg. Med. Chem., 2005, 13, 165–174. © 2005, with permission from Elsevier). | |
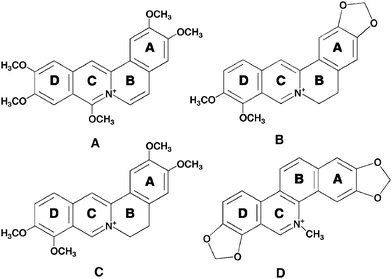
Due to extensive biological activities, alkaloids have occupied a lead position in medicinal chemistry. Medicinal plants may represent valuable, untapped source of drugs. Protoberberine alkaloids, which are readily extractable from Chinese and Korean medicinal plants, have been shown to possess diverse biochemical and pharmacological actions while being nontoxic to humans even at high dosages. Needless to say, protoberberinealkaloids that are most widely distributed in several botanical families have a long history of use world-wide in folk medicine. They exhibit myriad therapeutic applications.24 The naturally occurring berberine, palmatine, jatrorrhizine, sanguinarine and the synthetic coralyne are the representatives of the protoberberine group so far known to be medically important. These protoberberinealkaloids are among the most widely distributed alkaloids of the isoquinoline series and they are also the main active components of some Chinese herbal medicines such as Rhizoma coptidis and Cortex phellodendri.25Berberine and palmatine are distributed in many plants such as the Chinese herb huanglian (Coptis chinesis), goldensal (Hydrastis canadensis), Turkish berberis and the roots of species belonging to the Malagasy genus Burasaia, Menispermaceae.26,27Sanguinarine, on the other hand, is a benzophenanthridine alkaloid derived from the root of Sanguinaria canadenis and other poppy-fumaria species. Sanguinarine also has a long history of use world-wide in folk medicine. Berberine and palmatine bear the same tetracyclic structure but differ in the nature of the substituents on the benzo ring (ring A, Fig. 2), these being methylene dioxy for berberine and dimethoxy for palmatine. Unlike berberine and palmatine with buckled structure (due to partial saturation in the ring B, Fig. 2), coralyne and sanguinarine are planar molecules. The biological activities of these protoberberinealkaloids have been recently reviewed.28–32 This review summarizes the physico-chemical characterization on the interaction of these plant alkaloids with single-stranded poly(A) in the context of their biophysical and medicinal activities.
Protoberberine alkaloid–poly(A) interactions
Coralyne–poly(A) interaction
Coralyne [13-methyl [1,3]benzodioxolo[5,6-c]-1,3-dioxolo[4,5-i]phenanthridium] (Fig. 2A) is a synthetic protoberberinealkaloid and a close structural analogue of berberine. Coralyne has a planar structure due to saturated conjugated rings. This molecule exhibits excellent antileukemic activity and is particularly noted for its dual poisoning of topoisomerase I and II.33–36Coralyne has a high tendency to aggregate and this property was an inhibition in understanding its nucleic acid binding aspects for several years. The high tendency of aggregation was solved by the use of 30% alcoholic buffer by Maiti and colleagues that enabled detailed investigation on its DNA binding, establishing true intercalative binding with guanine–cytosinebase pair specificity, from a variety of studies.37,38Coralyne and its derivatives have been reported as low toxic and very high antitumor agents compared to other protoberberinealkaloids.19,35,36 The topoisomerase I poisoning and the DNA interaction properties of coralyne derivatives have been correlated where a model involving intercalation and groove binding to DNA, depending on the state of saturation of the ring system, has been suggested.33 Xing et al. have reported that coralyne binds to poly(A) non-cooperatively with a Ka of 1.8 × 106 M−1 at pH 7.19 They also showed that coralyne induced self-structure formation in poly(A) molecules whereby two poly(A) chains hydrogen bond through adenine residues.19,39 Self-structure formation of poly(A) is a reversible aggregation process giving rise to a physical non-covalent cross-linking effect between two poly(A) molecules. This involves a decrease of the absolute value of the macromolecule electrophoretic mobility and hence the neutralization of local charges. Once the self-structure is formed, the two strands are held together through H-bonding of the adenine residues with bound ligand, where the strand arrangement may be parallel or antiparallel. The presence of a planar aromatic ring is an essential condition to induce such an aggregation process through physical cross-linking. The high selectivity of this alkaloid to poly(A) was also revealed from competition dialysisassay where pronounced binding of coralyne to poly(A) was observed at equilibrium.19 The absorbance spectral change of coralyne in the absence and in presence of poly(A) is shown in Fig. 3A. The free coralynespectrum has a maximum around 420 nm, whereas the bound coralyne has two local maxima at 412 and 435 nm, respectively. These spectral changes on complexation with poly(A) were reported recently from our laboratory and indicated cooperative binding of coralyne to poly(A).39 The binding stoichiometry determined from the Job plot analysis was two base pairs per alkaloid indicating an intercalative mode of binding to self-structured poly(A) obeying the nearest neighbor exclusion principle. In the presence of coralyne, the circular dichroic spectrum of poly(A) underwent significant changes with generation of an induced circular dichroic spectrum for the bound coralyne molecules (Fig. 3B). A strong evidence for the self-structure induction was the cooperative circular dichroic melting profile of the complexed poly(A) with a melting temperature of about 60 °C (as also reported from the work of Xing et al.), shown in Fig. 3C.19,39 Our laboratory further showed cooperative UV melting of coralyne–poly(A) complexes (inset, Fig. 3A) and also the cooperative reversible melting in differential scanning calorimetry (Fig. 3D) where the calorimetric and van’t Hoff enthalpy was found to be identical for the complex. Furthermore, a cooperative binding of coralyne to poly(A) was elucidated from the Scatchard plots obtained from absorption spectra.39 The energetics of the binding was evaluated from isothermal titration calorimetry, which indicated that the binding was enthalpy driven (ΔH° = −8.3 ± 0.6 kcal mol−1) with a binding free energy of −8.4 kcal mol−1 (Table 1). The self-structure was proposed to be of antiparallel nature,19 which was supported further from recent work from our laboratory.39 It should be mentioned here that coralyne was reported to induce similar secondary conformations in poly(dA) and homo-adenine polymers also.40,41 Although the molecular basis underlying this self-structure formation by the ligand is still unknown, our recent work has suggested that reversible cooperative binding of coralyne to poly(A) may be a trigger for the self-structure formation.39 This was verified from a study where several aromatic and other molecules that bound poly(A) cooperatively and induced self-structure formation.39 A proposal from Hud’s laboratory recently also related the crescent shape of coralyne and a cooperative binding to be the necessary condition for the self-structure formation in poly(A).42 Further they also suggested that poly(A) sequence elements and appropriate ligands could be used to reversibly drive transitions in RNA based molecular structures by simply diluting or concentrating a sample about the poly(A)–coralyne critical concentration.42
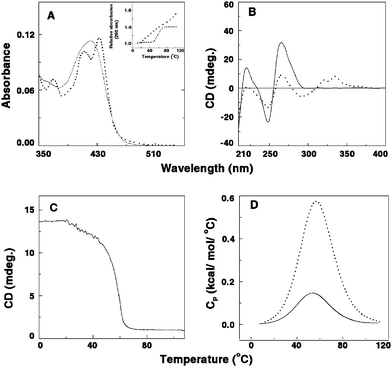 |
| | Fig. 3 (A) Representative absorption spectral changes of coralyne in the absence (solid curve) and presence (dashed curve) of poly(A). Inset: UV melting profile of poly(A) (□) and the poly(A)–coralyne complex (■). (B) Circular dichroic spectra of single-stranded poly(A) in the absence (solid curve) and presence (dashed curve) of coralyne. (C) Circular dichroic melting profile of a solution containing 55 μM of poly(A) and 13.75 μM of coralyne monitored at 274 nm. (D) DSC thermograms of poly(A) (solid curve) and the complex of poly(A) and coralyne (dashed curve). (Reprinted from Giri and Kumar,39Arch. Biochem. Biophys., 2008, 474, 183–192. © 2008, with permission from Elsevier). | |
|
Alkaloid
|
10−5Ka/M−1 |
N
|
ΔG°/kcal mol−1 |
ΔH°/kcal mol−1 |
TΔS°/kcal mol−1 |
T
m/°C |
ΔHv/kcal mol−1 |
ΔHcal/kcal mol−1 |
|
For references see 19, 39, 52, 63, 64 and 86.
K
a, binding affinity; N, binding stoichiometry; ΔG°, binding free energy change; ΔH°, binding enthalpy change; TΔS, entropy contribution towards binding; Tm, melting temperature; ΔHv, van’t Hoff enthalpy; ΔHcal, calorimetric enthalpy. |
|
Berberine
|
9.79 ± 0.45 |
0.34 |
−6.66 |
−6.22 ± 1.20 |
0.475 ± 0.015 |
65.2 ± 1.1 |
21.31 ± 1.90 |
18.75 ± 1.45 |
|
Palmatine
|
8.36 ± 0.26 |
0.63 |
−7.99 |
−8.61 ± 1.40 |
0.665 ± 0.020 |
63.4 ± 1.0 |
22.90 ± 1.78 |
19.67 ± 2.12 |
|
Coralyne
|
18.0 ± 3.10 |
0.24 |
−8.40 |
−8.30 ± 0.60 |
0.10 ± 0.02 |
57.8 ± 1.1 |
22.38 ± 1.32 |
22.30 ± 1.66 |
|
Sanguinarine
|
46.0 ± 2.80 |
0.29 |
−8.99 |
−14.40 ± 1.30 |
5.45 ± 0.85 |
58 ± 1.0 |
24.02 ± 1.50 |
24.27 ± 1.23 |
Berberine–poly(A) interaction
Berberine (7,8,13,13a-tetradehydro-9,10-dimethoxy-2,3-methylenedioxyberberinium) (Fig. 2B), is the most widely known alkaloid belonging to the protoberberine group, exhibiting several therapeutic behaviors such as antisecretory, antiinflammatory, antibacterial, antimalarial and anticancer activities with low cytotoxicity.43–45Berberine has also been established as a drug in the treatment of dermal leishmaniasis, gastroenteritis in children and cholera.46 The alkaloid induces apoptosis in HL-60 leukemia cells.45 Strong DNA binding affinity and adenine–thyminebase pair specificity of berberine has been reported.47–50 Several years ago a remarkably stronger affinity of this alkaloid to single-stranded poly(A) over double-stranded (ds) B-DNA and tRNA structures was revealed from absorption, fluorescence and circular dichroism studies by Maiti and co-workers.51 Further details of the specificity of this interaction was revealed from the recent work of Yadav et al.52 and Centikol and Hud42 although the competition dialysis technique of Chairs53 revealed no appreciable binding of berberine to poly(A). Berberine binding to poly(A) exhibited typical hypochromic and bathochromic effects in the absorption spectrum of berberine (Fig. 4A), several fold enhancement of its rather low intrinsic fluorescence intensity and increase of relative specific viscosity and perturbation of the circular dichroic spectrum of poly(A) with generation of strong induced circular dichroic bands for the bound berberine molecules (Fig. 4B). Non-cooperative UV melting (inset, Fig. 4A) and absence of CD melting pattern (Fig. 4C) indicated that there was no self-structure induction in poly(A) compared to the results obtained in coralyne–poly(A) complexation. The non-cooperative binding constant evaluated from the absorption spectral study was high and of the order of ∼106 M−1. The binding affinity of berberine–poly(A) complexation was shown to decrease with increasing salt concentration of the medium indicating a strong role for the electrostatic interaction in the binding process. A comparative fluorescence study of berberine with various polynucleotides indicated the strongest binding of the alkaloid with poly(A) over double-stranded calf thymus DNA and tRNA structures. More recent work of Hud’s laboratory also confirmed the hypochromic and bathochromic effects and circular dichroic changes in poly(A) on binding of berberine.42 Very recently, Giri and Kumar (2008) investigated the energetics involved in the binding by ultra-sensitive isothermal titration calorimetry, and revealed that berberine binds poly(A) with exothermically (ΔH° = −6.22 kcal mol−1) and with a binding free energy (ΔG°) of −6.66 kcal mol−1 (Table 1).39 The binding has very negligible contribution from the entropy term (TΔS° = 0.475 kcal mol−1) indicating that the binding was clearly enthalpy driven. The large negative binding enthalpy is generally typical for intercalative interaction of small molecules to nucleic acids.54–56 The non-cooperative non-reversible differential scanning calorimetric profile of poly(A) (ΔHcal = 5.82 kcal mol−1, ΔHv = 21.51 kcal mol−1) with the thermal melting temperature of 54.8 °C gets shifted towards to a higher melting temperature of 65.2 °C on complexation with berberine (Fig. 4D). The enthalpy of denaturation also became higher than that for uncomplexed poly(A) indicating appreciable stabilization of the complex.39 The mode of the interaction of this complex formation was investigated from the sensitive hydrodynamic studies.15 The relative specific viscosity of poly(A) increased gradually with an increase in concentration of berberine, consistent with an intercalative/partial intercalative binding, the later being the most probable mode considering the single-stranded nature of poly(A). In circular dichroism, no evidence for the self structure induction of poly(A) was observed. Similarly the differential scanning calorimetry profiles gave evidence for non-cooperative binding. However, Hud’s laboratory recently investigated the self-structure formation by diluting/concentrating a sample of poly(A) about the critical concentration and showed that by this protocol berberine also induced self-structure in poly(A).42 It was revealed that at least six berberine molecules are bound for the stabilization of the poly(A) self structure with half assembly at 25 μM and 71.4% assembly at the highest concentration (50 μM) measured. By this procedure, berberine also was detected to induce self-structure in poly(A) although it is pertinent to emphasize that melting studies in CD, UV and DSC did not confirm this. These different results from the same interaction may be due to the fact that small molecule–poly(A) binding is different from binding to other pre-structured nucleic acids. Binding of small molecules and induction of self-structure is a coupled event. By performing the dilution experiment it is possible to determine the minimum number of ligands required for assembly and the concentration below which the ligand driven poly(A) self-structure does not exist.42 It is likely that the ligand concentration used in thermal melting and CD melting studies may be lower than the minimum concentration for assembly (MCA) of ligand binding to an unstructured poly(A), so revealing no self-structure formation.
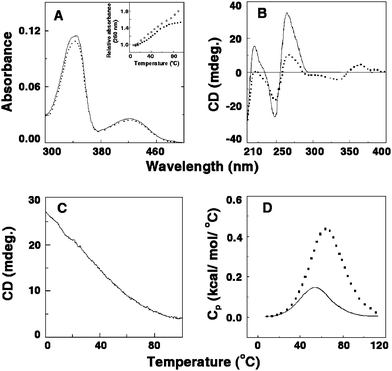 |
| | Fig. 4 (A) Representative absorption spectral changes of berberine in the absence (solid curve) and presence (dashed curve) of poly(A). Inset: UV melting profile of poly(A) (□) and the poly(A)–berberine complex (■). (B) Circular dichroic spectra of single-stranded poly(A) in the absence (solid curve) and presence (dashed curve) of berberine. (C) Circular dichroic melting profile of a solution containing 55 μM of poly(A) and 13.75 μM of berberine monitored at 274 nm. (D) DSC thermograms of poly(A) (solid curve) and the complex of poly(A) and of berberine (dashed curve). (Reprinted from Giri and Kumar,39Arch. Biochem. Biophys., 2008, 474, 183–192. © 2008, with permission from Elsevier). | |
Palmatine–poly(A) interaction
Palmatine (7,8,13,13a-tetradehydro-9,10-dimethoxyberberinium), is a naturally occurring alkaloid and a close analogue of berberine (Fig. 2C). Palmatine occurs to a lesser amount compared to berberine in plants. Palmatine, like berberine, has been reported to exhibit significant antitumor activity against HL-60 leukemic cells and has antimicrobial properties.24,44 One of the many reasons suggested for the antitumor activity of this alkaloid has been the inhibition of reverse transcriptase.57 Pharmacological activities of palmatine include antipyretic, antifungal, hepatoprotective and vasodilatory effects.58–61Palmatine, like berberine, also binds to DNA by partial intercalation with adenine–thyminebase pair specificity.62 Our laboratory was the first to report that palmatine selectively targeted poly(A) for binding from a comparative binding study of a variety of different single- and double-stranded polynucleotides using competition dialysis and comparative spectrofluorimetry studies.63,64 The striking result obtained from the study was the establishment of a poly(A) structure as the most preferred conformation for palmatine binding, followed by the double-stranded DNA and tRNA structures, respectively. Hypochromic and bathochromic effects were observed in the absorption spectrum of palmatine on complexation with poly(A) with significant enhancement of the rather small fluorescence intensity of palmatine, increase in relative specific viscosity and perturbation of circular dichroic spectrum of poly(A), with concomitant generation of induced circular dichroic bands for the bound hitherto achiral palmatine molecules.63,64 The binding of the alkaloid to poly(A) was non-cooperative and the binding constant evaluated by the analysis of the excluded site model of McGhee–von Hippel (1974) was of the order of ∼105 M−1 in 10 mM [Na+] concentration.64,65 Thus compared to the affinity of berberine, palmatine had one order lower affinity to poly(A). This may be attributed to the structural differences of the two alkaloids. The effect of [Na+] ion concentration on the binding process revealed the significant role of electrostatic forces in the complexation. Both van’t Hoff’s analysis of the temperature dependent binding constants and isothermal titration calorimetry were applied to reveal the energetics of the binding and a good correlation was obtained between them. The binding was revealed to be exothermic and enthalpy driven, similar to that of berberine. The thermodynamic parameters describing the binding reaction of palmatine to poly(A) may have contributions from the molecular interactions between the bound ligand and nucleic acid binding sites as a result of hydrogen bonding and hydrophobic interactions, arising from the conformational changes in either the nucleic acid or drug upon binding and or from processes such as ion release, proton transfer or change in the water structure. A partial intercalative binding mode of the alkaloid to poly(A) was suggested from viscosity measurements.64 Although palmatine has also a crescent shaped structure like coralyne and berberine, no self-structure induction in poly(A) has been reported.
The benzophenanthridine alkaloidsanguinarine [13-methylbenzodioxolo{5,6-c}-1,3-dioxolo{4,5-i}phenanthridinium] (Fig. 2D) is a phototoxic alkaloid derived from the root of Sanguinaria canadensis/Argemone mexicana and other poppy-fumaria species with a long history of use world-wide in folk medicine. Sanguinarine exhibits diverse pharmacological, photochemical and photobiological activities. It has potent anti-microbial, anti-inflammatory, antioxidant and anticancer activities and generates singlet oxygen.57,66–71 Recent results also confirmed the antiproliforative effect of this alkaloid against cancer cell lines .72,73 Again, studies correlate the cytotoxicity of sanguinarine to DNA damaging effects such as DNA intercalation, DNA strand breaks etc.74–77Sanguinarine has been reported to bind with various nucleic acid structures78–85 though the first report on the interaction of sanguinarine with single poly(A) structures have emerged recently from our laboratory. The high specificity of this alkaloid to poly(A) was confirmed through competition dialysisassay and fluorescence titration experiments.86 The binding of sanguinarine, examined by absorption spectral titration, revealed hypochromic and bathochromic shifts with an isosbestic point indicating equilibrium binding (Fig. 5A). The binding constant estimated from the Scatchard analysis of the titration data employing the excluded site model was (3.60 ± 0.24) × 106 M−1 and this strong binding was proposed to be due to intercalation. The binding stoichiometry evaluated from the Job plot was four nucleotides per bound sanguinarine. Circular dichroic spectral results revealed that the conformation of poly(A) was remarkably altered by the binding of sanguinarine. The large decrease in the 260, 250 and 210 nm bands of poly(A) circular dichroism with the concomitant development of strong conservative induced circular dichroic bands in the 300–400 nm region (Fig. 5B) for the bound hitherto optically inactive sanguinarine molecules clearly suggested the alteration of the poly(A) secondary structure on binding of sanguinarine on the one hand and the strong asymmetric environment of the bound sanguinarine in the helical organization of poly(A) on the other, presumably by intercalative binding. The circular dichroic melting profile (Fig. 5C) monitored at 274 nm was cooperative in nature with a melting temperature of ∼60 °C indicating the formation of self-structured poly(A) induced by the alkaloid.86 Further evidence on the self-structure induction in poly(A) by sanguinarine was obtained by ultraviolet melting experiments, where the melting transition in poly(A) alone was very broad while that of the complex was cooperative with a melting temperature of about 60 °C (inset, Fig. 5A), consistent with the circular dichroic melting data. The strongest evidence of the intercalation of the alkaloid to adenine–adeninebase pairs was obtained from fluorescence quenching experiments using potassium iodide as an anionic quencher , where free sanguinarine was quenched substantially but the bound drug molecules were completely protected from the quencher , revealing the true intercalation in self-structured poly(A). The energetics of the binding process was determined from isothermal titration calorimetry and differential scanning calorimetry. The ITCtitration profile revealed the binding as exothermic in nature (ΔH° = −14.40 kcal mol−1) with a binding affinity of 4.6 × 106 M−1, in excellent agreement with the spectroscopic result. The binding appeared to be enthalpy driven with a significant negative TΔS° value (−5.45 kcal mol−1) that can be rationalized due to the formation of ordered self-structure and the intercalation of sanguinarine, and the consequent loss of translational and rotational degrees of freedom. The large negative binding enthalpy is generally typical for intercalative interaction of small molecules to helical nucleic acid structures.54–56 The DSC melting profile (Fig. 5D) of the sanguinarine–poly(A) complex further reiterated the self-structure induction. In the presence of the alkaloid, the melting of poly(A) became cooperative and fully reversible with a cooperativity factor of unity (ΔHv = 24.02 ± 1.50 kcal mol−1, ΔHcal = 24.27 ± 1.23 kcal mol−1). Similar energetics were also obtained in the melting profile of the poly(dA)–sanguinarine complex indicating a similar type of anti-parallel duplex structure formation by sanguinarine (Giri and Kumar, unpublished). However, a parallel strand arrangement cannot be ruled out and high-resolution additional structural studies are required for unequivocal conformational assignment and the polarity of the strands of the duplex. It is pertinent to observe that this type of self-structure was obtained on interaction of coralyne with single-stranded poly(A) (vide supra).19 It is noteworthy that sanguinarine induced self-structure formation in poly(A) was the first report of the specific molecular recognition of poly(A) through self-structure formation by a natural alkaloid.
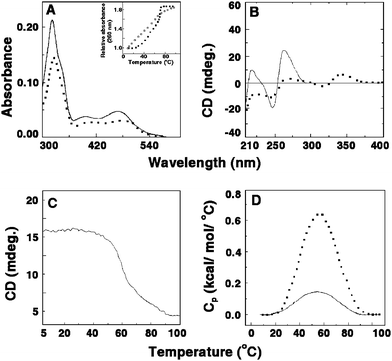 |
| | Fig. 5 (A) Representative absorption spectral changes of sanguinarine in the absence (solid curve) and presence (dashed curve) of poly(A). Inset: UV melting profile of poly(A) (□) and poly(A)–sanguinarine complex (■). (B) Circular dichroic spectra of single-stranded poly(A) in the absence (solid curve) and presence (dashed curve) of sanguinarine. (C) Circular dichroic melting profile of a solution containing 55 μM of poly(A) and 13.75 μM of sanguinarine monitored at 274 nm. (D) DSC thermograms of poly(A) (solid curve) and the complex of poly(A) and sanguinarine (dashed curve). (Reprinted from Giri and Kumar,78Biochim. Biophys. Acta, 2007, 1770, 1419–1426, © 2007, with permission from Elsevier). | |
Conclusion and perspectives
Protoberberine alkaloids represent an interesting group of the abundant natural products that exhibit remarkably significant and diverse biological activities. In order to exploit these alkaloids as futuristic therapeutic agents, their ability to modulate nucleic acid structure–function must be understood in detail. The DNA binding of these alkaloids has been studied for several years and more recently their RNA binding potential is getting unravelled. Coralyne was the first molecule discovered to induce self-structure in single-stranded poly(A). The natural analogues, berberine and palmatine could not induce similar effects as tested by circular dichroic melting and other experiments although a different indirect technique involving the dilution/concentration of ligand–poly(A) complexes below or above a critical concentration suggested self-structure formation with berberine also. On the other hand, sanguinarine easily induced self-structure formation in poly(A). All of these alkaloids have more or less similar crescent shaped structures. Although the differences in planarity was thought to be the primary cause to account for this, subsequent studies with a number of molecules have suggested more specific features to be responsible for this. Apparently, an interplay of size, shape and planarity inter alia appears to be critical features for self-structure induction. Of the protoberberinealkaloids discussed in this review, coralyne and sanguinarine have been reported to induce self-structure in poly(A) as revealed from cooperative melting in absorbance, CD and DSC. The recent observation of Hud with berberine suggests that planarity may not be a critical or dominating factor compared to the other factors discussed above. A summary showing different aspects of the interaction of the alkaloids with poly(A) structure through diverse biophysical tools is presented in Table 2. Additional structural studies with more derivatives of these alkaloids are required for a complete understanding of the mode and mechanism of this intriguing structural transition in poly(A). Rational drug design based on nucleic acid targeting has become an emerging area in medicinal chemistry. Single stranded poly(A) tails are the most important determinant in eukaryotic cells for mRNA maturation, stability and alternate protein production. Self-structure formation is very significant with respect to its biological importance. The possibility of the formation of double helical poly(A) in a cell can allow elucidation of the mechanisms of several biological processes that proceed with the participation of poly(A) sequences viz. the termination of mRNA polyadenylation, autoregulation of poly(A) bindingprotein I (PAB I) and stabilization of AU rich regions containing mRNA by embryonic lethal abnormal visual (ELAV)-like proteins.87 The rules governing the molecular recognition of double-stranded RNA have drawn increasing attention due to some important biological processes such as RNA interference (RNAi), the interferon -controlled antiviral response and pre mRNA editing.88–91 So the molecules that can specifically target the poly(A) tail can be promising leads for the design of new compounds that might recognize single-stranded nucleic acids for the development of therapeutic agents by medicinal chemists.92 The binding of small molecules to poly(A) is clearly different from the binding of small molecules to pre-structured nucleic acids such as double-stranded DNA or tRNA. The plant based alkaloids considered in this review are important in modulating nucleic acid structure, in understanding the structure–function relationship in nucleic acids, leading to the futuristic development of anti-viral as well as anti-cancer drugs.
Table 2 Comparative summary of different alkaloids on single-stranded polyadenylic acida
| Methods |
Systems |
|
Coralyne–poly(A) |
Berberine–poly(A) |
Palmatine–poly(A) |
Sanguinarine–poly(A) |
|
For references see 19, 39, 42, 63, 64 and 86.
NA = Data not available.
|
| Mode of interaction (nature of binding) |
Non-covalent binding (cooperative) |
Non-covalent binding (non-cooperative) |
Non-covalent binding (non-cooperative) |
Non-covalent binding (cooperative) |
| |
| Method of determination |
CD melting |
T
m studies |
DSC melting |
Dilution/concentration measurement |
CD melting |
T
m studies |
DSC melting |
Dilution/concentration measurement |
CD melting |
T
m studies |
DSC melting |
Dilution/concentration measurement |
CD melting |
T
m studies |
DSC melting |
Dilution/concentration measurement |
| Induction of self-structure |
Yes |
Yes |
Yes |
Yes |
No |
No |
No |
Yes |
No |
No |
No |
NAb |
Yes |
Yes |
Yes |
NAb |
| |
| Number of drugs/targets |
4 |
6 |
3 |
4 |
Acknowledgements
Support from the Council of Scientific and Industrial Research (CSIR), Government of India through the network project on “Comparative genomics and biology of non-coding RNA in human genome” (NWP0036) is gratefully acknowledged. We thank Prof. Siddhartha Roy, Director, IICB for his patronage and all the pre- and post-doctoral co-workers of the Biophysical Chemistry Laboratory who have contributed at different times to the work on the interaction of alkaloids with poly(A). P. G. would like to take this opportunity to pay his sincere respect to his parents for their constant support and encouragement during his post-doctoral research activity.
References
-
W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1984 Search PubMed.
- W. D. Wilson and K. Li, Curr. Med. Chem., 2000, 7, 73–98 CAS.
- J. Gallego and G. Varani, Acc. Chem. Res., 2001, 34, 836–843 CrossRef CAS.
- Y. Tor, ChemBioChem, 2003, 4, 998–1007 CrossRef CAS.
- Q. Vicens and E. Westhof, ChemBioChem, 2003, 4, 1018–1023 CrossRef CAS.
- D. Munroe and A. Jacabson, Mol. Cell. Biol., 1990, 10, 3441–3455 CAS.
- C. J. Decker and R. Parker, Genes Dev., 1993, 7, 1632–1643 CrossRef CAS.
- Z. Wang, N. Day, P. Trifillis and M. Kiledjian, Mol. Cell. Biol., 1999, 19, 4552–4560 CAS.
- Z. Chen, Y. Li and R. M. Krug, EMBO J., 1999, 18, 2273–2283 CrossRef CAS.
- B. Tian, J. Hu, H. Zhang and C. S. Lutz, Nucleic Acids Res., 2005, 33, 201–212 CrossRef CAS.
- A. W. B. Craig, A. Haghighat, A. T. K. Yu and N. Sonenberg, Nature, 1998, 392, 520–523 CrossRef CAS.
- T. Maniatis and R. Reed, Nature, 2002, 416, 499–506 CrossRef CAS.
- K. M. Neugebauer, J. Cell Sci., 2002, 115, 3865–3871 CrossRef CAS.
- O. Calvo and J. L. Manley, Genes Dev., 2003, 17, 1321–1327 CrossRef CAS.
- N. Proudfoot, Curr. Opin. Cell Biol., 2004, 16, 272–278 CrossRef CAS.
- M Fuke and M. Ohno, Nucleic Acids Res., 2007, 36, 1037–1049 CrossRef.
- S. L. Topalian, S. Kaneko, M. I. Gonzales, G. Bond, Y. Ward and J. L. Manley, Mol. Cell. Biol., 2001, 21, 5614–5623 CrossRef CAS.
- S. L. Topalian, M. I. Gonzales, Y. Ward, X. Wang and R. F. Wang, Cancer Res., 2002, 62, 5505–5509 CAS.
- F. Xing, G. Song, J. Ren and J. B. Chaires, FEBS Lett., 2005, 579, 5035–5039 CrossRef CAS.
- J. R. Fresco and P. Doty, J. Am. Chem. Soc., 1957, 79, 3928–3929 CrossRef CAS.
- D. N. Holcomb and I. Tinoco, Jr, Biopolymers, 1965, 3, 121–133 CrossRef CAS.
- M. Leng and G. Felsenfeld, J. Mol. Biol., 1966, 15, 455–466 CrossRef CAS.
- A. Petrovic and P. L. Polavarapu, J. Phys. Chem. B, 2005, 109, 23698–23705 CrossRef CAS.
- T. Schmeller, B. L. Bruning and M. Wink, Phytochemistry, 1997, 44, 257–266 CrossRef CAS.
- W. H. Chen, C. L. Chan, Z. Cai, G. A. Luo and Z. H. Jiang, Bioorg. Med. Chem. Lett., 2004, 14, 4955–4959 CrossRef CAS.
- R. J. Upton, The Journal of Alternative and Complementary Medicine, 2001, 7, 15 Search PubMed.
- L. Mambu, V. Ramanandraibe, M. T. Martin, A. Blond and F. Frappier, Planta Med., 2002, 68, 377–378 CrossRef CAS.
- J. Dostal and J. Slavik, Chem Listy, 2000, 94, 15–20.
- M. Chrzanowska and M. D. Rozwadowska, Chem. Rev., 2004, 104, 3341–3370 CrossRef CAS.
- A. Zdarilová, J. Malíkova, Z. Dvorak, J. Ulrichova and V. Simanek, Chem Listy, 2006, 100, 30–41 CAS.
- L. Grycová, J. Dostal and R. Marek, Phytochemistry, 2007, 68, 150–175 CrossRef CAS.
-
M. Wink, in Molecular Modes of Action of Cytotoxic Alkaloids: From DNA Intercalation, Spindle Poisoning, Topoisomerase Inhibition to Apoptosis and Multiple Drug Resistance, ed. G. A. Cordell, Elsevier Science, New York, The Alkaloids: Chemistry and Biology, 2007, vol. 64, pp. 1–47 Search PubMed.
- D. S. Pilch, C. Yu, D. Makhey, E. J. Lavoie, A. K. Srinivasan, W. K. Olson, R. R. Sauers, K. J. Breslauer, N. E. Geacintov and L. F. Liu, Biochemistry, 1997, 36, 12542–12553 CrossRef CAS.
- K. Y. Zee-Cheng and C. C. Cheng, J. Pharm. Sci., 1973, 62, 1572–1573 CrossRef CAS.
- R. S. Gupta, W. Murray and R. Gupta, Br. J. Cancer, 1988, 58, 441–447 CAS.
- M. Gatto, M. M. Sanders, C. Yu, H. Y. Yu, D. Makhey, E. J. LaVoie and L. F. Liu, Cancer Res., 1996, 56, 2795–2800 CAS.
- S. Pal, G. S. Kumar, D. Debnath and M. Maiti, Indian J. Biochem. Biophys., 1998, 35, 321–332 Search PubMed.
- B. Garcia, S. Ibeas, R. Ruiz, J. M. Leal, T. Biver, A. Boggioni, F. Secco and M. Venturini, J. Phys. Chem. B, 2009, 113, 188–196 CrossRef CAS.
- P. Giri and G. S. Kumar, Arch. Biochem. Biophys., 2008, 474, 183–192 CrossRef CAS.
- O. Persil, C. T. Santai, S. S. Jain and N. V. Hud, J. Am. Chem. Soc., 2004, 126, 8644–8645 CrossRef CAS.
- N. Micali, V. Villari, M. Cuusumano, M. L. Di Pietro and A. Giannetto, J. Phys. Chem. B, 2007, 111, 1231–1237 CrossRef CAS.
- O. P. Centikol and N. V. Hud, Nucleic Acids Res., 2009, 37, 611–621.
- N. Ivanovska and S. Philipov, Int. J. Immunopharmacol., 1996, 18, 553–561 CrossRef.
- C. L. Kuo, C. C. Chou and B. Y. M. Yung, Cancer Lett., 1995, 93, 193–200 CrossRef CAS.
- H. L. Wu, C. Y. Hsu, W. H. Liu and B. Y. M. Yung, Int. J. Cancer, 1999, 81, 923–929 CrossRef CAS.
-
M. Sufness and G. A. Cordell, The Alkaloids, Academic Press, Orlando, 1985, p. 25 Search PubMed.
- D. Debnath, G. S. Kumar, R. Nandi and M. Maiti, Indian J. Biochem. Biophys., 1989, 26, 201–208 Search PubMed.
- D. Debnath, G. S. Kumar and M. Maiti, J. Biomol. Struct. Dyn., 1991, 9, 61–79 CAS.
- A. Saran, S. Srivastava, E. Coutinho and M. Maiti, Indian J. Biochem. Biophys., 1995, 32, 74–77 Search PubMed.
- G. S. Kumar, S. Das, K. Bhadra and M. Maiti, Bioorg. Med. Chem., 2003, 11, 4861–4870 CrossRef CAS.
- R. Nandi, D. Debnath and M. Maiti, Biochim. Biophys. Acta, Gene Struct. Expression, 1990, 1049, 339–342 CrossRef CAS.
- R. C. Yadav, G. S. Kumar, K. Bhadra, P. Giri, R. Sinha, S. Pal and M. Maiti, Bioorg. Med. Chem., 2005, 13, 165–174 CrossRef CAS.
- J. Ren and J. B. Chaires, Biochemistry, 1999, 38, 16067–16075 CrossRef CAS.
- I. Haq, Arch. Biochem. Biophys., 2002, 403, 1–15 CrossRef CAS.
- J. B. Chaires, Arch. Biochem. Biophys., 2006, 453, 26–31 CrossRef CAS.
- J. B. Chaires, Annu. Rev. Biophys., 2008, 37, 135–151 Search PubMed.
- M. L. Sethi, J. Pharm. Sci., 1983, 72, 538–541 CrossRef CAS.
- Y. L. Chang, S. Usami, M. T. Hsieh and M. J. Jiang, Life Sci., 1999, 64, 597–606 CrossRef CAS.
- E. Küzpeli, M. Kosar, E. Yesilada, K. H. C. Baser and C. Baser, Life Sci., 2002, 72, 645–657 CrossRef CAS.
- A. Volleková, D. Kostalova, V. Kettmann and J. Toth, Phytother. Res., 2003, 17, 834–837 CrossRef CAS.
- F. Wang, H. Y. Zhou, L. Cheng, G. Zhao, J. Zhou, L. Y. Fu and W. X. Yao, World J. Gasteroenterol., 2003, 9, 329–333 Search PubMed.
- K. Bhadra, M. Maiti and G. S. Kumar, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 1071–1080 Search PubMed.
- P. Giri, M. Hossain and G. S. Kumar, Bioorg. Med. Chem. Lett., 2006, 16, 2364–2368 CrossRef CAS.
- P. Giri, M. Hossain and G. S. Kumar, Int. J. Biol. Macromol., 2006, 39, 210–221 CrossRef CAS.
- J. D. McGhee and P. H. von Hippel, J. Mol. Biol., 1974, 86, 469–484 CrossRef CAS.
- J. Lenfeld, M. Kroutil, E. Marsalek, V. Preininger and V. Simanek, Planta Med., 1981, 43, 161–165 CrossRef CAS.
-
V. Simanek, in The Alkaloids, ed. A. Brossi, Academic Press Inc., London, 1985, pp. 185–240 Search PubMed.
- K. C. Godowski, J. Clin. Dent., 1989, 1, 96–101 Search PubMed.
- J. T. Arnason, B. Guerin, M. M. Kraml, B. Mehta, R. W. Redmond and J. C. Scaiano, Photochem. Photobiol., 1992, 55, 35–38 CrossRef CAS.
- M. Maiti and A. Chatterjee, Curr. Sci., 1995, 68, 734–736 CAS.
- V. M. Adhami, M. H. Aziz, S. R. Reagan-Shaw, M. Nihal, H. Mukhtar and N. Ahmad, Mol. Cancer Ther., 2004, 3, 933–940 CAS.
- N. Ahmad, S. Gupta, M. M. Husain, K. M. Heiskanen and H. Mukhtar, Clin. Cancer Res., 2000, 6, 1524–1528 CAS.
- A. Vogt, A. Tamewitz, J. Skoko, R. P. Sikorski, K. A. Giuliano and J. S. Lazo, J. Biol. Chem., 2005, 280, 19078–19086 CrossRef CAS.
- V. O. Kaminskyy, M. D. Lootsik and R. S. Stoika, Central Eur. J. Biol., 2006, 1, 2–15 Search PubMed.
- N. P. S. Bajaj, M. J. McLean and M. J. Waring, J. Mol. Recognit., 1990, 3, 48–54 CAS.
- A. Sen and M. Maiti, Biochem. Pharmacol., 1994, 48, 2097–2102 CrossRef CAS.
- J. Urbanová, P. Lubal, I. Slaninova, E. Taborska and P. Taborsky, Anal. Bioanal. Chem., 2009, 394, 997 CrossRef CAS.
- M. Maiti and R. Nandi, J.
Biomol. Struct. Dyn., 1987, 5, 159–175 CAS.
- A. Sen, A. Ray and M. Maiti, Biophys. Chem., 1996, 59, 155–170 CrossRef CAS.
- S. Das, A. Banerjee, A. Sen and M. Maiti, Curr. Sci., 2000, 79, 82–87 CAS.
- M. Maiti, Indian J. Biochem. Biophys., 2001, 38, 20–26 Search PubMed.
- M. Vlckova, V. Kuban, J. Vicar and V. Simanek, Electrophoresis, 2005, 26, 1673–1679 CrossRef.
- L. P. Bai, Z. Z. Zhao, Z. Cai and Z. H. Jiang, Bioorg. Med. Chem., 2006, 14, 5439–5445 CrossRef CAS.
- M. Hossain and G. S. Kumar, J. Chem. Thermodyn., 2009, 41, 764–774 CrossRef CAS.
- L. P. Bai, M. Hagihara, Z. H. Jiang and K. Nakatani, ChemBioChem, 2008, 9, 2583–2587 CrossRef CAS.
- P. Giri and G. S. Kumar, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 1419–1426 Search PubMed.
- M. I. Zarudnaya and D. M. Hovorun, IUBMB Life, 1999, 48, 581–584 CAS.
- D. M. Dykxhoorn, C. D. Novina and P. A. Sharp, Nat. Rev. Mol. Cell Biol., 2003, 4, 457–467 CrossRef CAS.
- G. Meister and T. Tuschl, Nature, 2004, 431, 343–349 CrossRef CAS.
- C. C. Mello and D. Conte, Jr, Nature, 2004, 431, 338–342 CrossRef CAS.
- J. M. E. Withey and M. R. Crompton, Curr. Cancer Ther. Rev., 2005, 1, 11–17 Search PubMed.
- P. Giri and G. S. Kumar, Curr. Med. Chem., 2009, 16, 965–987 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |
Click here to see how this site uses Cookies. View our privacy policy here.